
Abstract
The maintenance of viable and functional islets is critical in successful pancreatic islet transplantation from cadaveric sources. During the isolation procedure, islets are exposed to a number of insults including ischemia, oxidative stress and cytokine injury that cause a reduction in the recovered viable islet mass. A novel approach was designed in which streptozotocin (STZ)-damaged rat pancreatic islets (rPIs) were indirectly cocultured with rat bone marrow-derived mesenchymal stem cells (rBM-MSCs) to maintain survival of the cultured rPIs. The results indicated that islets cocultured with rBM-MSCs secreted an increased level of insulin after 14 days, whereas non-cocultured islets gradually deteriorated and cell death occurred. The cocultivation of rBM-MSCs with islets and STZ-damaged islets showed the expression of IL6 and transforming growth factor-β1 in the culture medium, besides the expression of the antiapoptotic genes (Mapkapk2, Tnip1 and Bcl3), implying the cytoprotective, anti-inflammatory and antiapoptotic effects of rBM-SCs through paracrine actions.
Similar content being viewed by others


Main
Pancreatic islet transplantation has the potential to replace insulin-secreting β-cell mass and restore insulin-regulated glucose homeostasis in patients with type 1 diabetes mellitus. Despite the recent developments in the isolation of pancreatic islets, only a part of the total islet can be successfully recovered by the present protocols.1 Even more, a functionality loss was detected in more than a half of these isolated islets.2 To be successful in the β-cell islet transplantation, many problems associated with maintenance of viable, functional islets isolated from cadaveric sources should be solved. The isolation of islets is performed through mechanically enhanced enzymatic digestion of the pancreas, which dissociates the islets from the surrounding acinar and ductal tissues. During this process, islets are exposed to a number of insults including ischemia, oxidative stress and cytokine injury that may result in cellular damage and functional impairment, which ultimately leads to a reduction in the viable islet mass recovered.3 Therefore, different strategies were investigated for improving the results of experimental and clinical islet isolation.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Among these attempts, the antiapoptotic and protective effects of some compounds, functioning as cytoprotectives, such as nicotinamide,3 lisofylline,5 exendine-4,6 interleukin (IL)4,10 prolactin12 and protein kinase C epsilon,13 were examined on islet cells in vivo and/or in vitro. In other studies, many approaches of cellular application were used.4, 7, 9, 11, 14, 15, 16
The first study in promoting the survival and function of islet β-cells in tissue culture was realized by Rabinovitch et al.4 They cocultured (direct) islet cells from newborn rat pancreases with fibroblasts from a mouse embryonic cell line (3T3-L2). They concluded that fibroblastic cells produce a macromolecular factor(s) capable of enhancing the survival of functional islet β-cells in tissue culture.4 Similar approaches were tested using pancreatic ductal cells (DECs),16 MNNK-1 cells,7 bone marrow (BM) mononuclear cells9 and rat pancreatic extracts (RPEs)15 on the viability and function of pancreatic islets. In all these studies, the islets were directly cocultured with the cell components. As a result, cells, extracts and even stem cells could have a promising effect on the protection of islets from damage during culture.
Here, we designed a novel approach to maintain the viability and functionality of the cultured rat pancreatic islets (rPIs) by indirect coculturing of streptozotocin (STZ) injured and untreated rPIs with rat bone marrow-derived mesenchymal stem cells (rBM-MSCs). The rBM-MSCs in the coculture islets maintained their viability and functionality as shown by the increased level of insulin secretion in comparison with monocultures. A gradual destruction was observed in monocultured islets, especially in those injured with STZ. Ultimately, cell death occurred after 14 days of incubation.
Results
Light microscopy of cultured islets
Examinations of cultured islets under an inverted microscope showed that they displayed a regular cellular structure (Figure 1a). There were no disintegrated islets.

Morphology of PIs and rBM-MSCs. (a) Free-floating rat islets (arrows). (b) Rat islets stained with DTZ for specificity. (c) During the onset of growth (P0-13th day), isolated cells from the bone marrow formed single-cell-derived colonies (arrows). In later passages, most of these MSCs exhibited large, flattened or fibroblast-like morphology (P1-12th day) (d) (Original magnification: a, c × 40; b, d × 200)
Staining of islets with dithizone
After cultivation, some islets were immediately separated and treated with dithizone (DTZ). All the islets stained with DTZ became crimson red. No other cell types were observed with the islets, indicating that only islets were present in the preparation (Figure 1b).
Cell culture of rBM-MSC
MSCs attached to the culture flasks sparsely and a majority of the cells displayed a fibroblast-like, spindle-shaped morphology during the early days of incubation. These cells began to proliferate after 3–4 days, and gradually grew to form small colonies that were termed colony-forming units (Figure 1c). By days 6–8, the number of cellular colonies with different sizes had increased. As growth continued, colonies gradually expanded in size and the adjacent ones interconnected with each other. These primary cells reached monolayer confluence after plating for 12–15 days in their first passages. In later passages, most of these MSCs exhibited large, flattened or fibroblast-like morphology (Figure 1d). No changes in morphology or growth pattern were observed throughout 25 passages.
Flow cytometry: identification of rBM-MSCs
Both previous and present data indicated that rBM-MSCs expressed CD29 and CD90, but not CD45 (data not shown).17 rBM-MSCs maintained their phenotype throughout passages. Therefore, there was no evidence of hematopoietic precursors in the cultures. In contrast, the expression of other surface antigens agreed with the previous reports regarding murine MSCs, and indicated that the cells used in this study had the characteristics of MSCs reported in other studies.17, 18, 19
Immunocytochemistry: identification of rBM-MSCs
Immunohistochemical studies were performed to characterize the progeny of the rBM-MSCs by using a large panel of antibodies specific to known antigens associated with different phenotypes. The immunocytochemical staining results are shown in Supplementary Figures 1 and 2, and Supplementary Table 1.
The antigenic phenotype of MSCs is not unique and no single marker has been found to be specific for MSCs. However, in the past, certain antigens such as surface and cytoplasmic proteins have been used in attempts to characterize MSCs. Expression of some of these antigens was also examined for the rBM-MCSs in this study. Under standard culture conditions, α-smooth muscle actin (Acta2), actin-β (Actb), osteopontin (secreted phosphoprotein 1, Spp1), glial fibrillary acidic protein (GFAP), FBJ osteosarcoma oncogene (Fos), CD45, osteocalcin, osteonectin (secreted acidic cysteine-rich glycoprotein, Sparc), myosine IIa (Supplementary Figure 1), β-tubulin (Tubb3), nestin (Nes), fibronectin (FN) and BMP2 (Supplementary Figure 2), vimentin (Vim), desmin (Des), bone morphogenetic protein 4 (BMP4), myogenin (Myog), type I and II collagen, and CD105 (data not shown) were expressed. rBM-MSCs of this study did not express some surface markers including CD31 (endothelial) (Supplementary Figure 2C), CD34 (data not shown), CD45 (hematopoietic markers) (Supplementary Figure 1F) and CD71 (Supplementary Figure 2D).
Gene expression profiles of rBM-MSCs
Supplementary Table 2 summarizes the results of the RT-PCR analysis of rBM-MSCs. The expressions of some of the key transcripts for embryonic stem cell genes (Pou5f1, Zfp42) indicated the self-renewal capacity of rBM-MSCs. The cells were also found to be positive for Actb, glyceraldehyde 3-phosphate dehydrogenase (Gapdh), Vim, Acta2, Des, Myog, Nes, Gfap, Eno2, Tubb3, Sparc, Bmp4 and Spp1, showing that their expression is the indication of the differentiation to other tissues (Figure 2).

Agarose gel electrophoresis of RT-PCR products. Representative panel of RT-PCR analysis of rBM-MSCs. Internal control: glyceraldehyde 3-phosphate dehydrogenase (Gapdh). Negative control: PCR mix without template
The expression of myogenic (Des, Myog, MyosinIIa, Acta2), neurogenic (enolase-γ (Eno2), MAP2a,b, Fos, Nes, Gfap, Tubb3) and osteogenic (Sparc, Osteocalcin, Spp1, BMP2, BMP4, type I Collagen) markers by BM-MSCs without induction is the explanation for the possible reason of the fact that these cells can readily differentiate into functional multiple lineages both in vitro and in vivo.
It was also shown that rBM-MSCs expressed myogenic, neurogenic and osteogenic genes under basal conditions designating the diverse immunophenotypic characteristics of MSCs that might explain the reason why these cells could easily differentiate into cells of bone, cartilage, muscle and neural tissues. Consistently, the expression of precursor markers of mesoderm and ectoderm by MSCs indicates that they could differentiate into cells of three germ layers under permissive in vitro conditions.
MSCs were investigated for the presence of molecules involved in immunomodulation or suppression. Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2) is related to kinases that mediate a wide range of biological functions in response to mitogens and stress stimuli. Tnip1 (TNFAIP3 (tumor necrosis factor, α-induced protein 3)-interacting protein 1), which is previously known as A20, is another antiapoptotic gene (stress gene) like Mapkapk2 expressed by both cell lines (Supplementary Table 2, Figure 2). In addition, rBM-MSCs expressed Bcl3 (B-cell leukemia) (antiapoptotic gene) and interleukin 6 receptor-α (IL6ra) but failed to express IL10 and interferon-γ (Ifng) (Supplementary Table 2, Figure 2). IL6ra, also known as CD126, is a type I cytokine receptor. Interleukin 6 (IL6) is a potent pleiotropic cytokine that regulates cell growth and differentiation and has an important role in immune response.
Differentiation potential of rBM-MSCs
Adipogenic differentiation
rBM-MSCs at passage 3 (P3) were used in the adipogenic differentiation experiments, in which 80–90% sub-confluence was found to be optimal for cell differentiation that lasted for 2–3 weeks. At the beginning of the third week, lipid droplets enlarged and invaded the entire cytoplasm of the adipocyte-differentiated rBM-MSCs (Supplementary Figure 3).
Osteogenic differentiation
Histo- and/or immunohistochemical techniques for the detection of bone-specific markers were applied to cells grown in osteogenic differentiation medium and in control medium to assess the osteogenic differentiation of rBM-MSCs. Proliferation and differentiation of both cultures were compared using phase contrast microscopy. In both control and osteogenic cultures, cells proliferated and reached almost complete confluency after 8–10 days of incubation. Later, the cellular aggregates were observed in osteogenic differentiation culture plates and gradually increased until the completion of the experiment. These aggregates were characterized by the presence of amorphous material deposits (Supplementary Figure 4A). The staining of these nodular aggregates in osteogenic cultures by Alizarin red S showed that the amorphous deposits observed at the microscope were actually calcium deposits. Alizarin red positive nodular aggregates were already present at day 15 (Supplementary Figure 4B) and control cultures showed only minimal background staining (Supplementary Figure 4D). Cells that were incubated for 15 days were also strongly stained positive for BMP4 (Supplementary Figure 4C), osteopontin (Supplementary Figure 4C), osteocalcin (data not shown) and extracellular located matrix proteins BMP2 (data not shown).
Antiapoptosis ability of rBM-MSCs on STZ-induced pancreatic islets
After coculture experiments, quantitative analysis of apoptosis was conducted by FACS (AnnexinV/PI) and microscopic evaluations on the islets stained with fluorescein diacetate (FDA)/propidium iodide (PI).
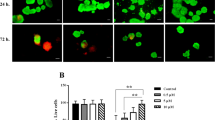
For determination of cell viability in normal and STZ-induced pancreatic islets cocultured with rBM-MSCs, FDA/PI staining was performed. In Figure 3, the images of rPIs cultured under different conditions are given. Dead cells stained red and viable cells stained green. The percentage viability of islet cells was calculated as described in the Materials and Methods section. As shown in Figure 4c, STZ induced a significant decrease in the viability rates of islets compared with untreated islets (10%±2.8, 60%±2.81, respectively; P<0.001). In contrast, coculture with rBM-MSCs induced a significant increase in viability rates in STZ-induced islets (72.5%±4.01; P<0.001).

Cell viability. For determination of cell viability in normal (a, b) and STZ-induced pancreatic islets cocultured with rBM-MSCs (c, d), PI/FDA staining was performed. The small bright spots in the fluorescent micrographs correspond to the intense red staining of PI-positive (dead) cells. The diffusely stained regions correspond to the FDA-stained (live) cells (Scale bars=50 μm)

Detection of apoptosis. (a) Apoptosis was quantified by FACS analysis after staining with Annexin V and PI at the end of the experiment (day 14) (islets were incubated with trypsin to obtain dispersed cells). Viable cells were Annexin V−/PI− (III), early apoptotic cells were Annexin V+/PI− (IV), late apoptotic cells were Annexin V+/PI+ (II) and necrotic cells were Annexin V−/PI+ (I). Representative examples are shown in b. (c) We also performed PI/FDA staining for determination of cell viability in normal and STZ-induced pancreatic islets cocultured with rBM-MSCs and calculated as described in Methods. STZ induced a significant decrease in the viability rates in islets compared with untreated islets (10%±2.8, 60%±2.81, respectively). Coculture with rBM-MSCs induced a significant increase in viability rates in STZ-induced islets (72.5%±4.01; P<0.001). (mean±S.D., n=3 each, **P<0.01, ***P<0.001)
Cell apoptosis was measured by Annexin V-FITC, which binds to phosphatidylserine residues that are redistributed from the inner to the outer leaflet of the cell membrane as an early event in apoptosis. After loss of membrane integrity, PI can enter the cell and intercalate into DNA.20 Figure 4a and b shows the percentages of Annexin V-PI-stained cells of islets. The average percentages of Annexin V+/PI+ (late apoptotic cells) were highest in STZ-induced islets (67.34%±1.8) and the percentage in islets cocultured with rBM-MSCs was the lowest (19.3%±1.4). Moreover, there was a statistical significance between STZ-induced islets and STZ-induced islets cocultured with rBM-MSCs (52.26%±1.04) (P<0.01) as well as the rate of Annexin V−/PI− (viable cells), Annexin V+/PI− (early apoptotic cells) and Annexin V−/PI+ (necrotic cells) (data not shown). Therefore, this suggested that islets cocultured with rBM-MSCs had a superior tolerance to STZ-induced cytotoxicity.
Insulin content
The content of total cellular insulin in experimental groups was evaluated and it was found that STZ-induced islets showed a significant decrease in cellular insulin content in comparison to the untreated islets when induced with L-DMEM and H-DMEM (3.38±0.79 ng/mg protein and 9.72±0.68 ng/mg protein in L-DMEM, and 5.25±1.43 ng/mg protein and 10.51±0.91 ng/mg protein in H-DMEM, with respect to STZ-induced islets and untreated islets; P<0.01) (Figure 5). The proportion of insulin release to insulin content in STZ-induced islets cocultured with rBM-MSCs was estimated to be approximately 9.77±0.66 ng/mg protein in L-DMEM and 10.53±0.64 ng/mg protein in H-DMEM (Figure 5).

Insulin release in response to glucose stimulation of islets in different groups. On day 14, normal islets, islets cocultured with rBM-MCSs, STZ-induced islets and STZ-induced islets cocultured with rBM-MSCs secreted insulin into the medium and the insulin was increased under glucose challenge. However, the insulin secretion level of STZ-induced islets was decreased more significantly than in other groups, similar to under H-DMEM conditions. Interestingly, injured islets cocultured with rBM-MSCs released insulin into culture medium under glucose stimulation, similar to normal islets. Data represent the mean±S.E. of values of three independent measurements. Data were analyzed for significant change by using two-way ANOVA and paired t-test (*P<0.05; **P<0.01)
Cytokine secretion
Transforming growth factor-β1 (TGF-β1), IL4 and IL6 were found to be secreted by rBM-MSCs independent of the cocultured islets. The analysis of three cytokines (TGF-β1, IL4 and IL6) in all three cell culture supernatants showed that all of the monocultures have detectable concentrations of these cytokines. Only the concentrations of IL4 were not altered in cocultures (data not shown). The coculturing of rBM-MSCs with islets and STZ islets led to an increase in secretion of IL6 and TGF-β1 in comparison to monocultures of rBM-MSCs, islets and STZ islets (Figure 6).

Cytokine secretion evaluated by ELISA. The expressions of TGF-β1 and IL-6 were quantified in various monocultures including islets, STZ-induced islets and rBM-MSCs and cocultures including islets+rBM-MSCs and STZ-induced islets+rBM-MSCs. Data represent the mean±S.E. of values of three independent measurements. Data were analyzed for significant change by using two-way ANOVA and paired t-test (*P<0.05; **P<0.01)
Discussion
Therapeutic effects of MSCs are believed to occur not only by direct differentiation into injured tissue but also by production of paracrine and autocrine factors. MSCs at the injured tissue environments can promote the secretion of a variety of cytokines and growth factors that have both paracrine and autocrine activities.21, 22 On this line, several studies have been performed. For instance, in probing the mechanism of treating effects of MSCs transplanted into the infracted heart, several researchers noticed that MSCs undergoing hypoxia environments stimulated the infracted heart local microenvironment to secrete more amounts of cardioprotective vital growth factors to inhibit cardiomyocyte apoptosis compared with MSCs in vitro cultured under normoxia.23 Gatto et al.16 reported that DECs are putative stem cells for islets, which might secrete specific factors supporting islet growth and function. Therefore, the effects of direct coculturing with DECs on the viability of fresh and cryopreserved human pancreatic islets were studied. The results indicated that the coculture process represents a valuable tool to improve the survival and functional activity of islets.16 Miki et al.7 showed that coculture of the mouse, rat and pig islets with MNNK-1 cells that served as a feeder cell in the coculture maintained their viability and insulin secretion with glucose responsiveness.7 Luo et al.9 examined the reparative effects of allogeneic BM mononuclear cells that were placed in culture with isolated human islets. Coculture with BM showed the ability of damage repair by human islets resulting in functional insulin release from islets in the long term. They suggested that BM increases survival of islets and function with the eventual formation of pancreatic endocrine tissue capable of sustaining β-cell function.9 Chao et al.11 designed an approach to maintain the cultured rat pancreatic islet-like cell cluster (ICC) survival and function, when cocultured with human umbilical cord mesenchymal stem cells (HUMSCs). Their findings suggest that coculture with HUMSCs may have a significant potential to protect ICCs from damage.11 Similarly, Xu et al.15 studied the impact of RPE on cytokine secretion by MSCs and examined the influence of administration of conditioned media of MSCs treated with RPE on blood glucose levels in diabetic rats. Their report indicated that RPE enhanced the secretion of cytokines by MSCs. MSCs in the pancreatic microenvironment might exert indirect salutary effects through paracrine mediators on injured pancreatic cells in an STZ-induced diabetic animal model. The secreted factors may exert their therapeutic benefits by preventing apoptosis of pancreatic β-cells.15 More recently, Solari et al.14 examined the immunoregulatory effect of autologous MSCs on sub-optimal numbers of islets co-transplanted into an omental pouch to enhance insulin secretion and sustained normoglycemia in a model of STZ-induced diabetic rats. They have shown that allogeneic islets with autologous MSCs induced insulin secretion and promoted long-term islet allograft survival.14
In this study, we used a novel approach to maintain viability and functionality of the cultured rPIs. In this approach, cultures were separated by a membrane preventing any direct interaction between islets and rBM-MSCs. In routine cultures, STZ induction reduced the viability and functional activity of rPIs compared with non-inductive islets (fresh tissue). However, it was found that rBM-MSCs in coculture had the ability to maintain islet viability and functionality, proven by prolonged insulin secretion by islets for 14 days. Unlike protected islets, a gradual destruction was observed in unprotected islets and a significant loss of function was observed in islets injured by STZ ultimately leading to cell death after 14 days of incubation.
It has also been shown that in vitro expanded and purified rat MSCs spontaneously secrete TGF-β1, hepatocyte growth factor and IL6, but not IFNG, IL4, IL5 or IL10.24, 25 It was considered rather that MSCs promote tissue repair by secreting soluble factors that modulate inflammation and angiogenesis.26 In this study, the cocultivation of rBM-MSCs with islets and STZ islets led to an increase in IL6 and TGF-β1 into the culture supernatant in comparison with monocultures of rBM-MSCs, islets and STZ islets. Secretion of these cytokines by MSCs could have a role in their anti-inflammatory or antiapoptotic function.
Oh et al.27 showed that human MSCs secreted small amounts of IL6 while secreting large amounts of TGF-β1 into the culture medium.27 Moreover, increased IL6 secretion was shown when hMSCs were cocultured with chemically damaged human corneal epithelial cells (hCECs). This is in agreement with the data reporting upregulation of IL6 in stimulated MSCs.28, 29 In a recent study, it has been suggested that suppressive effects of MSCs can be through IL6 by inhibiting lymphocyte apoptosis.30 Their findings pointed out that MSCs inhibit apoptosis of lymphocytes, and that soluble factors, mainly IL6 secreted by MSCs after direct interaction with lymphocytes, have an important role in their antiapoptotic function. In another report, IL6 mediated antiapoptotic effects and drug resistance mechanisms through both STAT3 and bcl-xL pathways in prostate cancer cells were revealed.31 In this study, IL6 and its specific receptor (IL6RA) were expressed by rBM-MSCs, and these data might show the antiapoptotic effect of MSCs. In addition, it has been shown that MSCs suppress various immune functions through a release of immunosuppressive soluble factor, TGF-β1.32
SPP1 is a highly hydrophilic and negatively charged sialo-protein of approximately 298 amino acids that contains a Gly-Arg-Gly-Asp-Ser motif. SSP1 exists both intra- and extracellularly. It has been shown that Ssp1 is expressed in the pancreatic islets of the rat and that its pancreatic levels are acutely upregulated in response to STZ-induced diabetes. It was also shown that SSP1 protects the islets from STZ-induced no production.33 SSP1 also prevents IL-1β -induced rat islet dysfunction.8 In this study, it was shown that rBM-MSCs expressed SSP1 at the levels of gene and protein without stimulation. Therefore, it was considered that SSP1 could be secreted by rBM-MSCs into coculture medium and could protect islets from destruction. Moreover, it has been observed that FN treatment significantly inhibited lipopolysaccharide (LPS)-induced apoptotic activities in FN-treated MSCs-derived hepatocyte-like cells and improved the survival rates of LPS-induced SCID mice.34 This study showed that FN was expressed by rBM-MSCs at both the gene and protein levels.
Genes with known antiapoptotic functions, and those expressed under stress include Mapkapk2, Tnip1 and Bcl3. The expressions of these genes by stem cells were examined in this study. MAPKAPK2 is a direct substrate of p38 MAP kinase in response to cellular stress such as mechanical stress, heat shock, osmotic stress, UV irradiation, bacterial LPS, several inflammatory cytokines and growth factors.35 TNIP1 was originally described as an antiapoptotic TNF-α-induced gene in endothelial cells.36 BCL3, in complex with p52, can promote transcription of the genes encoding the cell cycle regulator cyclin D1 and the antiapoptotic BCL2 protein.37 The presented data showed the expression of all three genes in rBM-MSCs. Expression of these genes by rBM-MSCs might imply the role of rBM-SCs and highlight the antiapoptotic influence mechanisms that could have a protective role on pancreatic islet cells.
Overall, the underlying mechanisms modulating pancreatic islet viability might be attributed to the paracrine mediators IL6, TGF-β1, SSP1 and FN secreted by MSCs. Coculturing with rBM-MSCs might have a significant potential to protect STZ-induced injured pancreatic islets through paracrine actions such as cytoprotective, anti-inflammatory and antiapoptotic effects. In this study, an indirect coculture approach was used for the first time to protect and improve islet viability and its functionality, although several other studies, in which islets were directly cocultured with cellular extracts or with various cell types (e.g., fibroblasts, pancreatic ductal cells, MNNK-1 cells BM-mononuclear cells) to reach the same target, bring about the requirement for separation afterwards.4, 7, 9, 11, 14, 15, 16 The uniqueness of our approach is the elimination of mixing the different cell populations or contaminating cell extracts by placing a separator (insert) between the islets and the cells/cell extracts. Therefore, the approach used in this study would be very important in allogeneic islet transplantations and clinical practices.
Materials and Methods
Animals
Wistar male rats (8–10 weeks old) were obtained from the Experimental Animal Center of Kocaeli University (Kocaeli, Turkey). Animal housing and experiments were approved by the local animal care committee according to the institutional guidelines and national animal welfare. They were housed under standard conditions for a week before use.
Isolation of islets
Islets were isolated as described38 and cultured in RPMI 1640 (Invitrogen/GIBCO, Grand Island, NY, USA) with glucose 2 g/l supplemented with 10% fetal bovine serum (FBS; Invitrogen/GIBCO), 100 IU/ml penicillin-100 μg/ml streptomycin (Invitrogen/GIBCO) and glutamine (2 mmol/l; Invitrogen/GIBCO) at 37°C in a humidified air atmosphere containing 5% CO2.
Islets were then tested for their specificity by DTZ staining. DTZ staining was carried out by adding 10 μl DTZ stock solution to islets suspended in 1 ml Krebs-Ringer bicarbonate buffer (pH 7.4) with HEPES (10 mM) (KRBH) and incubated at 37°C for 10–15 min. The stained islets appeared as bright red under the inverted microscope (CKX41, Olympus, Tokyo, Japan). Non-islet tissue remained unstained.
Isolation and culturing of rBM-MSCs
To establish the MSC culture, the animals were anesthetized with Ketalar (Pfizer, Kırklareli, Turkey) and killed by cervical dislocation method. Under sterile conditions, both femur and tibiae from each rat were excised. Muscle and the entire connective tissue were detached. The ends of the bones were removed and a 21-gauge needle, inserted into the shaft of the BM, was extruded by flushing with RPMI 1640 supplemented with 10% FBS and 100 IU/ml penicillin-100 μg/ml streptomycin that was also used as the growth medium. Marrow plug suspension was dispersed by pipetting, successively filtered through 70 μm mesh nylon filter (BD Biosciences, Bedford, MA, USA), and centrifuged at 200 g for 10 min. Supernatant containing thrombocytes and erythrocytes was discarded, and the cell pellet was resuspended in the medium. The cells from one rat were seeded onto two 25 cm2 plastic tissue culture flasks (BD Biosciences) and incubated at 37°C in a humidified atmosphere containing 5% CO2 for 3 days. The mesenchymal stem cells were isolated on the basis of their ability to adhere to the culture plates. On the third day, red blood cells and other non-adherent cells were removed and fresh medium was added to allow further growth. The adherent cells grown to 70% confluency were defined as passage zero (P0) cells. The P0 MSCs were washed with Ca2+-Mg2+ free phosphate-buffered saline (PBS) (Invitrogen/GIBCO) and detached by incubating with 0.25% trypsin-EDTA solution (Invitrogen/GIBCO) for 5–10 min at 37°C. Complete medium was added to inactivate the trypsin. The cells were centrifuged at 200 g for 10 min, resuspended in 1 ml complete medium and counted manually in duplicate using the Thoma chamber. Then the cells were plated as P1 in 75 cm2 flasks (BD Biosciences) at densities of 1 × 106 cell/flask. Complete medium was replaced once every 3 days over a 10–14 day period. For each passage, the cells were plated similarly and grown to a confluency of 70%.
Phenotype identification of rBM-MSCs
Flow cytometry
To confirm that rBM-MSCs maintain their phenotypic characteristics after growth in culture, undifferentiated MSCs were subjected to flow cytometry analysis. Flow cytometry was performed on an FACS Calibur (BD Immunocytometry Systems, San Jose, CA, USA) and the data were analyzed with the Cell Quest software (BD Immunocytometry Systems). Immunophenotyping of rBM-MSCs was performed with antibodies against rat antigens CD29 (Integrin-β1 chain; Ha2/5;FITC), CD45 and CD90 (Thy-1/Thy-1.1-FITC), as well as their isotype controls (IgG2aK; FITC) (BD Biosciences, San Diego, CA, USA).
Immunohistochemistry
The immunohistochemistry studies on the P0, P1, P3, P4, P7, P9 and P10 cells were performed as previously described by us and used primary antibodies were listed in Supplementary Table 1.17 The mounted cells were examined under a light/fluorescence microscope (Leica DMI 4000B, Wetzlar, Germany).
Reverse transcription-polymerase chain reaction
Total RNA was isolated from rat islets and P3 cell cultures (rBM-MSCs) by the High Pure RNA Isolation Kit (Roche, Mannheim, Germany) and cDNA was synthesized by using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche). The PCR mix consisted of 1 × PCR buffer, 0.2 mM dNTPs, 0.5 μM of each primer (Supplementary Table 2), 1.25 mM MgCl2 and 1.5 U of Taq DNA polymerase. The PCR products were analyzed by agarose gel (2%) electrophoresis. Sequences and annealing temperatures of each primer pair are listed on Supplementary Table 2. The studied genes were selected: (i) to show that MSCs have diverse immunophenotypic characteristics and could be characterized by studying the expression of Spp1, Sparc, Bmp4, Gfap, tubulin beta 3 (Tubb3), Eno2, smooth muscle α-actin (Acta2), actin-β (Actb), Myog, Des, Nes, Fos and Vim genes; (ii) to relate the antiapoptotic and anti-inflammatory effects of MSCs cocultured with STZ-injured islets following the expression of apoptosis-related genes, such as IL6ra, TNFAIP3 interacting protein 1 (Tnip1), Bcl3, Ifng and IL10. In the study, the housekeeping gene, Gapdh, was selected as the internal control.
In vitro differentiation of rBM-MSCs
The in vitro studies of adipogenic and osteogenic differentiations were performed according to the methods described previously by Karaoz et al.17
Apoptosis induction in pancreatic islets
In vitro STZ treatment to apoptosis induction
STZ was prepared 1 min before use by dissolving in 0.1 mol/l sodium citrate (pH 4.5) at 4°C a concentration of 200 mmol/l. A volume of 10 μl of this solution was added to the islets to obtain 1 mmol/l STZ. Islets were then incubated overnight (14 h).39 In the next experimental set STZ-induced islets+MSCs group, STZ induction was performed in the presence of MSCs. Later, STZ-exposed islets were transferred into the upper section of the wells separated by an insert that was previously prepared by the addition of MSCs.
rBM-MSCs/rPIs indirect coculture experiments
All cocultures were conducted in triplicate in 12-well plates (Becton Dickinson Labware Europe, Le Pont De Claix, France) using 0.4-μm pore size, high pore density and polyethylene terephthalate track-etched cell culture inserts (Becton Dickinson Labware Europe). rBM-MSCs (1 × 105) were seeded in the lower chamber of the coculture system, and incubated in RPMI 1640 medium including 10% FBS and 1% antibiotic. Once cultured MSCs reached confluence, pancreatic islets (normal and STZ-induced) (20 per well) were then placed on the upper surface of the membrane of the cell culture inserts. Cocultured cells and islets were maintained for 14 days in RPMI 1640 at 37°C and 5% CO2 in a humidified atmosphere with the medium being changed every 2 days. Control MSCs without pancreatic islets were prepared in the same conditions as cocultured rBM-MSCs.
Apoptosis detection in pancreatic islets with/without coculture
Apoptosis detection by FDA-PI viability staining
Islet preparations were assessed for islet cell viability using cell membrane exclusion dyes. Viability of islets was assessed using fluorescein diacetate (FDA) (Sigma-Aldrich, St. Louis, MO, USA) and PI (Sigma-Aldrich) staining.3 Briefly, islets in the inserts were transferred in PBS to a Petri dish (35 mm). FDA and PI stock solutions were added to the sample at a final concentration of 0.67 and 75 μM, respectively. Using a fluorescent microscope (Leica DMI 4000B), a procedure for the assessment of intact pancreatic islet viability was used. After 30 s of adding the stain, it was placed in a Petri dish containing 2.0 ml of PBS. The sample was placed on the fluorescent microscope, visualized and photographed using the filter FITC for fluorescein (em ∼530 nm) and rhodamine (em >600 nm). Dead cells were stained red and viable cells were stained green. The objective of the test was to assess, as accurately as possible, the percentage of islet cells that were viable. The cells were assigned to categories as follows: 0: few or no cells were green and the majority were red (average viability=non-viable), 1: approximately 75% of the cells were red (average viability=25%); 2: approximately 50% of the cells were red (average viability=50%); 3: approximately 25% of the cells were red (average viability=75%); 4: few or no cells were red (average viability=100%).
Calculations
Total viable=0.25 (sum of category 1)+0.5 (sum of category 2)+0.75 (sum of category 3)+(sum of category 1)
Total number of islets=(sum of category 1)
Percent viable=(total viable × 100)/total number of islets
Apoptosis detection by flow cytometry
Following coculture experiments, islets were incubated for 5 min with trypsin-EDTA. 0.25% trypsin, 1 mM EDTA.4Na in Hanks’ balanced salt solution without Ca+2 and Mg+2 at 37°C, and islet cells were gently dispersed.20 After washing with PBS, apoptotic cell percentage was detected by flow cytometry with Annexin-V-FITC Apoptosis Detection Kit (BD Pharmingen).
Measurement of insulin secretion by ELISA
At the end of the 14th day, inserts including islets were transferred into new 12-well plates and the culture media were switched to serum-free low glucose (L)-DMEM (5.5 mmol/l) containing 0.5% BSA for 2 h, washed twice with PBS, and then stimulated by high glucose (H)-DMEM (25 mmol/l) for 2 h. At the end of this second 2 h incubation, the culture media were collected and frozen at −70°C until they were assayed for insulin content. To estimate total cellular insulin levels, the total cell protein content was tested using BCA Protein Assay Kit (Sigma Aldrich). Insulin release was detected by rat insulin enzyme-linked immunosorbent assay (ELISA kit, Linco) according to the manufacturer's instructions.
Cytokine quantification
Secretions of rat interleukin (IL4, IL6, IL10) and transforming growth factor (TGF-β1) were determined in supernatants by ELISA (ELISA kit, Invitrogen, Camarillo, CA, USA) according to the manufacturer's recommendations. The minimum detectable dose (MDD) was 2 pg/ml for IL4, 7 pg/ml for IL6, 5 pg/ml for IL10, and 15.6 pg/ml for TGF-β1. All the experiments were performed in triplicate.
Statistical analysis
All experiments were repeated a minimum of three times. Data are reported as means±S.D. All statistical analyses were performed using SPSS 10.0 (SPSS Inc., Chicago, IL, USA). Data were analyzed using one-way ANOVA and paired t-test. Differences between the experimental and control groups were regarded as statistically significant when P<0.05.
Abbreviations
- Tubb3:
-
β-tubulin
- Actb:
-
actin-β
- Acta2:
-
α-smooth muscle actin
- Bcl3:
-
B-cell leukemia
- rBM-MSCs:
-
bone marrow-derived mesenchymal stem cells
- Bmp4:
-
bone morphogenetic protein 4
- DEC:
-
pancreatic ductal cells
- Des:
-
desmin
- DTZ:
-
dithizone
- Eno2:
-
enolase-γ
- Fos:
-
FBJ osteosarcoma oncogene
- FDA:
-
fluorescein diacetate
- FN:
-
fibronectin
- Gapdh:
-
glyceraldehyde 3-phosphate dehydrogenase
- Gfap:
-
glial fibrillary acidic protein
- ∣HUMSCs:
-
human umbilical cord mesenchymal stem cells
- IFNG:
-
interferon γ
- ICCs:
-
islet-like cell clusters
- IL6:
-
interleukin 6
- IL6ra:
-
interleukin 6 receptor-α
- IL10:
-
interleukin 10
- LPS:
-
lipopolysaccharide
- MAPKAPK2:
-
mitogen-activated protein kinase-activated protein kinase 2
- Myog:
-
myogenin
- Nes:
-
nestin
- PBS:
-
phosphate-buffered saline
- PI:
-
propidium iodide
- RPE:
-
rat pancreatic extract
- rPIs:
-
rat pancreatic islets
- Sparc:
-
secreted acidic cysteine-rich glycoprotein/osteonectin
- Spp1:
-
secreted phosphoprotein/osteopontin
- STZ:
-
streptozotocin
- Tnip1:
-
TNFAIP3 (tumor necrosis factor, α-induced protein 3)-interacting protein 1
- TGF-β1:
-
transforming growth factor-β1
- Vim:
-
vimentin
References
Ricordi C . Islet transplantation: a brave new world. Diabetes 2003; 52: 1595–1603.
Paraskevas S, Maysinger D, Wang R, Duguid TP, Rosenberg L . Cell loss in isolated human islets occurs by apoptosis. Pancreas 2000; 20: 270–276.
Ichii H, Wang X, Messinger S, Alvarez A, Fraker C, Khan A et al. Improved human islet isolation using nicotinamide. Am J Transplant 2006; 6: 2060–2068.
Rabinovitch A, Russell T, Mintz DH . Factors from fibroblasts promote pancreatic islet B cell survival in tissue culture. Diabetes 1979; 28: 1108–1113.
Yang Z, Chen M, Carter JD, Ellett JD, Smith KM, Nadler JL . Inflammation blockade improves pancreatic islet function. Transplant Proc 2004; 36: 2864–2865.
Sharma A, Sörenby A, Wernerson A, Efendic S, Kumagai-Braesch M, Tibell A . Exendin-4 treatment improves metabolic control after rat islet transplantation to athymic mice with streptozotocin-induced diabetes. Diabetologia 2006; 49: 1247–1253.
Miki A, Narushima M, Okitsu T, Takeno Y, Soto-Gutierrez A, Rivas-Carrillo JD et al. Maintenance of mouse, rat, and pig pancreatic islet functions by coculture with human islet-derived fibroblasts. Cell Transplant 2006; 15: 325–334.
Arafat HA, Katakam AK, Chipitsyna G, Gong Q, Vancha AR, Gabbeta J et al. Osteopontin protects the islets and beta-cells from interleukin-1 beta-mediated cytotoxicity through negative feedback regulation of nitric oxide. Endocrinology 2007; 148: 575–584.
Luo L, Badiavas E, Luo JZ, Maizel A . Allogeneic bone marrow supports human islet beta cell survival and function over six months. Biochem Biophys Res Commun 2007; 361: 859–864.
Kaminski A, Kaminski ER, Morgan NG . Pre-incubation with interleukin-4 mediates a direct protective effect against the loss of pancreatic beta-cell viability induced by proinflammatory cytokines. Clin Exp Immunol 2007; 148: 583–588.
Chao KC, Chao KF, Chen CF, Liu SH . A novel human stem cell coculture system that maintains the survival and function of culture islet-like cell clusters. Cell Transplant 2008; 17: 657–664.
Yamamoto T, Ricordi C, Mita A, Miki A, Sakuma Y, Molano RD et al. beta-Cell specific cytoprotection by prolactin on human islets. Transplant Proc 2008; 40: 382–383.
Kvezereli M, Vallentin A, Mochly-Rosen D, Busque S, Fontaine MJ . Islet cell survival during isolation improved through protein kinase C epsilon activation. Transplant Proc 2008; 40: 375–378.
Solari MG, Srinivasan S, Boumaza I, Unadkat J, Harb G, Garcia-Ocana A et al. Marginal mass islet transplantation with autologous mesenchymal stem cells promotes long-term islet allograft survival and sustained normoglycemia. J Autoimmun 2009; 32: 116–124.
Xu YX, Chen L, Hou WK, Lin P, Sun L, Sun Y et al. Mesenchymal stem cells treated with rat pancreatic extract secrete cytokines that improve the glycometabolism of diabetic rats. Transplant Proc 2009; 41: 1878–1884.
Gatto C, Callegari M, Folin M, Conconi M, Paolin A, Di Falco G et al. Effects of cryopreservation and coculture with pancreatic ductal epithelial cells on insulin secretion from human pancreatic islets. Int J Mol Med 2003; 12: 851–854.
Karaoz E, Aksoy A, Ayhan S, Sariboyaci AE, Kaymaz F, Kasap M . Characterization of mesenchymal stem cells from rat bone marrow: ultrastructural properties, differentiation potential and immunophenotypic markers. Histochem Cell Biol 2009; 132: 533–546.
Yu Y, Yao AH, Chen N, Pu LY, Fan Y, Lv L et al. Mesenchymal stem cells over-expressing hepatocyte growth factor improve small-for-size liver grafts regeneration. Mol Ther 2007; 15: 1382–1389.
de Macedo Braga LM, Lacchini S, Schaan BD, Rodrigues B, Rosa K, De Angelis K et al. In situ delivery of bone marrow cells and mesenchymal stem cells improves cardiovascular function in hypertensive rats submitted to myocardial infarction. J Biomed Sci 2008; 15: 365–374.
Huang YC, Yang ZM, Chen XH, Tan MY, Wang J, Li XQ et al. Isolation of mesenchymal stem cells from human placental decidua basalis and resistance to hypoxia and serum deprivation. Stem Cell Rev 2009; 5: 247–255.
Xu YX, Chen L, Wang R, Hou WK, Lin P, Sun L et al. Mesenchymal stem cell therapy for diabetes through paracrine mechanisms. Med Hypotheses 2008; 71: 390–393.
Ichim TE, Alexandrescu DT, Solano F, Lara F, Campion Rde N, Paris E et al. Mesenchymal stem cells as anti-inflammatories: implications for treatment of Duchenne muscular dystrophy. Cell Immunol 2010; 260: 75–82.
Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J 2006; 20: 661–669.
Aksu AE, Horibe E, Sacks J, Ikeguchi R, Breitinger J, Scozio M et al. Co-infusion of donor bone marrow with host mesenchymal stem cells treats GVHD and promotes vascularized skin allograft survival in rats. Clin Immunol 2008; 127: 348–358.
Boumaza I, Srinivasan S, Witt WT, Feghali-Bostwick C, Dai Y, Garcia-Ocana A et al. Autologous bone marrow-derived rat mesenchymal stem cells promote PDX-1 and insulin expression in the islets, alter T cell cytokine pattern and preserve regulatory T cells in the periphery and induce sustained normoglycemia. J Autoimmun 2009; 32: 33–42.
Phinney DG, Prockop DJ . Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair-current views. Stem Cells 2007; 25: 2896–2902.
Oh JY, Kim MK, Shin MS, Wee WR, Lee JH . Cytokine secretion by human mesenchymal stem cells cocultured with damaged corneal epithelial cells. Cytokine 2009; 46: 100–103.
Aggarwal S, Pittenger MF . Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005; 105: 1815–1822.
Djouad F, Charbonnier LM, Bouffi C, Louis-Plence P, Bony C, Apparailly F et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells 2007; 25: 2025–2032.
Xu G, Zhang Y, Zhang L, Ren G, Shi Y . The role of IL-6 in inhibition of lymphocyte apoptosis by mesenchymal stem cells. Biochem Biophys Res Commun 2007; 361: 745–750.
Pu YS, Hour TC, Chuang SE, Cheng AL, Lai MK, Kuo ML . Interleukin-6 is responsible for drug resistance and anti-apoptotic effects in prostatic cancer cells. Prostate 2004; 60: 120–129.
Puissant B, Barreau C, Bourin P, Clavel C, Corre J, Bousquet C et al. Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol 2005; 129: 118–129.
Katakam AK, Chipitsyna G, Gong Q, Vancha AR, Gabbeta J, Arafat HA . Streptozotocin (STZ) mediates acute upregulation of serum and pancreatic osteopontin (OPN): a novel islet-protective effect of OPN through inhibition of STZ-induced nitric oxide production. J Endocrinol 2005; 187: 237–247.
Kao CL, Lin HT, Chen YW, Chen YC, Tsai FT, Chang YL et al. Fibronectin suppresses lipopolysaccharide-induced liver damage and promotes the cytoprotection abilities of hepatocyte-like cells derived from human bone marrow mesenchymal stem cells. Transplant Proc 2007; 39: 3444–3445.
Ono K, Han J . The p38 signal transduction pathway: activation and function. Cell Signal 2000; 12: 1–13.
Opipari Jr AW, Boguski MS, Dixit VM . The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 1990; 265: 14705–14708.
Kashatus D, Cogswell P, Baldwin AS . Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev 2006; 20: 225–235.
Omer A, Duvivier-Kali VF, Aschenbach W, Tchipashvili V, Goodyear LJ, Weir GC . Exercise induces hypoglycemia in rats with islet transplantation. Diabetes 2004; 53: 360–365.
Xu J, Han J, Long YS, Lock J, Weir GC, Epstein PN et al. Malic enzyme is present in mouse islets and modulates insulin secretion. Diabetologia 2008; 51: 2281–2289.
Acknowledgements
We thank S Ayhan and A Okcu for their excellent technical assistance in the experiments and M Kasap for his contributions to the writing of the article. This study was supported by a grant (107S276) from the Scientific and Research Council of Turkey (TUBITAK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This article is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 license. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Karaoz, E., Genç, Z., Demircan, P. et al. Protection of rat pancreatic islet function and viability by coculture with rat bone marrow-derived mesenchymal stem cells. Cell Death Dis 1, e36 (2010). https://doi.org/10.1038/cddis.2010.14
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2010.14
Keywords
This article is cited by
-
Recapitulating pancreatic cell–cell interactions through bioengineering approaches: the momentous role of non-epithelial cells for diabetes cell therapy
Cellular and Molecular Life Sciences (2021)
-
Combined signaling of NF-kappaB and IL-17 contributes to Mesenchymal stem cells-mediated protection for Paraquat-induced acute lung injury
BMC Pulmonary Medicine (2020)
-
Fibroblasts as a practical alternative to mesenchymal stem cells
Journal of Translational Medicine (2018)
-
Co-culture of rat luteal cells with islet cells enhances islet viability and revascularization
In Vitro Cellular & Developmental Biology - Animal (2018)
-
Co-encapsulation and co-transplantation of mesenchymal stem cells reduces pericapsular fibrosis and improves encapsulated islet survival and function when allografted
Scientific Reports (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}