
Abstract
Accumulating data indicates that following anti-cancer treatments, cancer cell death can be perceived as immunogenic or tolerogenic by the immune system. The former is made possible due to the ability of certain anti-cancer modalities to induce immunogenic cell death (ICD) that is associated with the emission of damage-associated molecular patterns (DAMPs), which assist in unlocking a sequence of events leading to the development of anti-tumour immunity. In response to ICD inducers, activation of endoplasmic reticulum (ER) stress has been identified to be indispensable to confer the immunogenic character of cancer cell death, due to its ability to coordinate the danger signalling pathways responsible for the trafficking of vital DAMPs and subsequent anti-cancer immune responses. However, in recent times, certain processes apart from ER stress have emerged (e.g., autophagy and possibly viral response-like signature), which have the ability to influence danger signalling. In this review, we discuss the molecular nature, emerging plasticity in the danger signalling mechanisms and immunological impact of known DAMPs in the context of immunogenic cancer cell death. We also discuss key effector mechanisms modulating the interface between dying cancer cells and the immune cells, which we believe are crucial for the therapeutic relevance of ICD in the context of human cancers, and also discuss the influence of experimental conditions and animal models on these.
Similar content being viewed by others

Facts
-
Cell death in the presence of pathogen-associated molecular patterns (PAMPs) can elicit potent immune responses. However, in the absence of PAMPs, damage-associated molecular patterns (DAMPs) emitted by dying cancer cells can elicit cancer antigen-directed anti-tumour immunity.
-
The ultimate outcome of an immune response to cancer cell death (i.e., anti-tumourigenic, pro-tumourigenic or autoimmunity or different combinations of these) tends to be complex and may depend on a number of factors like the type of the cancer cells that die and their in vivo location, the type of cell death pathway they follow to die, the types of immune cells that phagocytose them or interact with them and, last but not the least, whether a cancer antigen is recognized or not.
-
Tolerogenicity towards cell death, as happens predominantly when cancer cells undergo physiological apoptosis (after treatment with most anti-cancer therapies), depends on a number of factors including the presence of immunosuppressive factors, absence or inactivation of DAMPs, induction of tolerogenic dendritic cells (DCs), ‘suboptimal’ activation of CD8+ T cells only and apoptotic ‘mimicry’.
-
Accentuated immunogenicity exhibited by cancer cells undergoing immunogenic cell death (ICD; after treatment with selected anti-cancer therapies), depends on a number of factors like emission of DAMPs (i.e., surface exposure of certain chaperones, secretion or release of certain nucleotides and endokines), presence of immunostimulatory factors, induction of DC maturation (both phenotypic and functional) and optimal activation of CD4+ αβ, CD8+ αβ and γδ T-cell responses.
-
Certain DAMPs are actively trafficked during ICD by danger signalling pathways, which are instigated and regulated by a complex interplay between endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) production and certain metabolic/biosynthetic processes (e.g., autophagy, caspase activity and secretory pathway).
Open Questions
-
As ICD is apoptotic in nature, does a ‘gray area’ exist due to the ‘overlap’ between DAMP-based immunogenicity of ICD and the apoptosis-associated tolerogenicity that could negatively influence anti-tumour immunity?
-
As currently known ICD-associated DAMPs only partially account for its exhibition of anti-tumour immunity; do as-yet-unknown DAMPs or certain known but non-ICD associated DAMPs (e.g., uric acid, intact nucleic acids, interleukin (IL)-33) exist that might be mediating its immunogenicity?
-
Apart from the complex interplay between ER stress and ROS production; are there other regulators or initiators of danger signalling during ICD? For instance, could viral response-like gene expression profile mediate ICD-associated danger signalling?
-
Does an ‘ideal ICD inducer’ exist that could efficiently impede pro-tumourigenic processes and therapy-resistant cancer microevolution while aiding anti-tumourigenic processes? Can combinatorial therapies involving ICD inducers with treatments like anti-cancer vaccines, anti-CTLA-4 or anti-PD1 antibodies and Toll-like receptor (TLR) agonists help us achieve such ideal properties?
-
Can ICD help us to characterize biomarkers that are good at predicting cancer patient’s therapy responses?
-
As most parameters used for ICD characterization are detected in vitro or ex vivo; can we characterize surrogate in vivo markers of ICD that can be detected robustly in preclinical as well as clinical set-ups?
Millions of cells die in our body on a daily basis to maintain normal ‘wear and tear’ and homeostasis, through ‘physiological apoptosis’1, 2 (see Box 1). During physiological apoptosis, various intracellular constituents of cells, including the majority of those that can act as danger signals, are proteolytically cleaved or inactivated by enzymes, such as caspases.3 This process is accompanied by exposure of specific ‘eat me’ and ‘find me’ signals4 (Box 1) to mediate an immunologically silent clearance of the dying cell’s material and antigens by scavenging immune cells (e.g., macrophages or DCs);3, 5 (Box 1). Considering the amount of cells that die in our body regularly, it is essential that they do not activate the immune system and therefore this process has ‘evolved’ to stay ‘silent’3, 4 (Box 1). However, problems arise when cancer cells (along with their antigens) follow the same physiological pathway to die or tend to exhibit apoptotic ‘mimicry’—all of which can induce tolerization towards cancer antigens (Box 1). Most chemotherapeutic agents used for anti-cancer treatment kill cancer cells through the process of non-immunogenic or tolerogenic apoptosis6, 7 (Box 1). Thus, just like normal cells, these dying cancer cells also get cleared ‘silently’—a scenario that compromises the efficacy of anti-cancer treatment3, 6, 8, 9, 10 (Box 1). Interestingly, it was recently discovered that certain chemotherapeutics, radiotherapy and photodynamic therapy (PDT)11, 12 (Table 1) can induce a form of apoptosis in cancer cells that is characterized by the presence of immunogenicity-accentuating DAMPs/danger signals, which assist in instigating anti-tumour immunity, known as, ‘immunogenic cell death’ (ICD).6, 7, 11
Danger signals or DAMPs have a very important role in enhancing cancer cell immunogenicity over physiological apoptosis.13 Their spatiotemporally defined emission provides the immune system with sequential or simultaneous ‘pulses’ of ‘impending danger’,1 thereby keeping the immune system in a pro-inflammatory state.2, 7, 11 DAMPs are molecules that are normally sequestered within live cells (where they perform predominantly non-immunological functions), which acquire immunomodulatory activities14 once secreted or surface exposed by dying or stressed/damaged cells.13 In cancer cells, specific inducers (Table 1) cause ICD by activating efficient danger signalling pathways that help in the trafficking of DAMPs either toward the surface or the extracellular region,6, 7, 15 where they gain their danger signalling properties (Figure 1). These danger signals in the company of cancer cell constituents and antigens cause maturation of DCs, which ultimately ‘cross-prime’ and activate anti-tumourigenic CD4+/CD8+ T-cell immunity6, 7, 9, 16 (Figure 1). In the present review, we will discuss the nature, origins and regulation of the danger signalling pathways during ICD. We also explore the significant plasticity that exists in danger signalling and sensing on account of different ICD inducers and experimental systems or models.

A schematic representation of the ICD concept in cancer. Cancer cells undergoing ICD expose various danger signals or DAMPs in different stages of apoptosis, including surface-tethered CRT, HSP70 or HSP90; actively or passively secreted ATP; endokines like HMGB1; and end-stage degradation products like nucleic acids. These DAMPs bind their respective receptors like CD91/various scavenger receptors; P2X7R/P2Y2R, TLR2/TLR4/RAGE; and other TLRs on the surface of DCs. These DAMPs along with cancer cell ‘cargo’ and antigens cause maturation of DCs, characterized by cell-surface upregulation of MHC-II, CD86, CD83, CD80 and a distinctly pro-inflammatory cytokine pattern (IL-12high, IL-6high, IL-1βhigh, NOhigh and IL-10low/absent). Altogether, these processes cause IL-1β-dependent activation of IL-17-producing γδT cells and increased proliferation of IFN-γ producing CD4+ or CD8+ αβT cells. Such potent T-cell immunity ultimately mediates a number of anti-tumourigenic processes that strive to impede the progression of therapy-resistant residual cancer cells or of an established tumour (in a curative set-up) or challenge by new tumour cells (in a prophylactic set-up). CD, cluster of differentiation; MHC, major histocompatibility complex; NO, nitric oxide; RAGE, receptor for advanced glycation end-products; S.R., scavenger receptors; TLR, Toll-like receptor
ER Stress and ROS: at the Origins of Danger Signalling?
The ability of the selected anti-cancer agents to induce ICD, along with a protective anti-cancer immune response in vivo, was initially shown to rely on their capability to evoke ER stress and ROS production in the succumbing cancer cell.6, 17 The reliance on the joint induction of ROS and ER stress for the efficient emission of DAMPs is perhaps not surprising considering that ER homeostasis governs protein folding and secretory machinery8, 10, 11 while oxidative modification of biomolecules (in particular proteins and lipids) represents an important mechanism in accentuating immunogenicity18, 19, 20 and enabling danger signalling.21 Notably, silencing of molecular effectors of the ER stress pathway triggered by ICD inducers blunts DAMPs (i.e., calreticulin (CRT) and adenosine triphosphate (ATP)) emission by the dying cancer cells and reduces their immunogenicity in vivo.7, 11, 15, 22 These data reinforce the concept that a robust ER stress response preferably accompanied by or induced by ROS production is a salient biochemical prerequisite for danger signalling and ICD. How exactly these two signalling components should operate to efficiently induce ICD is still elusive. ER stress could be secondary to mitochondria dysfunction and ROS production ((i.e., cardiac glycosides, UVC, wogonin1, 12, 23, 24) or to the local ROS generation at the ER ((i.e., hypericin-based photodynamic therapy (Hyp-PDT)7, 11, 25) and be propagated by lipid peroxidation signalling ((i.e., doxorubicin, mitoxantrone, ionizing irradiation, Hyp-PDT7, 11, 12) or be secondary to the activation of the unfolded protein response ((i.e., shikonin, bortezomib and CVB3 virus12).
Viral Response and ICD: A New Connection?
Emerging evidence suggest that there might be more to the danger signalling associated with ICD than just ER stress and ROS. As visible from Table 1, apart from various anti-cancer modalities, certain oncolytic viruses are also capable of inducing ICD.26 Interestingly, viral infection and replication activates not only ER stress (due to accumulation of un-/mis-folded viral proteins, overloading the ER) but also interferon (IFN) and cytokine responses (due to production of viral double-stranded RNA and PKR (protein kinase RNA-activated) activation),27 eliciting an ‘antiviral response’-associated gene expression programme in the target cells that may communicate a state of ‘danger’ to the host immune system.28 Could the induction of a viral response-like gene expression (VRGE) programme be a general requisite of ICD? Some preliminary evidence29 or retrospective analysis30 indeed suggest that certain known ICD inducers, like doxorubicin and Hyp-PDT (Table 1), might instigate in the cancer cells a gene signature that resembles the VRGE programme29, 30 (Figure 2). The protein products of various VRGE-related genes upregulated by doxorubicin (Figure 2a) or Hyp-PDT (Figure 2b) form an intricate evidence-based network; which is possibly a part of the host cell’s viral response-like reaction during ICD.

Activation of a VRGE profile in cancer cells undergoing ICD. The VRGE programme entails activation of various anti-viral processes like instigation of the type I IFN-regulatory system consisting of transcription factors like, IFN regulatory factor 3 (IRF3), IRF7, IRF5, IRF1112, 113 and secretion of IL-1 family cytokines like IL-1β and IL-18.114, 115 (a) This is an evidence-based network of the proteins whose genes showed an upregulated expression in the CD45-negative cells (predominantly tumour cells) retrieved from a doxorubicin-treated murine tumour, 2 days post treatment.29 (b) This is an evidence-based network of the proteins whose genes showed an upregulated expression in the T24 human bladder cancer cells retrieved 8 h after hypericin-based photodynamic therapy (Hyp-PDT).30 The types of evidence behind the network and the corresponding color code scheme are mentioned in the inset. The genes were considered to have role in a viral response based on data available in the GeneCards database version 3.09 (http://www.genecards.org/), wherever applicable. The networks were constructed through the tools available in the STRING database version 9.05 (http://string-db.org/)
In evolutionary sense, this still-to-be established possibility, is an attractive notion as response to a viral infection is one of the most ancient host cell reactions capable of communicating danger.27, 28 In line with this, recent experimental evidence suggests that a viral-response like stimulation of immune cells might be critical for enhanced antigen persistence and induction of anti-tumour immunity resulting in tumour rejection.31, 32, 33, 34
In the later sections, we discuss the nature, danger signalling-mediated trafficking and immunogenic relevance of DAMPs identified in the context of ICD.
Surface exposure of chaperones
Surface exposure (ecto-) of intracellular chaperones such as CRT, heat-shock protein 90 (HSP90) and HSP70 is crucial for the immunogenicity of dying cancer cells5, 6, 17, 35 (Figure 1). Ecto-CRT docks on lipid rafts and LRP1 in an inducer-dependent fashion,7 whereas HSP90 binds to LRP1.36 To date nothing is known about HSP70’s docking patterns, although its relatively homogenous surface distribution as patches and small clumps,22 which differs from the unevenly distributed patches observed for ecto-CRT,7, 22, 37 suggests a different docking entity.
In a functional sense, these chaperones have been reported to bind to various receptors on immune cells, like CD91 and certain scavenger receptors.2, 7 Pre-apoptotic ecto-CRT seems to be an important mediator of ICD’s immunogenicity, by acting as a potent pre-apoptotic ‘eat me’ signal that assists in phagocytic uptake of dying cancer cells.6, 7, 37 Moreover, ecto-CRT can incite the production of both IL-6 and tumour necrosis factor (TNF) from DCs and facilitate T helper type 17 (Th17) polarization.38 Similarly, ecto-HSP90 has been demonstrated to be a crucial mediator of immunogenicity, especially in the case of bortezomib-induced ICD.35 During anti-cancer DC vaccination based on ICD, ecto-HSP90 correlates better (or at least as strongly as ecto-CRT) with the ability of patients to respond to vaccination.39 The role of ecto-HSP70 in ICD has not been strongly elucidated; however, at least in the case of PDT11, 22 ecto-HSP70 might favour nitric oxide (NO) production from innate immune cells.7, 22, 40
In terms of capacity to mediate phagocytosis of dying cells by acting as an ‘eat me’ signal; an in silico analysis revealed that CRT harbours close homologues of crucial phagocytosis-assisting motifs, whereas HSP70/90 did not.22 In agreement, the presence of ecto-CRT correlates better with increased phagocytosis of dying cancer cells than either ecto-HSP70/90.41
Secretion of nucleotides or release of nucleic acids
Active secretion of the nucleotide ATP42 and release of nucleic acids43 from dying cells are essential for successful instigation of ICD44, 45 and anti-tumour immunity,46 respectively. Secreted ATP, a potent short-range ‘find me’ signal,42 can bind ionotropic (P2X) as well as metabotropic (P2Y) purinergic receptors42, 44 (Figure 1) and stimulate phenotypic DC maturation.44 During ICD, secreted ATP mainly binds the P2X7 receptors (Figure 1), causing activation of the NLRP3 inflammasome,44 which in turn leads to caspase-1-mediated processing and secretion of active IL-1β, a crucial cytokine for the propagation of anti-tumour immune responses.44 Apart from ATP, secretion of uric acid (UA)47 can activate the NALP3 inflammasome.48 Although UA is a danger signal associated with injury-induced necrosis,47 yet whether it is actively secreted by cells undergoing apoptosis is still elusive. However, considering that tumours exhibiting high levels of apoptosis have been associated with increased UA production, which was found to mediate tumour immune rejection;49 the link between UA release/secretion and apoptosis or ICD warrants an urgent analysis.
Interestingly, it has been recently discovered that release of nucleic acids, like DNA, as DAMPs from cells dying under chemotherapeutic stress can efficiently stimulate an antigen-specific anti-tumour immune response, accompanied by the production of IL-1β and IL-12.46 Whether such responses or other end-stage degradation products, like mitochondrial DNA and N-formyl peptides,50 are crucial for ICD remains an enigmatic question that needs to be answered in the near future.
Secretion or release of endokines
Endokines (or endogenous cytokines) are pro-inflammatory cytokines that are playing DNA-associated functions intracellularly (in a general cellular context), but perform cytokine-like DAMP activities, when released due to cellular stress or death. A critical endokine for ICD and immunogenicity includes passively released high-mobility group box 1 (HMGB1) protein.1, 5, 16 During ICD, extracellular HMGB1 has been found to bind TLR-4 on DCs (Figure 1) and activate the production of pro-inflammatory cytokines on one hand and help in proper antigen presentation on the other.2, 16 However, there are several contradictory observations regarding the HMGB1’s role in tumourigenesis, owing both to its redox-dependent switch in functionality and its pleiotropic rather than specific effects, making it hard to draw reliable conclusions5, 7, 51, 52 as discussed in the later section and in recent reviews.7, 13 Finally, it would also be imperative to analyze whether other endokines like high-mobility group nucleosome-binding domain,53 IL-1α54 and IL-3355 could also mediate immunogenicity of dying cancer cells.
The Plasticity and Regulation of Danger Signalling and Sensing
Plasticity in danger signalling due to different ICD inducers
Of the known ICD-associated DAMPs, danger signalling pathways have been mainly revealed for ecto-CRT and secreted ATP.11 Recent studies7, 11, 15, 22, 56 have shown that there exists significant plasticity in the trafficking pathways of these two DAMPs (Figure 3).

A schematic representation of plasticity and regulation of danger signalling and sensing. Various factors like the type of ICD inducers, differences in human and mice immune systems and differences between in vitro and in vivo experimental set-up can all lead to observations of plasticity or regulatory differences in danger signalling and its sensing. For more, please refer to the text. CNX, calnexin; Ecto, surface exposed; KDEL-R, KDEL receptor
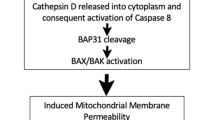
Chemotherapy (mitoxantrone or oxaliplatin)-induced pre-apoptotic ecto-CRT trafficking pathway has been found to be mediated by co-interaction with ERp57, protein kinase RNA-like endoplasmic reticulum kinase (PERK)-induced eukaryotic initiation factor 2 α (eIF2α) phosphorylation (eIF2α-P), ER-to-Golgi transport, the classical secretory pathway (Figure 3), and caspase-8-induced BAP31-mediated activation of BAX/BAK proteins.15, 56 A recent systematic analysis of the essential molecular components of the CRT trafficking pathway during ICD induced by chemotherapy or Hyp-PDT (Table 1) revealed that PERK-mediated proximal secretory pathway, ER-to-Golgi transport, BAX/BAK, classical secretory pathway and PI3K-mediated plasma membrane trafficking are shared by the two ICD inducers11, 25, 57 (Figure 3), whereas caspase-8 activity, ERp57 and eIF2α-P are not.7, 11, 22 Recently, wogonin-induced early apoptotic ecto-CRT (Table 1) was also found to be mediated by PERK and PI3K.58
Hence, in spite of this plasticity, the reliance of several ICD inducers on the ER stress sensor PERK as central ‘DAMP-trafficking master’ is noteworthy. Whether this is related to the archetypical induction of a viral response in the target cell (as described above), requiring the interface between PKR-mediated IFN response and PERK as ER stress sensor,59, 60 needs to be further explored. Both PKR and PERK are eIF2α phosphorylating kinases and eIF2α phosphorylation is required for some,11, 15 albeit not all ICD inducers,7, 11 suggesting that a certain degree of divergence in the trafficking mechanisms occurs downstream of PERK (Table 1). Intriguingly, the ability to mount an antiviral response do not always implicate eIF2α phosphorylation,61 suggesting that other still unknown PERK-dependent mechanisms may be recruited in a ICD-dependent manner. On the other hand, UV-induced early apoptotic ecto-CRT is mediated by phosphatidylserine-based scrambling/flipping and externalization37 (Figure 3). Finally, certain inducers of apoptosis have been shown to incite ecto-CRT, as a result of general exposure of ER chaperones or ER and Golgi membranes on the cell surface62 (Figure 3).
Mechanisms behind secretion of ATP by the dying cells are also dependent on the type of ICD or cell death inducers. Chemotherapy (mitoxantrone or oxaliplatin)-induced early apoptotic (at the blebbing stage) ATP secretion was recently demonstrated to be autophagy dependent45 (Figure 3). In addition, UV and anti-CD95 (also known as FAS) antibodies-induced early apoptotic ATP secretion were mediated by the pannexin-1 hemichannels63 (Figure 3). Notably, in the cases of both pannexin-1-dependent and autophagy-dependent pathways, caspases were required for ATP secretion.42, 45 Although the exact molecular mechanisms linking autophagy to ATP secretion are still unknown, it is remarkable that chemotherapy-mediated extracellular mobilization of ATP is concomitant to and requires caspase activation. This raises the intriguing possibility that functional modification of autophagy effectors by caspase-mediated processing, rather than the autophagy machinery per se, could be involved. However, Hyp-PDT-induced pre-apoptotic ATP secretion is autophagy independent64 and is mediated by the classical (Figure 3) and PERK-regulated proximal secretory pathway, as well as PI3K-dependent exocytosis.7 Instead, cancer cell-associated autophagy tends to suppress pre-apoptotic ecto-CRT induced by Hyp-PDT, by heightening the clearance of oxidatively damaged proteins within the ER.64
Thus, the biochemistry of an ICD inducer’s effect could be an important instigator of plasticity in danger signalling pathways that assist in trafficking of DAMPs.
Plasticity in sensing due to differences in mice and human immune systems
Murine models have been the mainstay of in vivo research. However, the two species diverged somewhere between 65 and 75 million years ago and differ strongly on a number of crucial levels.65 In fact, recently it was reported that the mouse models of acute and severe inflammation have very little in common with human pathogenesis.66 The authors of this study found that, during inflammation, the human genes that exhibited altered expression were almost never similar to the murine versions of the genes that showed differences in expression.66 In fact, these differences as we discuss further, could also influence the existing paradigms of DAMP sensing and ICD. In Table 2, we have summarized the main molecule-specific differences between human and mice immune systems, along with the most crucial ICD-sensing immunological components.2 Although these differences might appear subtle (Table 2), larger differences do exist (as discussed subsequently) that could ultimately impact ICD.
In the case of chemotherapy-induced ICD, apart from the activity of IFN-γ-producing αβT cells, the activity of IL-17 producing γδT cells has also been found to be crucial67 (Figure 1). Interestingly, it has been reported that the mice and human systems differ markedly when it comes to γδT-cell biology (Figure 3). Although γδT cells represent the predominant T-cell population found in the mouse cutaneous region, the same is not true for humans where the αβT cells predominate in both the cutaneous and subcutaneous regions.65 Considering that the above study linking ICD with γδT-cell activity was exclusively carried out in subcutaneously transplanted tumours in mice,67 one might understand that this could have created a bias for specific enrichment of γδT cells in this model. Thus with this background, the decisive role of γδT cells in chemotherapy-induced anticancer immune responses might have been overestimated; and thus may differ in a model, where the tumours do not have a privilege of predominantly receiving γδT cells. Therefore, such differences between the two species could have a significant impact on anti-cancer immunity mediated by γδT cells.
Moreover, differences exist in the biology of αβT-cell polarization between humans and mice (Figure 3). It has been recognized that the polarization of αβT cells towards type 1 (Th1) cytokine profile is anti-tumourigenic while a polarization towards type 2 (Th2) profile is pro-tumourigenic.3, 8 In fact, we had recently proposed a preferentially Th1-inducing effect (while, if possible, impeding Th2 induction) as one of the properties that an ideal ICD inducer should possess.7 However, although such distinct T-cell polarizations might be relatively easy to observe in mice, such distinctions are not as straightforward in humans.65 It has been reported that, while Th1 and Th2 cells are found in human diseases,68 a distinction is seldom visible between them, such that there is a strong probability for the generation of both T-cell polarizations simultaneously.65, 69 For instance, while in mice, IL-10 is a Th2 cytokine, in humans it can be produced by both Th1 and Th2 cells.65, 70
The impact that such species-to-species differences can have should not be underestimated; as made apparent by the situation concerning the anti-CTLA-4 antibody-based cancer immunotherapy.71 Research has demonstrated that treatment with this antibody shows better tumour-impeding results in mice than in humans, because the co-stimulatory receptor CD28 (whose T–cell-activating property is inhibited due to CD28-CTLA-4 interaction) is expressed in close to 100% of mice CD4+/CD8+ αβT cells,65 yet it is expressed in only 80% CD4+ and 50% CD8+ of human αβT cells.65 To this end, it is crucial to account for the differences that exist between mice and human immune systems, wherever applicable, while investigating the impact and sensing of ICD in cancer.
Regulation of anti-tumour immunity and differences in in vitro and in vivo conditions
Investigations that could be carried out in vitro have had a tremendous role in the progress of biology. However, they do not represent or reproduce the dynamic changes in various parameters that exist in vivo.72 Such differences may negatively influence the in vivo reproducibility of in vitro/ex vivo anti-tumour immunity experiments.
Tumour-associated immunosuppression is an important parameter that needs to be impeded during ICD induction by danger signalling in order to instigate potent anti-tumour immunity. Production of indoleamine 2,3-dioxygenase (IDO) enzyme (by immune cells or cancer cells) is an important immunosuppressive factor during tumourigenesis.30, 73 IDO causes degradation of tryptophan, thereby suppressing proliferation and differentiation of effector T cells and provoking enhanced suppressor activity of Treg cells.73, 74 Decrease in tryptophan concentrations <0.5–1 μM, in vitro, has been observed to inhibit T-cell proliferation,74 but such thresholds have not been established in vivo. Culture medium in vitro has supra-physiological concentrations of amino acids (200- to 500-fold higher than those present in normal human or mouse plasma),72 which are constant and only change due to in vitro degradation or manual replenishment of new nutrients. This does not represent the in vivo scenario, where concentrations of amino acids show dynamic and oscillatory changes due to constant lymph or interstitial fluid circulation or pressure.72 This scenario can also affect the experimental interpretation; as demonstrated by IFN-γ-induced IDO expression leading to strong tryptophan degradation in cell culture medium, resulting in a cell proliferation arrest that was reversed by replenishing the media with tryptophan.74
Interestingly, the bovine calf’s or foetal bovine serum (FBS) used in culture medium also pose problems. FBS (even when diluted to 10% of its original concentration) can have considerable amounts of placental hormones (like prolactin, oestrogen, progesterone) which not only create an in vitro environment of pregnancy but can also compromise/create background aberrations in ex vivo anti-tumour immunity experiments72, 75 (Figure 3). This is because such hormones increase IL-10 or IL-4 production, which can destabilize or interfere with IFN-γ levels, inhibit IL-1β levels, decrease T-cell proliferation and either promote overall Th2 polarization or create background aberrations in Th1/Th2 polarization levels.76 Curiously, even phenol red exhibits significant estrogenic activity at typical concentrations found in culture medium.72, 77
In conclusion, it is clear that the plasticity of the parameters in an experimental set-up, in vitro, can have a profound influence on the experimental outcome. All of this can affect the way we perceive and interpret the pre-clinical effects of ICD and anti-tumour immunity. This is especially relevant because most ICD parameters are tested in vitro,2, 7, 24 and at least in the case of PDT, it has been reported that the DAMP emission spectra can change for the same cancer cell line depending on whether it was treated in vivo or in vitro.30 Hence, in future, efforts should be made to bring experimental systems as close as possible to in vivo realities.
Future Perspectives on Plasticity and Regulation of Danger Signalling
Effect of tumour cell-extrinsic factors
There are a number of tumour cell-extrinsic factors that influence the outcome of danger signalling. These variables affect the outcome of ICD in terms of inducing either a beneficial anti-tumour response or a negative, tumour-promoting activity. For instance, recognition of ICD-associated signals can be affected by the type of the polymorphic variants of genes encoding danger-sensing receptors. Several allelic variants of genes encoding danger receptors or molecules associated with signalling pathways from these receptors have been described.78, 79, 80 Although complete deficiency of these molecules strongly compromises immune response, these are generally rare events. By contrast, genes for TLRs are highly polymorphic and encode many variants with either higher or lower activity in terms of ligand binding or signal transduction.81 Clinical response to radiotherapy or chemotherapy with anthracyclines or oxaliplatin was poorer in patients with loss-of-function alleles of TLR4 and P2R7 receptors for HMGB1 and ATP, respectively.16, 82, 83 It remains to be investigated whether functional consequences of these polymorphisms are of significance in determining therapeutic outcomes of ICD-inducing therapies.
Recognition of ICD-associated signals can also be influenced by various types of post-translational modification or association of DAMPs with other molecules that modulate binding to receptors. For example, HMGB1 can stimulate various inflammatory responses through multiple signalling pathways depending on its redox status1 or its association with CXCL12 chemokine can help it function as a ‘find-me’ signal to recruit myeloid cells through CXCR4.84 Another emerging regulatory mechanism involved in shaping responses to danger signals is associated with specific microRNA expression. A recent study revealed that exposure of leukocytes to DAMPs induces expression of miR-34c and miR-214 and downregulates miR-34a.85 miR-34c is involved in silencing IKKγ, an essential signalling molecule within NF-κB pathway, indicating that fine tuning of inflammatory pathways is extremely complex.
Degradation or turnover of danger signals
Factors that can affect danger signals’ capability to trigger anti-tumour immunity are the rate and extent of their degradation or turnover. For instance, the detection of extracellularly secreted or released ATP is modulated by extracellular or surface-associated ectonucleotidases that catalyse its degradation. Although ADP retains some of the immunostimulatory activities, adenosine is potently immunosuppressive.86 Therefore, the rate of ATP release from tumour cells as well as the activity of metabolic pathways involved in ATP degradation might determine the final outcome. Overexpression of CD39—an ATP-degrading enzyme—abolishes the immunogenicity of dying tumour cells rendering cancers resistant to chemotherapy.87 Anakinra, an IL-1 receptor antagonist has been shown to mitigate the anti-tumour efficacy of anthracyclines.44, 67, 82 Modulation of inflammasome activity can also affect HMGB1 signalling. Inflammasomes have been shown to participate in HMGB1 release,88 which can induce the expression of pro-IL-1β and other proinflammatory mediators.89
Danger signals: their ‘alter egos’ and unexpected complexity
It is frequently overlooked that tumour-associated danger signals might also contribute in a negative way to immunoregulation, by suppressing anti-tumour immunity rather than promoting it or providing pro-survival signals for tumour cells. Accordingly, HMGB1 can bind to TIM3 receptors on DCs to suppress development of anti-tumour immunity.46 Similarly, in a transgenic mouse model of prostate cancer, the release of HMGB1 was associated with priming of adaptive immunoregulatory response that facilitated malignant progression.90 Gemcitabine and 5-fluorouracil were shown to activate the proapoptotic protein BAX in myeloid-derived suppressor cells (MDSC), inducing lysosomal destabilization and release of cathepsin B.91 The latter triggers NLRP3 inflammasomes resulting in the secretion of proinflammatory cytokines (including IL-1β) that may curtail anti-tumour immunity by promoting IL-17 secretion from CD4+ T cells.91 Many chemotherapeutics although capable of promoting ICD exert at the same time potent immunosuppressive effects due to their cytotoxicity towards immune cells.
Another level of complexity in danger signalling is exosome-based communication. Exosomes are 30–100 nm vesicles92 released into the extracellular environment by cancer cells.93 Although exosomes express proteins in a cell-type dependent manner, numerous proteins frequently arise, such as MHC class I/II molecules, adhesion molecules, chaperones, cytoskeletal, ESCRT proteins, as well as transport and fusion proteins.94 Tumour-derived exosomes (TDEs) have been shown to elicit immunogenicity, vascular homeostasis and antigen presentation, as well as tumour growth, migration, survival, degradation of danger signals and immunoevasion.95, 96, 97, 98 Therefore, exosomal content may influence tumour outcome. TDEs have been shown to contain FASL or TRAIL, which can bind and activate T-cell killing.99 Furthermore, exosomal expression of TGFβ1 can interfere with NK and CD8+ T-cell activation by downregulating the NKG2D receptor,100 as well as inducing MDSCs.101 Exosomal CD39 and CD73 enhance extracellular adenosine concentration by degrading ATP and negatively modulating T-cell activity102, 103 (discussed previously). Moreover, TRYP2 and MET expression within TDEs have been implicated in the formation of a pre-metastatic niche, promoting the survival and outgrowth of disseminated tumour cells.104, 105
Conversely, exosomal expression and localization of danger signals (HSP70/72/90) may directly affect tumour outcome106, 107, 108 by activating an NK specific anti-tumour response.106 Moreover, cellular stresses that promote ecto-HSP70 induction and ultimately its derived exosomes, enhance the immunogenic response, due to greater activation of NK cell’s cytolytic capacity.109 HSPs chaperone antigens and peptides into TDE membranes, which can enhance CTL anti-tumour response, significantly delaying tumour growth.110 TDE expression of TNF can not only stimulate HMGB1 release from immune cells, inducing IFNγ secretion but can also decrease T-cell activation-induced death, generating T cells resistant to apoptosis.111 Therefore, understanding the dynamics of exosomal signalling and its role in general danger signalling in a cancer type and in a chemo/radio-therapeutic set-up may enable us to design therapeutic strategies based around modulating the TDE mechanics, kinetics and content.
Conclusion
Danger signalling has emerged recently as a promising mechanism to instigate within the dying or stressed cancer cells the emission of DAMPs, which allows the host immune cells to perceive these cells as ‘dangerous’ and ‘non-self’. Evidence accumulated over the past decade shows that this strategy works—as substantiated by the ability of DAMP-emitting ICD sub-routine to incite potent anti-tumour immunity. Emission of DAMPs from tumour cells might not be sufficient to elicit a productive immune response; of equal significance is the ability of immune cells to detect, transmit and to respond to these signals. In other words, the seeds (from tumour cells) must fall on the fertile ground (responsive immune cells) to give life (effective anti-tumour immunity). However, the danger signalling patterns behind DAMP trafficking and their immune sensing systems seem to be very plastic in nature and regulated in a highly multi-factorial manner. This means that every ICD or danger signalling inducer, animal model and experimental condition have to be treated in a context-dependent fashion when it comes to translation into the clinic. Considering that most parameters for characterization of ICD are in vitro or ex vivo in nature; it is crucial in the coming years to characterize at least surrogate in vivo markers that can be detected robustly in preclinical/clinical set-ups. Moreover, cancer cell intrinsic processes like oncogene-driven metabolic reprogramming, perturbations in mitochondrial homeostasis, autophagy, viral response-like gene signature and their link with danger signalling needs to be completely unravelled.
Abbreviations
- ATP:
-
adenosine triphosphate
- CRT:
-
calreticulin
- DAMP:
-
damage-associated molecular pattern
- DC:
-
dendritic cell
- ER:
-
endoplasmic reticulum
- HMGB1:
-
high-mobility group box 1
- HSP:
-
heat shock protein
- ICD:
-
immunogenic cell death
- IDO:
-
indoleamine 2,3-dioxygenase
- IFN:
-
interferon
- IL:
-
interleukin
- MDSC:
-
myeloid-derived suppressor cell
- PAMP:
-
pathogen-associated molecular pattern
- PDT:
-
photodynamic therapy
- PERK:
-
protein kinase RNA-like endoplasmic reticulum kinase
- PKR:
-
protein kinase RNA-activated
- ROS:
-
reactive oxygen species
- TDE:
-
tumour-derived exosome
- Th:
-
T helper
- TLR:
-
Toll-like receptor
- UA:
-
uric acid
- VRGE:
-
viral response-like gene expression
References
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P . Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12: 860–875.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L . Immunogenic cell death in cancer therapy. Annu Rev Immunol 2012; 31: 51–72.
Green DR, Ferguson T, Zitvogel L, Kroemer G . Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363.
Gregory CD, Pound JD . Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol 2010; 223: 177–194.
Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P . Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta 2010; 1805: 53–71.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31: 1062–1079.
Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P . ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med 2012; 18: 589–598.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202: 1691–1701.
Verfaillie T, Garg AD, Agostinis P . Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett 2010; 332: 249–264.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P . The emergence of phox-ER stress induced immunogenic apoptosis. OncoImmunology 2012; 1: 786–788.
Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P . Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev 2013; S1359-6101: 00006–3.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P . DAMPs and PDT-mediated photo-oxidative stress: exploring the unknown. Photochem Photobiol Sci 2011; 10: 670–680.
Matzinger P . Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045.
Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28: 578–590.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13: 1050–1059.
Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15: 3–12.
Callahan MK, Chaillot D, Jacquin C, Clark PR, Menoret A . Differential acquisition of antigenic peptides by Hsp70 and Hsc70 under oxidative conditions. J Biol Chem 2002; 277: 33604–33609.
Chiang CL, Ledermann JA, Aitkens E, Benjamin E, Katz DR, Chain BM . Oxidation of ovarian epithelial cancer cells by hypochlorous acid enhances immunogenicity and stimulates T cells that recognize autologous primary tumor. Clin Cancer Res 2008; 14: 4898–4907.
Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med 2004; 200: 1359–1370.
Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res 2011; 108: 235–248.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P . Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012; 61: 215–221.
Yang Y, Li XJ, Chen Z, Zhu XX, Wang J, Zhang LB et al. Wogonin induced calreticulin/annexin A1 exposure dictates the immunogenicity of cancer cells in a PERK/AKT dependent manner. PLoS One 2012; 7: e50811.
Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med 2012; 4: 143ra99.
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19: 1880–1891.
Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res 2012; 72: 2609–2621.
He B . Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 2006; 13: 393–403.
Tong AW, Senzer N, Cerullo V, Templeton NS, Hemminki A, Nemunaitis J . Oncolytic viruses for induction of anti-tumor immunity. Curr Pharm Biotechnol 2012; 13: 1750–1760.
Zitvogel L, Kroemer G, Delahaye N, Ma Y, Kepp O . Compounds and uses thereof to induce an immunogenic cancer cell death in a subject In: Office USPT (ed). http://www.freepatentsonline.com/y2013/0052160.html. US Patent Trademark Office: USA. URL: http://www.freepatentsonline.com/y2013/0052160.html. Institut Gustave Roussy: Villejuif, France, 2013.
Garg AD, Nowis D, Golab J, Agostinis P . Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis 2010; 15: 1050–1071.
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011; 208: 1989–2003.
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011; 208: 2005–2016.
Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M et al. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol 2011; 186: 5142–5150.
Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF . Type I interferon response and innate immune sensing of cancer. Trends Immunol 2013; 34: 67–73.
Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV . Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007; 109: 4839–4845.
Gopal U, Bohonowych JE, Lema-Tome C, Liu A, Garrett-Mayer E, Wang B et al. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS One 2011; 6: e17649.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123: 321–334.
Pawaria S, Binder RJ . CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat Commun 2011; 2: 521.
Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res 2010; 70: 9062–9072.
Song S, Zhou F, Chen WR, Xing D . PDT-induced HSP70 externalization up-regulates NO production via TLR2 signal pathway in macrophages. FEBS Lett 2012; 587: 128–135.
Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res 2011; 71: 4821–4833.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286.
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D . mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004; 279: 12542–12550.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15: 1170–1178.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334: 1573–1577.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 2012; 13: 832–842.
Behrens MD, Wagner WM, Krco CJ, Erskine CL, Kalli KR, Krempski J et al. The endogenous danger signal, crystalline uric acid, signals for enhanced antibody immunity. Blood 2008; 111: 1472–1479.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J . Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241.
Hu DE, Moore AM, Thomsen LL, Brindle KM . Uric acid promotes tumor immune rejection. Cancer Res 2004; 64: 5059–5062.
Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011; 32: 157–164.
Jube S, Rivera Z, Bianchi ME, Powers A, Wang E, Pagano IS et al. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res 2012; 72: 3290–3301.
Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol 2004; 164: 441–449.
Yang D, Postnikov YV, Li Y, Tewary P, de la Rosa G, Wei F et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J Exp Med 2012; 209: 157–171.
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA 2010; 107: 2574–2579.
Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009; 31: 84–98.
Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ 2008; 15: 1499–1509.
Garg AD, Bose M, Ahmed MI, Bonass WA, Wood SR . In vitro studies on erythrosine-based photodynamic therapy of malignant and pre-malignant oral epithelial cells. PLoS One 2012; 7: e34475.
Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol 2012; 189: 3209–3220.
Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N et al. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol 2004; 78: 12747–12761.
Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR . Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J Virol 2003; 77: 3578–3585.
Krishnamoorthy J, Mounir Z, Raven JF, Koromilas AE . The eIF2alpha kinases inhibit vesicular stomatitis virus replication independently of eIF2alpha phosphorylation. Cell Cycle 2008; 7: 2346–2351.
Franz S, Herrmann K, Furnrohr BG, Sheriff A, Frey B, Gaipl US et al. After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ 2007; 14: 733–742.
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER et al. Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature 2010; 467: 863–867.
Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013 (accepted).
Mestas J, Hughes CC . Of mice and not men: differences between mouse and human immunology. J Immunol 2004; 172: 2731–2738.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 2013; 110: 3507–3512.
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med 2011; 208: 491–503.
Romagnani S . Human TH1 and TH2 subsets: doubt no more. Immunol Today 1991; 12: 256–257.
Gor DO, Rose NR, Greenspan NS . TH1-TH2: a procrustean paradigm. Nat Immunol 2003; 4: 503–505.
Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S . Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J Immunol 1993; 150: 353–360.
Pardoll DM . The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Matarese G, La Cava A, Horvath TL . In vivo veritas, in vitro artificia. Trends Mol Med 2012; 18: 439–442.
Munn DH . Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012; 4: 734–745.
Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G . IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res 2010; 3: 113–120.
Huang W, Ramsey KM, Marcheva B, Bass J . Circadian rhythms, sleep, and metabolism. J Clin Invest 2011; 121: 2133–2141.
Zen M, Ghirardello A, Iaccarino L, Tonon M, Campana C, Arienti S et al. Hormones, immune response, and pregnancy in healthy women and SLE patients. Swiss Med Wkly 2010; 140: 187–201.
Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS . Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci USA 1986; 83: 2496–2500.
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 2000; 25: 187–191.
Verma D, Lerm M, Blomgran Julinder R, Eriksson P, Soderkvist P, Sarndahl E . Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis Rheum 2008; 58: 888–894.
Verma D, Sarndahl E, Andersson H, Eriksson P, Fredrikson M, Jonsson JI et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1beta and IL-18 production. PLoS One 2012; 7: e34977.
Netea MG, Wijmenga C, O'Neill LA . Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol 2012; 13: 535–542.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15: 1170–1178.
Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29: 482–491.
Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 2012; 209: 551–563.
Unlu S, Tang S, Wang E, Martinez I, Tang D, Bianchi ME et al. Damage associated molecular pattern molecule-induced microRNAs (DAMPmiRs) in human peripheral blood mononuclear cells. PLoS One 2012; 7: e38899.
Junger WG . Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11: 201–212.
Michaud M, Sukkurwala AQ, Martins I, Shen S, Zitvogel L, Kroemer G . Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. OncoImmunology 2012; 1: 393–395.
Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012; 488: 670–674.
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000; 192: 565–570.
He Y, Zha J, Wang Y, Liu W, Yang X, Yu P . Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res 2013; 73: 629–639.
Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 2013; 19: 57–64.
Thery C, Amigorena S, Raposo G, Clayton A . Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3 (Unit 3): 22.
Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ . Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci 2000; 113 (Pt 19): 3365–3374.
Thery C, Zitvogel L, Amigorena S . Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002; 2: 569–579.
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10: 1470–1476.
Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res 2010; 70: 1668–1678.
Marleau AM, Chen CS, Joyce JA, Tullis RH . Exosome removal as a therapeutic adjuvant in cancer. J Transl Med 2012; 10: 134.
Ichim TE, Zhong ZH, Kaushal S, Zheng XF, Ren XB, Hao XS et al. Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J Transl Med 2008; 6: 37.
Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 2002; 195: 1303–1316.
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z . Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 2008; 180: 7249–7258.
Xiang XY, Poliakov A, Liu C, Liu YL, Deng ZB, Wang JH et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer 2009; 124: 2621–2633.
Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z . Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol 2011; 187: 676–683.
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204: 1257–1265.
Peinado H, Lavotshkin S, Lyden D . The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 2011; 21: 139–146.
Alderton GK . Metastasis. Exosomes drive premetastatic niche formation. Nat Rev Cancer 2012; 12: 447.
Elsner L, Muppala V, Gehrmann M, Lozano J, Malzahn D, Bickeboller H et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J Immunol 2007; 179: 5523–5533.
Smalheiser NR . Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol Direct 2007; 2: 35.
Eustace BK, Jay DG . Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 2004; 3: 1098–1100.
Radons J, Multhoff G . Immunostimulatory functions of membrane-bound and exported heat shock protein 70. Exerc Immunol Rev 2005; 11: 17–33.
Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 2001; 7: 297–303.
Zhang HG, Liu C, Su K, Yu S, Zhang L, Zhang S et al. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J Immunol 2006; 176: 7385–7393.
Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 2013; 9: e1003118.
Barnes BJ, Field AE, Pitha-Rowe PM . Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem 2003; 278: 16630–16641.
Pien GC, Satoskar AR, Takeda K, Akira S, Biron CA . Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J Immunol 2000; 165: 4787–4791.
Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol 2007; 81: 2736–2744.
Garrido G, Rabasa A, Sanchez B, Lopez MV, Blanco R, Lopez A et al. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J Immunol 2011; 187: 4954–4966.
Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R et al. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS One 2012; 7: e31732.
Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res 2011; 71: 768–778.
Weibel S, Raab V, Yu YA, Worschech A, Wang E, Marincola FM et al. Viral-mediated oncolysis is the most critical factor in the late-phase of the tumor regression process upon vaccinia virus infection. BMC Cancer 2011; 11: 68.
Mahoney DJ, Lefebvre C, Allan K, Brun J, Sanaei CA, Baird S et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011; 20: 443–456.
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G . The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov 2012; 11: 215–233.
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I . Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 2008; 112: 2340–2352.
Beutler B . Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 2000; 12: 20–26.
Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF . Mammalian P2 × 7 receptor pharmacology: comparison of recombinant mouse, rat and human P2 × 7 receptors. Br J Pharmacol 2009; 157: 1203–1214.
Liu Z, Miner JJ, Yago T, Yao L, Lupu F, Xia L et al. Differential regulation of human and murine P-selectin expression and function in vivo. J Exp Med 2010; 207: 2975–2987.
Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA . Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett 2009; 283: 168–175.
Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002; 360: 295–305.
Connell PP, Weichselbaum RR . A downside to apoptosis in cancer therapy? Nat Med 2011; 17: 780–782.
Acknowledgements
This work was supported by grants from the Fund for Scientific Research Flanders (FWO-Vlaanderen; G.0661.09, G.0728.10 and G.0584.12N) and KU Leuven (GOA/11/009) to PA; ADG is a postdoctoral fellow supported by the BOF Postdoctoral Mandate (PDM) from KU Leuven (PDMK/12/146). This paper presents research results of the IAP7/32, funded by the Interuniversity Attraction Poles Programme, initiated by the Belgian State, Science Policy Office. The research in the lab of JG is supported by European Regional Development Fund through Innovative Economy grant POIG.01.01.02-00-008/08. JG is a member of TEAM Programme co-financed by the Foundation for Polish Science and the EU European Regional Development Fund. JG is a recipient of the Mistrz Award from the Foundation for Polish Science. Some of the figures were produced using Servier Medical Art (www.servier.com), for which we would like to acknowledge Servier.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Garg, A., Martin, S., Golab, J. et al. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 21, 26–38 (2014). https://doi.org/10.1038/cdd.2013.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.48
Keywords
This article is cited by
-
An ICD-Associated DAMP Gene signature predicts survival and immunotherapy response of patients with lung adenocarcinoma
Respiratory Research (2023)
-
Identification of an immunogenic cell death-related gene signature predicts survival and sensitivity to immunotherapy in clear cell renal carcinoma
Scientific Reports (2023)
-
Immunogenic cell death (ICD)-inducers in non-small-cell lung carcinoma (NSCLC): current knowledge and future perspective
Clinical and Translational Oncology (2022)
-
Transmissible ER stress between macrophages and tumor cells configures tumor microenvironment
Cellular and Molecular Life Sciences (2022)
-
Cancer nanotechnology: current status and perspectives
Nano Convergence (2021)
