
Abstract
Dying mammalian cells emit numerous signals that interact with the host to dictate the immunological correlates of cellular stress and death. In the absence of reactive antigenic determinants (which is generally the case for healthy cells), such signals may drive inflammation but cannot engage adaptive immunity. Conversely, when cells exhibit sufficient antigenicity, as in the case of infected or malignant cells, their death can culminate with adaptive immune responses that are executed by cytotoxic T lymphocytes and elicit immunological memory. Suggesting a key role for immunogenic cell death (ICD) in immunosurveillance, both pathogens and cancer cells evolved strategies to prevent the recognition of cell death as immunogenic. Intriguingly, normal cells succumbing to conditions that promote the formation of post-translational neoantigens (for example, oxidative stress) can also drive at least some degree of antigen-specific immunity, pointing to a novel implication of ICD in the etiology of non-infectious, non-malignant disorders linked to autoreactivity.
Similar content being viewed by others
Main
Mammalian cells respond to microenvironmental perturbations by activating signaling pathways that (at least initially) attempt to restore cellular homeostasis1. However, when such perturbations exceed cellular repair capacities in magnitude or duration, the same molecular cascades that initially support cytoprotection shift to a cytotoxic mode and ultimately promote regulated cell death (RCD)2. Depending on multiple parameters (for example, nature of the initiating perturbation, genetic/epigenetic cellular profile), RCD can proceed via distinct mechanisms3. Thus, while most human and mouse cells exposed to tumor necrosis factor (TNF) succumb to caspase 3 (CASP3)-dependent apoptosis, specific cell lines, such as mouse L929 fibroblasts, activate an alternative RCD mechanism commonly known as necroptosis4. Importantly, along with the evolution of multicellularity, the signaling pathways involved in adaptation to stress—be it successful (that is, recovering homeostasis) or not (that is, culminating with RCD or other terminal fates like cellular senescence)—have extended their reach beyond the domain of single cells and have acquired the capacity to inform the host about a potential danger1.
Specifically, stressed and dying mammalian cells release numerous bioactive molecules, including small metabolites (for example, ATP), nucleic acids (for example, mitochondrial DNA), proteins (for example, cytokines and damage-associated molecular patterns, DAMPs) and lipids (for example, oxidized cardiolipin), that interact with the immune system to dictate the immunogenic correlates of cellular stress and death5,6. Notably, some RCD variants, such as CASP3-dependent apoptosis, tend to occur in an immunologically silent manner, while others, such as necroptosis, generally have pronounced inflammatory potential7. However, the ability of specific instances of RCD to elicit bona fide adaptive immunity targeting dead-cell-associated antigens depends on multiple factors other than the specific pathway precipitating cellular demise8.
In 2005, the concept of ICD was introduced to differentiate cases of RCD that drive antigen-specific immune responses culminating in immunological memory from (1) (generally necrotic) RCD instances that only engage innate immune mechanisms (falling within the broad concept of ‘necroinflammation’)9, and (2) (generally apoptotic) RCD cases that elicit active immunosuppression10. This term is now widely employed in the scientific literature and has recently been used to define the mode of action of two anticancer agents approved for use in humans, belantamab mafodotin11 and lurbinectedin12,13. Indeed, various treatments commonly employed for cancer management—including specific chemotherapeutics, radiation therapy (RT) and some targeted anticancer agents14,15,16—cause bona fide ICD and hence engage tumor-targeting immune responses in support of treatment efficacy. RCD driven by infection is also highly immunogenic, and not only pathogens but also malignant cells harness various strategies to subvert the emission or detection of ICD-relevant signals to evade immunosurveillance8. These observations point to ICD as a highly desirable event in the context of malignant and infectious disorders. However, accumulating evidence suggests that normal cells succumbing to specific insults may also elicit antigen-specific immune responses that contribute to disease etiology, pointing to a novel implication for ICD in (at least some) non-infectious, non-malignant disorders linked to autoreactivity17 (Box 1).
Here, we critically discuss key events linking intracellular stress responses to the sensing of RCD as immunogenic, while emphasizing their subversion by malignant cells and pathogens during immunoevasion. We also propose a possible scenario for the evolution of ICD, suggest potential strategies to reinforce or restore ICD for therapeutic purposes and summarize emerging data on the role of ICD in non-infectious, non-malignant disorders associated with autoreactivity.
Hallmarks of ICD: antigenicity, adjuvanticity and environment
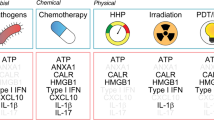
Immunogenicity arises from two factors: antigenicity and adjuvanticity. Moreover, adaptive immune responses driven by ICD can be properly executed only in the presence of permissive microenvironmental conditions8 (Fig. 1).

The ability of RCD to initiate an adaptive immune response culminating with an effector phase and associated with the establishment of immunological memory involves three key parameters: antigenicity, adjuvanticity and a permissive microenvironment. In the absence of antigens not covered by central or peripheral tolerance, RCD can drive only inflammatory responses that cannot engage adaptive immunity. In the presence of antigenicity, RCD can initiate adaptive immune responses only when accompanied by sufficient adjuvanticity. Conversely, the presentation of antigenic determinants to T cells in the context of poor adjuvanticity actively promotes tolerance. Finally, the microenvironment dictates whether T cells properly primed by DCs responding to ICD can access neoplastic lesions to mediate effector functions and establish a memory response. ECM, extracellular matrix; M1-like, proinflammatory; M2-like, anti-inflammatory; TAM, tumor-associated macrophage; TDLN, tumor-draining lymph node.
The mature conventional αβ T cell repertoire can recognize only peptides that are presented on autologous MHC class I and II molecules, implying that such peptides must structurally differ, at least to some degree, from peptides presented on the surface of healthy cells18. Accordingly, adaptive immunity driven by ICD is only possible when the immunopeptidome of dying cells contains such peptides to display sufficient antigenicity. This is obviously the case with infected cells, largely reflecting the abundant expression of foreign proteins encoded by the pathogen genome19. Malignant cells can also express genetically encoded antigens not covered by peripheral and central tolerance, either as a consequence of somatic mutations that generate novel antigenic determinants (‘tumor neoantigens’) or upon the derepression of genes normally silenced in the adult (‘oncofetal antigens’)20. Moreover, cancer cells can be recognized by mature T cells when they express lineage-specific antigens that are shared with normal cells but are covered by leaky peripheral tolerance (and hence are prone to driven autoimmune responses)20. These antigens may be more clinically relevant than previously thought, possibly explaining why neoplasms originating from stem cells that also generate adult tissues that are dispensable for organismal survival (for example, melanocytes, thyrocytes) exhibit superior curability20. Finally, the immunopeptidome of malignant cells can comprise novel antigenic determinants that are formed by enzymatic and non-enzymatic post-translational modifications of genetically normal proteins, including oxidized, phosphorylated and citrullinated antigens21,22.
Notably, cellular stress induced by chemotherapy, RT and targeted anticancer agents can also alter the immunopeptidome, not only by causing mutations that may ultimately generate additional neoantigens, but also (more commonly) by (1) favoring the expression of otherwise silenced genes containing non-synonymous mutations23,24 or otherwise non-transcribed RNAs encoding ‘cryptic’ peptides that appear to incorporate into nascent MHC class I molecules with high efficacy25,26; and (2) by altering signal transduction pathways that may ultimately generate post-translational neoantigens27. Moreover, both immunogenic chemotherapy and RT can upregulate the expression of MHC class I and II molecules on the surface of tumor cells, thus enhancing their antigenicity28,29.
Antigenicity is not sufficient to elicit adaptive immunity. Rather, the presentation of antigenic peptides to T cells in the absence of co-stimulatory signals generally results in T cell anergy linked to peripheral tolerance30. Importantly, the ability of antigen-presenting cells (APCs) to provide co-stimulation and proinflammatory cytokines in support of T cell priming originates from the ability of APCs to take up antigenic material for presentation as they gauge the microenvironment for signals of danger31. In the context of pathogen infection, such signals are provided by so-called microbe-associated molecular patterns (MAMPs), which are broadly conserved components of bacteria and viruses that are not shared with mammalian cells, such as lipopolysaccharide (LPS) and double-stranded RNA (dsRNA)32,33. Conversely, malignant cells experiencing immunogenic stress and death drive APC maturation by emitting a series of endogenous adjuvant signals that are cumulatively referred to as DAMPs5,6. DAMPs encompass both stress-associated molecular patterns (SAMPs), which emerge during stress responses irrespective of ultimate cellular fate34, and signals that are specifically associated with RCD. Importantly, both MAMPs and DAMPs are sensed by an evolutionarily conserved set of receptors highly expressed by myeloid cells that are cumulatively referred to as pattern recognition receptors (PRRs)6.
Although little is known about the spatiotemporal regulation of MAMP release by microbes and infected cells, considerable work has been devoted to the deconvolution of DAMP emission by malignant cells responding to immunogenic stressors35. Indeed, not all cell-death inducers can elicit the intracellular stress pathways that operate upstream of DAMP release in dying cancer cells (see below). Clinically actionable ICD inducers include specific conventional chemotherapeutics (for example, cyclophosphamide, anthracyclines, oxaliplatin and other platinum derivatives, but not cisplatin)10,36,37,38,39,40, RT41 and select targeted anticancer agents (for example, bortezomib, crizotinib)42,43,44,45,46,47,48, as well as some variants of endocrine therapy49, extracorporeal photochemotherapy50, photodynamic therapy (PDT)51,52,53,54 and oncolytic virotherapy55,56. As a general principle, the release of DAMPs by malignant cells succumbing to these agents involves either alterations of the cell surface (for example, owing to the translocation of intracellular proteins to the external leaflet of the plasma membrane) or changes of the extracellular microenvironment (for example, owing to the active secretion or passive release of metabolites and proteins that are normally secluded within live cells)57.
A large amount of work has mechanistically linked DAMP emission and detection to the initiation of adaptive immunity by cancer cells undergoing ICD57. The pannexin 1 (PANX1)- and lysosome-dependent secretion of ATP by dying cells, as well as its passive release upon plasma membrane permeabilization (PMP), elevates extracellular ATP levels to promote the purinergic receptor P2Y2 (P2YR2)-dependent recruitment of APC precursors cells into the tumor bed58,59. The passive liberation of the ubiquitously expressed cytosolic protein annexin A1 (ANXA1) guides APCs, in particular dendritic cells (DCs), into the vicinity of dying cancer cells via a formyl peptide receptor 1 (FPR1)-dependent mechanism, ultimately facilitating their physical interaction60. Importantly, dying cells undergoing ICD actively expose CALR and other endoplasmic reticulum (ER) chaperones, such as protein disulfide isomerase family A member 3 (PDIA3) on the cell surface61,62. This event, which occurs early in ontogeny and phylogeny (it is involved in oocyte–spermatocyte fusion, as well as in fusion among haploid yeast cells during sexual reproduction)63,64,65,66, provides an ‘eat me’ signal that facilitates the uptake of dying cancer cells by DCs, presumably through an interaction with LDL receptor related protein 1 (LRP1, best known as CD91)61,67.
Both extracellular ATP signaling via purinergic receptor P2X 7 (P2RX7) and the activation of Toll-like receptor 4 (TLR4) by high mobility group box 1 (HMGB1), which is passively released by cancer cells undergoing ICD, trigger DC maturation68. A similar effect also ensues the liberation of soluble F-actin, which is detected by C-type lectin domain containing 9A (CLEC9A)69. CLEC9A is predominantly expressed by type 1 conventional DCs (cDC1s), where it favors the rerouting of exogenous antigens to presentation on MHC class I molecules by disrupting phagosomes70. This may explain, at least in part, the superior cross-presenting capacity of CLEC9A-expressing cDC1s as compared with that of other DC subsets71.
An important immunostimulatory effect also emerges from intracellular DAMPs that accumulate or relocalize within malignant cells in the course of ICD, such as aberrant RNA molecules, which are detected by endosomal TLR3 (ref. 72), as well as micronuclei and mitochondrial DNA (mtDNA), which upon cytosolic accumulation promote the sequential activation of cyclic GMP–AMP synthase (CGAS) and stimulator of interferon response cGAMP interactor 1 (STING1)73,74. These latter pathways culminate with the NF-κB- and interferon responsive factor 3 (IRF3)-dependent secretion of multiple cytokines, including type I interferon (IFN), which further supports the initiation of adaptive immunity downstream of RCD75. The premortem activation of NF-κB as a consequence of receptor interacting serine/threonine kinase 1 (RIPK1) signaling may also orchestrate an immunostimulatory genetic program in the context of apoptotic or necroptotic ICD29,76,77.
Not all DAMPs are immunostimulatory, which contributes to the inability of various instances of RCD to drive adaptive immunity. For instance, prostaglandin E2 (PGE2), which is released abundantly upon CASP3 activation, mediates potent immunosuppressive effects78. Along similar lines, CASP3 signaling promotes the secretion of lysophosphatidylcholine, which recruits phagocytes to sites of cell death, and the surface exposure of phosphatidylserine,which promotes the tolerogenic removal of cell corpses79,80,81. Finally, the release of tumor protein, translationally-controlled 1 (TPT1) by dying cancer cells has been shown to promote the recruitment of myeloid-derived suppressor cells (MDSCs) to the tumor microenvironment (TME) in support of local immunosuppression and disease progression82. The mechanisms underlying the liberation of TPT1, however, remain obscure.
Logically, the aforementioned cascade of events can drive robust tumor infiltration by myeloid and lymphoid cells, shifting neoplastic lesions from a ‘cold’ to a ‘hot’ phenotype83. Of note, the attack of cancer cells by CD8+ cytotoxic T lymphocytes (CTLs) may also result in ICD84,85, suggesting a mechanism of self-amplification that might facilitate antigen spreading during the local immune response86. However, several features of the TME influence the propensity of RCD to efficiently prime adaptive immune responses that culminate with an effector phase associated with the establishment of immunological memory8. For instance, tumors exhibiting abundant infiltration by DCs expressing the immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1) are expected to offer a TME less permissive to the initiation of anticancer immunity than malignancies with scarce IDO1+ DC levels87. Similarly, abundant infiltration of neoplastic lesions by CD4+CD25+FOXP3+ regulatory T (Treg) cells, which mediate powerful immunosuppressive effects linked to cytotoxic T lymphocyte 4 (CTLA4) expression and interleukin-10 (IL10) secretion, prevents not only the effector functions of robustly primed CTLs but also priming per se88, as the latter appears to occur at least partially in tertiary lymphoid structures (TLSs) generated within the tumor bed89. Finally, tumors characterized by an elevated fibrotic response offer considerable resistance to infiltration by circulating effector CTLs primed within tumor-draining lymph nodes, de facto evading the effector phase of adaptive immunity driven by ICD90. Importantly, while these considerations emerge from the oncology field, it is likely that ICD driven by pathogen infection is also influenced by microenvironmental features, including (but not limited to) type and abundance of immune cells, cytokine milieu and vascularization.
In summary, antigenicity, adjuvanticity and a permissive microenvironment are invariably required for RCD to be perceived as immunogenic and elicit the effector phase of adaptive immunity in conjunction with the establishment of immunological memory (Fig. 1).
Stress-responsive pathways underlying ICD
Only some cellular stressors can induce bona fide ICD, including select agents commonly used in clinical practice (Table 1). However, while artificial-intelligence algorithms can predict the ability of specific agents to elicit ICD on the basis of their physicochemical properties14,91, in vivo experiments are required to formally validate such predictions8. At the mechanistic level, it appears that a common (but not universal) feature of ICD-inducing molecules is their capacity to inhibit transcription92. At this stage, however, how transcriptional inhibition (as opposed to the blockade of DNA replication, translation or vital bioenergetic pathways) triggers ICD-relevant stress pathways remains unclear. Another frequent mechanism for ICD induction involves microtubular disruption, as exemplified by taxanes93 and dimethyl aurostatin F11. Hence, it appears plausible that distinct ICD inducers act on different proximal targets to trigger stress-responsive pathways that enable ICD initiation.
The mechanisms linking stress responses in malignant cells to increased antigenicity have been scarcely characterized, although transcriptional and translational stress appear to be particularly efficient at generating potential neoantigens20. That said, some (if not the majority) of malignant cells are expected to express a sufficient amount of antigenic determinants not covered by central or peripheral tolerance at baseline94, suggesting that the key difference between ICD inducers and agents that elicit non-immunogenic RCD relates to DAMP release and adjuvanticity, an aspect that has been extensively investigated.
The ‘integrated stress response’ (ISR) consists of the phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1, best known as eIF2α) by any of four distinct eukaryotic translation initiation factor 2 alpha kinases (EIF2AK1–4)95. EIF2AK2 (best known as PKR) and EIF2AK3 (best known as PERK) appear to be particularly important for ICD induction92,96. eIF2α phosphorylation (which is also a conserved response to viral infection)97 blocks cap-dependent translation but favors the translation of proteins relying on internal ribosome entry sites (IRESs)95. In contrast to cytotoxic agents that do not elicit ICD, most ICD-inducing interventions efficiently stimulate eIF2α phosphorylation, which is indeed a pathognomonic biomarker of ICD91,92. In patient-derived tumor specimens, eIF2α phosphorylation correlates with CALR exposure and tumor infiltration by DCs and CTLs, as well as good prognosis98. Moreover, a non-phosphorylatable version of eIF2α (eIF2αS51A) abolishes ICD-associated CALR exposure99 as well as autophagy activation (which is involved in ATP release, see below) driven by anthracyclines92, pointing to the central role for the ISR in this context. It appears that transcriptional inhibitors are particularly efficient at inducing the ISR92. However, the underlying molecular mechanisms remain elusive. Moreover, it appears that in response to specific stimuli, for instance in the context of PDT, PERK can promote ICD irrespective of eIF2α phosphorylation52.
The ISR is part of the ER stress response100, ultimately leading to the upregulation of activating transcription factor 4 (ATF4), which can be transcribed despite eIF2α phosphorylation thanks to an IRES101. However, pharmacological ICD inducers fail to activate the canonical ER stress response elicited when unfolded proteins accumulating in the ER lumen displace heat shock protein family A (Hsp70) member 5 (HSPA5) from various signal transducers91. Such sensors of the unfolded protein response (UPR) include PERK (which promotes the ISR and ATF4 activation), ATF6 (which operates as stress-responsive transcription factor upon nuclear translocation) and endoplasmic reticulum to nucleus signaling 1 (ERN1, best known as IRE1)101. The latter functions by splicing the messenger RNA coding for X-box binding protein 1 (XBP1), which is yet another stress-responsive transcription factor101. Of note, the selective experimental inactivation of the IRE1–XBP1 pathway abolishes ICD-associated CALR exposure driven by cetuximab42, suggesting that this process relies on specific components of the ER stress response. Whether the apparent discrepancy in the involvement of the PERK–ATF4 versus IRE1–XBP1 axes in CALR exposure driven by different agents reflects specific features of the ICD inducer remains unclear.
The ISR is also required for induction of autophagy, a cytoprotective mechanism that is intimately connected to DAMP emission102. However, the ultimate impact of autophagy on adaptive immunity driven by RCD exhibits considerable context dependency. For instance, autophagy limits MHC class I exposure on the surface of pancreatic cancer cells (de facto limiting their antigenicity)103, the exposure of CALR by cancer cells undergoing PDT-driven ICD51 and type I IFN secretion by breast cancer cells succumbing to ICD upon irradiation74. Consistently, genetic signatures of proficient autophagy in diagnostic biopsies correlate with inhibited type I IFN and interferon gamma (IFN-γ) signaling as well as with poor disease outcomes in patients with (mostly luminal) breast cancer74. However, autophagy has also been mechanistically connected to optimal ATP release in the course of chemotherapy-driven ICD58,104. Accordingly, in samples from people with triple-negative breast cancer, signs of autophagic proficiency correlated with good prognosis and a favorable CTL/Treg cell ratio105. Moreover, both autophagy-stimulatory dietary regimens (such as intermittent fasting or ketogenic diets) as well as drugs (which have been dubbed caloric restriction mimetics) have been shown to promote tumor control by chemotherapy in a variety of preclinical tumor models104,106. While this beneficial effect was mechanistically linked to the activation of autophagy in malignant cells104, it may also involve autophagy induction in multiple immune compartments, including DCs and memory T cells107. However, direct evidence supporting a major role for autophagic responses in DCs or T cells for the elicitation of adaptive immunity downstream of ICD is missing.
Taken together, these observations exemplify the critical role of stress-responsive pathways in the adjuvanticity of cells undergoing ICD and delineate strong parallels between the mechanisms whereby mammalian cells respond to pathogen infection and to immunogenic drugs.
Hypothetical evolution of ICD
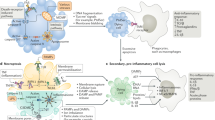
Innate immunity precedes its adaptive counterpart in both phylogeny and ontogeny. Thus, it appears plausible, although speculative, that the ability of mammals to mount antigen-specific responses to RCD has developed in subsequent waves (Fig. 2).

The ability of dying cells to initiate adaptive immunity has presumably emerged in the context of host–pathogen co-evolution through multiple stages. First, unicellular (and presumably colonial) eukaryotes acquired the ability to detect invading pathogens and mount defensive responses, such as the ISR and autophagy. Early during evolution, such responses may have been connected to the communication of a danger status to hitherto uninfected neighboring cells. Moreover, RCD may have emerged as a way to limit pathogen dissemination and/or to provide nutrients to healthy neighbors. The phagocytosis of infected cells (including cells dying in response to infection) stands out as a plausible subsequent step in the evolution of the ICD-sensing apparatus, with CALR emerging early a pro-phagocytic signal. Ultimately, a complex system involving innate only (first) and then adaptive (later) immune cells of multiple types has evolved to interpret the signals emitted by dying cells in support of the activation (or inhibition) of antigen-specific immunity. Ig, immunoglobulins; P, phosphate; TCR, T cell receptor.
The very first sets of responses to infectious pathogens affecting unicellular eukaryotes108 likely involved two core stress-responsive pathways: (1) the ISR, attenuating the translation of microbial (especially viral) mRNAs109 and (2) autophagy, mediating the sequestration and clearance of cytosolic pathogens110. Presumably, both these pathways would operate downstream of the detection of foreign components within infected cells, suggesting that at least some PRRs have also emerged early during evolution111,112. In support of this notion, TLRs are highly conserved across the eukaryotic domain113. Beyond a critical level of infection-imposed stress, unicellular eukaryotes would probably activate primordial variants of RCD to limit pathogen replication and/or provide nutrients to viable, potentially uninfected, neighbor cells114.
In a second step accompanying the acquisition of multicellularity, phagocytosis of infected and damaged cells by their healthy neighbors (initially non-professional phagocytes and later macrophage-like cells) has developed, allowing for the controlled destruction of microbial threats prior to dissemination as a consequence of RCD115. It is tempting to speculate that CALR exposure is (one of) the first phagocytic signal(s) that emerged during evolution, because the structure of CALR and its exposure pathway are phylogenetically conserved64,65,67. It is only much later in the course of evolution that multiple myeloid (and even later lymphoid) cell types came into action to build a sophisticated immune system yielding highly orchestrated phagocytic/innate (and later adaptive) responses to pathogens, sterile damage and malignant disorders116.
In the aforementioned scenario, mechanisms must have developed to link intracellular stress responses to danger management at the organismal level1. Such links explain why perturbations of intracellular homeostasis elicit signals, including ‘come get me’ and ‘eat me’ cues that attract phagocytes and ensure the removal of stressed cells even when they are still alive or shortly after they die117,118. In mammals, the nature of the signals emitted by stressed/dying cells and their contexture appear to determine the identity of phagocytes involved (for example, non-professional phagocytes versus macrophages versus DCs) as well as their activation and polarization, ultimately dictating the immunological outcome of the process119,120. Such outcomes can range from silent corpse removal (which has been dubbed efferocytosis)118,121 to sterile inflammation6 or the ICD-driven elicitation of adaptive immunity89.
Notably, RCD driven by sterile damage of normal cells (due to physical, chemical or nutritional stress) is most often handled by efferocytosis (if individual cells are affected) or a transient phase of inflammation (if a wound-healing response is required to compensate for the loss of supracellular units), largely reflecting poor antigenicity118,122. Conversely, the presence of infectious pathogens (or other sources of antigenicity) must give rise to an acute response that not only limits local perturbations of homeostasis, but also ignites adaptive immunity57. In this scenario, ICD may have emerged during host–pathogen co-evolution to facilitate the immune recognition of cells that are succumbing to intracellular pathogens. Interestingly, specific anticancer agents that have been empirically selected for clinical use on the basis of their efficacy apparently mimic multiple effects of pathogen infection, as they elicit intracellular stress pathways that are related to the host. This implies that the general principle underlying the perception of RCD driven by non-infectious challenges as immunogenic is a state of ‘viral mimicry’72 that recapitulates not only the antigenicity but also the adjuvanticity of RCD driven by pathogens.
If correct, this hypothetical scenario of ICD evolution implies that: (1) the DAMPs and PRRs involved in the immune response against pathogens and non-infectious ICD inducers overlap, and (2) the DAMP–PRR interactions and their downstream signals that are intercepted by pathogens, highlighting their biological importance, are also determinants of the outcome of ICD-driving cancer therapies.
Subversion of ICD by viruses and cancer cells
As a result of host–pathogen co-evolution, viral pathogens have developed numerous strategies to suppress the antigenicity and adjuvanticity of RCD and hence evade immune recognition. Most of these strategies are also harnessed by malignant cells, which must similarly escape immunosurveillance to progress into life-threatening neoplasms (Supplementary Table 1).
Rodent herpesvirus Peru (RHVP) encodes an ubiquitin ligase that causes degradation of MHC class I proteins123, while the Molluscum contagiosum virus protein MC80 targets tapasin for degradation to inhibit the loading of antigenic peptides onto MHC class I (ref. 124). Similarly, many cancer cells lose beta-2-microglobulin (B2M), which is required for MHC class I exposure on the plasma membrane125, or experience promoter hypermethylation or loss of heterozygosity at the HLA locus126,127. Finally, under the selective pressure of treatments, some tumors can selectively lose the expression of specific antigens, such as CD19 in the case of hematological malignancies treated with CD19-targeting chimeric-antigen-receptor-modified (CAR) T cells128. All these strategies exemplify mechanisms whereby pathogens and cancer cells evade (ICD-associated) immunosurveillance by minimizing antigenicity.
One obvious strategy for avoiding adaptive immunity downstream of ICD consists of subverting RCD itself. Indeed, multiple viruses encode inhibitors of key components of the RCD apparatus, including BCL2 associated X, apoptosis regulator (BAX)129, RIPK3 (a necroptosis mediator)130 and both pre-mitochondrial and post-mitochondrial caspases131,132,133. Moreover, various viral strains express proteins that resemble mammalian RCD inhibitors, such as BCL2 apoptosis regulator (BCL2)134, upregulate endogenous caspase inhibitors, like CASP8 and FADD like apoptosis regulator (CFLAR)135, or actively promote the degradation of caspases136,137 or RIPK3 (ref. 138). Similarly, most malignant cells exhibit the upregulation of one or more antiapoptotic members of the BCL2 family139,140 or escape RCD driven by CTLs (which is a form of ICD) by acquiring loss-of-function mutations in CASP8 (ref. 125). Moreover, malignancies exhibiting high CFLAR levels or reduced expression of RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL, another component of the necroptotic apparatus) exhibit poor prognosis linked to deficient immunosurveillance141,142. Similar strategies are also employed by intracellular bacteria. As a standalone example, Shigella flexneri encodes an ubiquitin ligase that targets core components of pyroptosis (another RCD variant)143 for degradation144.
A second more subtle strategy for subverting ICD relies on direct interference with DAMPs or the pathways that underlie their emission. Multiple viruses, including hepatitis C virus (HCV) and SARS-CoV-2, evolved stratagems to prevent PKR activation and consequent eIF2α phosphorylation145,146,147. HCV also favors the upregulation of endogenous protein phosphatase 1 regulatory subunit 15A (PPP1R15A, best known as GADD34), which promotes eIF2α deposphorylation145, while both coronaviruses and picornaviruses encode competitive inhibitors of phospho-eIF2α (ref. 148). Moreover, numerous viral strains, including SARS-CoV-2, encode CD47-like molecules or elicit the expression of endogenous CD47 as anti-phagocytic signals that antagonize CALR149,150. Along similar lines, malignant cells can subvert CALR signaling by (1) preventing PERK activation via V-set domain containing T cell activation inhibitor 1 (VTCN1, best known as B7-H4)151, (2) retaining CALR intracellularly upon interactions with stanniocalcin 1 (STC1)152, (3) limiting CALR binding sites on the cell surface (syalylated glycans)153; and (4) secreting a truncated version of CALR that act as decoy for CD91 (ref. 154). Supporting a key role for the ISR and consequent CALR exposure in cancer immunosurveillance, reduced eIF2α phosphorylation correlates with reduced CALR exposure, scarce immune infiltration by activated DCs and T cells and poor prognosis in people with acute myeloid leukemia155,156, non-small-cell lung cancer157 and ovarian carcinoma158. Conversely, multiple neoplasms overexpress CD47, generally correlating with poor disease outcome159,160,161.
HCV, as well as herpes simplex virus 1 (HSV1) and Epstein–Barr virus (EBV), encode autophagy-inhibiting factors or favor the upregulation of endogenous autophagy suppressors to avoid not only lysosomal degradation, but also the release of immunostimulatory ATP during pathogen-driven ICD162,163,164. Similarly, malignant cells (especially early during malignant transformation) exhibit suboptimal autophagic responses, at least in part because most oncosuppressor genes (which are lost during tumorigenesis) promote autophagy, while most oncogenes (which are amplified/hyperactivated during carcinogenesis) inhibit it165. Corroborating the notion that autophagy inactivation supports immunoevasion (at least in some clinical settings), markers of inhibited autophagy correlated with reduced CTL/Treg cell ratios and poor survival in cohorts of triple negative breast cancer (TNBC)105. As an alternative approach, cancer cells circumvent ICD-associated ATP release by upregulating (or favoring the upregulation by immune cells) of two ectonucleotidases that sequentially catalyze the conversion of extracellular ATP into adenosine (which is highly immunosuppressive)166, namely, ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39) and 5′-nucleotidase ecto (NT5E, best known as CD73)59,167. Elevated levels of CD39 or CD73 have been linked to immunosuppression and poor disease outcomes in various cohorts59,167. Malignant cells have also been shown to minimize the expression of proteins such as ANXA1 and HMGB1, which is associated with scarce immune infiltration in several cancers105,168. Moreover, cancer-cell-derived gelsolin (GSN) competitively disrupts the interaction between extracellular F-actin and CLEC9A on cDC1s, correlating with disrupted immunosurveillance and poor prognosis169.
A large panel of viral proteins inhibits PRR signaling, presumably reflecting the ancient evolutionary pressure on this aspect of pathogen–host co-evolution170. These factors encompass inhibitors of dsRNA sensors171,172 and their transducers173, dsDNA sensors174 and their transducers175,176,177,178 and the transcription factors executing the cellular response to foreign nucleic acids, including IRF3 and IRF7 (refs. 176,177,179,180,181,182). Along similar lines, cancer cells harness strategies to limit nucleic-acid-driven innate immune signaling in support of immunoevasion. For instance, malignant cells exposed to RT above 10–12 Gy per fraction upregulate three prime repair exonuclease 1 (TREX1), resulting in quenched type I IFN signaling downstream of accrued cytosolic dsDNA degradation183. Moreover, some malignant cells prevent optimal expression of CGAS, STING1 or signal transducers thereof by promoter hypermethylation184. Reflecting the advantage provided by these mechanisms to progressing neoplasms, low levels of nucleic acid sensors, their transducers or effectors have been linked to reduced immune infiltration and poor disease outcome in multiple patient cohorts72,185,186.
Taken together, these observations not only reinforce the parallels between the mechanisms employed by pathogens and malignant cells for evading ICD-driven immunity, but also highlight the critical relevance of these pathways for the immunosurveillance of infectious and malignant disorders.
Systemic defects compromising the immunogenicity of RCD in cancer
Multiple systemic defects limit the ability of RCD to elicit adaptive immunity culminating with an effector phase linked to immunological memory. Many such defects relate to germline single-nucleotide polymorphisms (SNPs) affecting PRRs or their transducers, which have been comprehensively reviewed elsewhere, especially in the context of infectious disorders187. Here, we will discuss organismal alterations impacting ICD driven by malignant cells.
Cancer diagnosis and treatment cause symptoms that meet the full clinical criteria of post-traumatic stress disorder (PTSD) in up to ~20% of cases, leading to long-term alterations in neuroendocrine circuitries including increased glucocorticoid tonus188. Stress-induced endogenous glucocorticoids, as well as the administration of synthetic glucocorticoid analogs (which is common in people with advanced cancer), not only promote metastatic dissemination of some malignant cells via intrinsic mechanisms189, but also compromise the immune response to malignant cells undergoing ICD owing to nuclear receptor subfamily 3 group C member 1 (NR3C1, best known as GR) activation and consequent upregulation of the immunosuppressive factor TSC22 domain family member 3 (TSC22D3) in DCs190. Accordingly, elevated TSC22D3 levels in the TME are a marker of poor prognosis in individuals with colorectal, gastric and lung cancer190.
In mice, some malignancies cause a systemic increase in catecholamines that (upon binding to β-adrenergic receptors) ultimately cause an ileopathy compromising the intestinal barrier function and perturbing the local microflora191. Intestinal dysbiosis is not only common among people with cancer, but also associated with poor prognosis, most likely as a consequence of inhibited immunosurveillance192. Multiple studies indicate that the prolonged use of broad-spectrum antibiotics, which decrease ecological diversity of gut microbes, is associated with limited immunotherapy efficacy in individuals with cancer192. Gnotobiotic or antibiotic-treated mice also manifest immune defects that compromise responses to antiviral vaccination193, as well as the anticancer effects of immune checkpoint inhibitors (ICIs)194,195. The latter defect can be reversed by fecal microbiome transplantation, establishing a cause–effect relationship between the reduction of microbial diversity and failing immunosurveillance194,195.
In this context, it is interesting to note that the necrotic demise of endocrine cells that occurs during autoimmune disorders is accompanied not only by DAMP release, but also by the liberation of hormones196. Although not necessarily driven by malignant cells, this process, which has been dubbed damage-induced release of endocrine factors (DIRE), may also compromise the initiation of tumor-targeting immune responses downstream of ICD via systemic alterations of hormonal tone196.
Yet another mechanism through which progressing neoplasms can compromise the ability of RCD to drive adaptive immunity is systemic inflammation. Thus, an increase in proinflammatory (and immunosuppressive) cytokines and circulating MDSCs is common in patients with advanced or terminal malignancies197,198. In this context, the repeated administration of cytotoxic agents that are commonly employed for cancer management can contribute to myelosuppression and lymphopenia, adding an iatrogenic dimension to the problem199,200. In addition, it appears plausible (but remains to be demonstrated) that the expansion of the tumor mass beyond a critical threshold is coupled to the progressive (and irreversible) anergy/exhaustion of increasingly outnumbered tumor antigen-specific CTLs despite the fact that large tumors contain a considerable fraction of cells undergoing RCD201.
An unbiased screen for potential ICD-relevant immune defects revealed that a loss-of-function SNP in FPR1 (rs867228) reduces progression-free and overall survival in individuals with breast cancer receiving adjuvant anthracycline-based chemotherapy60. Such an effect of rs867228 was epistatic to other loss-of-function SNPs in TLR3 and TLR4 (refs. 60,202), which have also been linked to differential responsiveness to ICD-inducing chemotherapeutics in a variety of clinical cohorts161. When present in the heterozygous state, rs867228 also compromises the clinical response to oxaliplatin-based chemotherapy in individuals with colorectal cancer60, as well as the 5-year overall survival of individuals with rectal cancer treated by neoadjuvant chemoradiotherapy203. Notably, rs867228 is also associated with the premature manifestation of all carcinomas, with major effects on luminal B breast cancer as well as colorectal and esophageal carcinomas204,205. In line with this notion, whole-body deletion of Fpr1 accelerates the development of carcinogen-driven luminal B-like mammary carcinomas in mice204.
Although formal evidence for this conjecture is still lacking, it appears plausible that rs867228 accelerates the development of epithelial cancers through an effect of immunosurveillance. Indeed, DCs from individuals harboring rs867228 exhibit reduced interaction with stressed/dying cancer cells60. Moreover, DCs from Fpr1−/− mice exhibit positional defects in vivo, as they fail to localize in the proximity of malignant cells undergoing ICD60, and have a reduced capacity to present soluble antigens to T cells in vitro204. Of note, FPR1 is the receptor for Yersinia pestis, the agent causing bubonic plague206, and it promotes inflammatory injury of the lung and the brain in preclinical models203,207, as well as immunosuppression in individuals with septic shock208. Thus, depending on the circumstances, loss-of-function polymorphisms of FPR1 may increase genetic fitness, providing a plausible explanation for the maintenance of rs867228 in the general population.
In summary, there is abundant evidence that both inherited and acquired systemic immune defects may compromise ICD-ignited adaptive immunity.
Restoring and reinforcing ICD in support of cancer immunosurveillance
Infected or malignant cells undergoing ICD may go unnoticed to immunosurveillance because of local or systemic defects. Each of these defects must be detected and counteracted to restore or reinforce ICD-driven immunity in a personalized fashion (Fig. 3).

Malignant cells have evolved a plethora of strategies to circumvent the detection of RCD as immunogenic. Each of these stratagems, encompassing altered autophagic responses coupled to defective ATP or type I IFN secretion, accrued extracellular ATP degradation leading to superior adenosinergic signaling, insufficient CALR exposure or ANXA1 secretion, CD47 upregulation, inhibited PRR signaling, as well as other strategies for local and systemic immunosuppression, offer specific targets to develop combinatorial therapeutic regimens in support of restored cancer immunosurveillance. COX, cyclooxygenase; mAbs, monoclonal antibodies.
Defective ATP secretion can be stimulated by autophagy induction104, and accrued ATP degradation can be counteracted by monoclonal antibodies or small molecules targeting CD39 and/or CD73 (refs. 59,167). Similarly, adenosine receptor antagonists may be used to limit adenosine-driven immunosuppression downstream of CD39 and CD73 (ref. 59). The sensitivity of ANXA1-defective tumors to immunogenic chemotherapy can be restored by intratumoral administration of recombinant CALR168 or systemic treatment with the TLR3 ligand polyinosinic:polycytidylic acid (polyI:C)204. Similarly, tumors developing in Fpr1–/– mice only respond to ICD-inducing chemotherapy plus polyI:C but not to either agent alone, consistent with polyI:C being able to restore the positional defect of tumor-infiltrating Fpr1–/– DCs204. Thus, polyI:C appears to overcome chemotherapy resistance in ANXA1- and FPR1-deficient contexts.
Several strategies have been proposed to overcome deficient ICD-associated CALR exposure, including: (1) ISR induction by intratumoral injection of ER stressors (such as thapsigargin)209, (2) ISR restoration via agents that stimulate B7-H4 degradation151, and (3) inhibition of the ISR-terminating eIF2α phosphatase, which can be achieved by disrupting the interaction between GADD34 and protein phosphatase 1 catalytic subunit alpha (PPP1CA, best known as PP-1A)210. Conversely, deficient type I IFN responses can be rescued by intratumoral injections of (1) PRR agonists (including TLR3, TLR9 and STING activators, as well as virus-like particles) or (2) recombinant type I IFN75,211,212. Moreover, autophagy inhibitors, as well as inhibitors of post-mitochondrial caspases (notably CASP3), have been shown to boost type I IFN secretion in the context of RT-driven ICD74,213,214. Such measures should not only improve the recruitment of immune effectors downstream of restored type I IFN signaling, but also upregulate MHC class I molecules on the surfaces of malignant cells75,103, rendering them recognizable by CTLs.
Alternative approaches to restore or reinforce ICD signaling involve the inhibition of endogenous suppressors of adaptive immunity elicited by ICD. Such targets include catecholamine signaling (with β blockers)191, gelsolin169, glucocorticoid receptors (with mifepristone)190, PGE2 (with cyclooxygenase inhibitors)215 and CD47 (with specific monoclonal antibodies)160,216. CD47-blocking antibodies are indeed being developed by several biotech firms and have undergone preliminary clinical evaluation for the treatment of hematological and solid malignancies.
Ever more studies report the development of complex delivery platforms that combine rapid and slow-release formulations to provide the sequential, temporally controlled (and sometimes light-, heat- or magnetic-field-induced) release of immunogenic chemotherapeutics and additional immunostimulatory molecules, most often PRR agonists217,218. The purpose of such formulations (which are generally delivered intratumorally) is to elicit local anticancer immune responses that, in an ideal scenario, can achieve systemic outreach and target other, non-treated lesions (in thus far resembling out-of-field, abscopal responses to RT)219. In some instances, such mixtures include viral or liposomal vectors coding for cytokines or ICI-like polypeptides (most often targeting PD-1 or its main ligand CD274, best known as PD-L1) aimed at locally amplifying the immune response218,220.
Combinatorial regimens encompassing ICD-inducing chemotherapeutics and Food and Drug Administration-approved ICIs targeting PD-1 or PD-L1 (and less so CTLA4) are being tested in hundreds of clinical trials, on the basis of the rationale that initially ‘cold’ tumors will become ‘hot’ upon ICD-eliciting chemotherapy and hence will respond to ICIs14,221. In such trials, chemotherapy cycles are usually reduced with respect to the clinical routine, with the aim to minimize side effects that would compromise anticancer immune responses, including myelosuppression and lymphopenia. Several excellent reviews cover this approach in detail222,223. However, it is important to note that the ISR, which is central to ICD signaling, reportedly causes the upregulation of PD-L1 on tumor cells224,225, providing yet another explanation for the beneficial effects of combining ICD-inducing chemotherapeutics with PD-1 or PD-L1 blockers.
Taken together, these observations identify multiple strategies that can be harnessed for restoring or reinforcing ICD to inform the development of combinatorial anticancer regimens with superior efficacy.
Conclusions and outlook
The term ICD has become part of the standard oncology vocabulary, spurring the development of new therapeutic agents, treatment combinations and personalization strategies. Although it appears highly plausible that ICD emerged during host–pathogen co-evolution, the term is not yet employed by the scientific community working on infectious disorders. Moreover, while ICD may be implicated in a variety of non-infectious, non-malignant disorders (Box 1), the term has not yet been widely accepted by this field. The future will tell whether the term ICD (or at least its underlying concept) will gain traction in disciplines other than oncology.
It is now clear that the immune system does not only recognize cells that harbor pathogen-encoded neoantigens or mutated/modified self-antigens, but also targets cells expressing stress-induced antigens20. Accordingly, normal breast epithelial cells treated with anthracyclines or RT in vitro can be administered to mice as a prophylactic vaccine to retard carcinogen-induced mammary oncogenesis226. Similarly, oxaliplatin-killed normal ileal cells administered with specific bacteria as adjuvants can elicit a protective immune response against colon cancer cells227. Finally, induction of an autoimmune disease against cholangiocytes can protect mice against oncogene-driven and transplanted cholangiocarcinomas228. These examples suggest that, if appropriately stressed, even non-transformed cells can elicit tumor-targeting immune responses evoking a sort of ‘beneficial autoimmunity’ that contributes to cancer immunosurveillance. Identifying the precise antigenic determinants of this phenomenon and determining whether a similar strategy can be employed in the clinics, for instance for immunoprophylaxis in genetically cancer-prone individuals, remain challenges for future investigation.
While many DAMPs emanating from cells that have succumbed to ICD have already been characterized, it appears possible that the list of ICD-relevant DAMPs is not yet exhaustive. For instance, multiple metabolites other than ATP are released from dying cancer cells, and one among these factors, spermidine, mediates local anti-inflammatory effects229, but can also stimulate the immune response in the context of immunogenic chemotherapy104, suggesting that it might also contribute to ICD. Finally, cells continue protein synthesis (at least for some time) even in the context of mild PMP, provided that ATP remains available229,230. While ICD is generally linked to transcription and translation inhibition, the impact of post-PMP protein synthesis on the secretion of ICD-relevant DAMPs and cytokines remains to be elucidated. Altogether, these observations call for additional experiments aimed at the unbiased identification of novel signals linked to the perception of RCD as immunogenic in multiple settings, followed by their functional validation.
It is our hope that spatially and temporally resolved omics technologies, as well as the ex vivo/in vitro examination of infected tissues and living tumors by three-dimensional videomicroscopy, will resolve this complexity, yielding important information that can be harnessed to modulate ICD for the treatment of multiple human disorders.
References
Galluzzi, L., Yamazaki, T. & Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 19, 731–745 (2018).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Bedoui, S., Herold, M. J. & Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 21, 678–695 (2020).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012).
Gong, T., Liu, L., Jiang, W. & Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112 (2020).
Fuchs, Y. & Steller, H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 16, 329–344 (2015).
Galluzzi, L. et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 8, e000337 (2020).
Sarhan, M., Land, W. G., Tonnus, W., Hugo, C. P. & Linkermann, A. Origin and consequences of necroinflammation. Physiol. Rev. 98, 727–780 (2018).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Montes de Oca, R. et al. Belantamab mafodotin (GSK2857916) drives immunogenic cell death and immune-mediated antitumor responses in vivo. Mol. Cancer Ther. 20, 1941–1955 (2021).
Kepp, O., Zitvogel, L. & Kroemer, G. Lurbinectedin: an FDA-approved inducer of immunogenic cell death for the treatment of small-cell lung cancer. Oncoimmunology 9, 1795995 (2020).
Xie, W. et al. Lurbinectedin synergizes with immune checkpoint blockade to generate anticancer immunity. Oncoimmunology 8, e1656502 (2019).
Galluzzi, L., Humeau, J., Buqué, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Petroni, G., Buqué, A., Zitvogel, L., Kroemer, G. & Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 39, 310–345 (2021).
Rodriguez-Ruiz, M. E., Vitale, I., Harrington, K. J., Melero, I. & Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 21, 120–134 (2020).
Clement, C. C. et al. Pleiotropic consequences of metabolic stress for the major histocompatibility complex class II molecule antigen processing and presentation machinery. Immunity 54, 721–736 (2021).
Klein, L., Kyewski, B., Allen, P. M. & Hogquist, K. A. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 14, 377–391 (2014).
Georgieva, M., Buckee, C. O. & Lipsitch, M. Models of immune selection for multi-locus antigenic diversity of pathogens. Nat. Rev. Immunol. 19, 55–62 (2019).
Zitvogel, L., Perreault, C., Finn, O. J. & Kroemer, G. Beneficial autoimmunity improves cancer prognosis. Nat. Rev. Clin. Oncol. 18, 591–602 (2021).
Katayama, H. et al. Protein citrullination as a source of cancer neoantigens. J. Immunother. Cancer 9, e002549 (2021).
Engelhard, V. H. et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 8, e000262 (2020).
Lhuillier, C. et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Invest. 131, e138740 (2021).
Melacarne, A. et al. Identification of a class of non-conventional ER-stress-response-derived immunogenic peptides. Cell Rep. 36, 109312 (2021).
Reits, E. A. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 203, 1259–1271 (2006).
Apavaloaei, A., Hardy, M. P., Thibault, P. & Perreault, C. The origin and immune recognition of tumor-specific antigens. Cancers 12, 2607 (2020).
Winter, M. et al. Deciphering the acute cellular phosphoproteome response to irradiation with X-rays, protons and carbon ions. Mol. Cell Proteomics 16, 855–872 (2017).
Galaine, J. et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 145, 3112–3125 (2019).
Zhou, Y. et al. Activation of NF-κB and p300/CBP potentiates cancer chemoimmunotherapy through induction of MHC-I antigen presentation. Proc. Natl Acad. Sci. USA 118, e2025840118 (2021).
Jhunjhunwala, S., Hammer, C. & Delamarre, L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 21, 298–312 (2021).
Paludan, S. R., Pradeu, T., Masters, S. L. & Mogensen, T. H. Constitutive immune mechanisms: mediators of host defence and immune regulation. Nat. Rev. Immunol. 21, 137–150 (2021).
Wolf, A. J. & Underhill, D. M. Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 18, 243–254 (2018).
Rehwinkel, J. & Gack, M. U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020).
Rubartelli, A. & Sitia, R. Stress as an intercellular signal: the emergence of stress-associated molecular patterns (SAMP). Antioxid. Redox. Signal 11, 2621–2629 (2009).
Aaes, T. L. & Vandenabeele, P. The intrinsic immunogenic properties of cancer cell lines, immunogenic cell death, and how these influence host antitumor immune responses. Cell Death Differ. 28, 843–860 (2021).
Bugaut, H. et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS ONE 8, e65181 (2013).
Schiavoni, G. et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 71, 768–778 (2011).
Tesniere, A. et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29, 482–491 (2010).
Yamazaki, T., Buqué, A., Ames, T. D. & Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology 9, 1721810 (2020).
Wang, Z. et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J. Clin. Invest. 129, 4850–4862 (2019).
Golden, E. B. et al. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 3, e28518 (2014).
Pozzi, C. et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 22, 624–631 (2016).
Liu, P. et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 10, 1486 (2019).
Petrazzuolo, A. et al. Pharmacological inhibitors of anaplastic lymphoma kinase (ALK) induce immunogenic cell death through on-target effects. Cell Death Dis. 12, 713 (2021).
Goel, S. et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017).
Petroni, G., Formenti, S. C., Chen-Kiang, S. & Galluzzi, L. Immunomodulation by anticancer cell cycle inhibitors. Nat. Rev. Immunol. 20, 669–679 (2020).
Spisek, R. et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 109, 4839–4845 (2007).
Gulla, A. et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2, 468–483 (2021).
Sequeira, G. R. et al. Enhanced antitumor immunity via endocrine therapy prevents mammary tumor relapse and increases immune checkpoint blockade sensitivity. Cancer Res. 81, 1375–1387 (2021).
Tatsuno, K. et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 10, 578 (2019).
Garg, A. D. et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 9, 1292–1307 (2013).
Garg, A. D. et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 31, 1062–1079 (2012).
Gomes-da-Silva, L. C. et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. EMBO J. 37, e98354 (2018).
Choi, J. et al. Visible-light-triggered prodrug nanoparticles combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. https://doi.org/10.1021/acsnano.1c03416 (2021).
Bommareddy, P. K., Zloza, A., Rabkin, S. D. & Kaufman, H. L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 8, 1591875 (2019).
Shekarian, T. et al. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci. Transl. Med. 11, eaat5025 (2019).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Kepp, O. et al. ATP and cancer immunosurveillance. EMBO J. 40, e108130 (2021).
Vacchelli, E. et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350, 972–978 (2015).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Sprooten, J. & Garg, A. D. Type I interferons and endoplasmic reticulum stress in health and disease. Int. Rev. Cell Mol. Biol. 350, 63–118 (2020).
Park, B. J. et al. Calreticulin, a calcium-binding molecular chaperone, is required for stress response and fertility in Caenorhabditis elegans. Mol. Biol. Cell 12, 2835–2845 (2001).
Madeo, F. et al. Phylogenetic conservation of the preapoptotic calreticulin exposure pathway from yeast to mammals. Cell Cycle 8, 639–642 (2009).
Sukkurwala, A. Q. et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 21, 59–68 (2014).
Tokuhiro, K. et al. Calreticulin is required for development of the cumulus oocyte complex and female fertility. Sci. Rep. 5, 14254 (2015).
Gardai, S. J. et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 (2005).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Ahrens, S. et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645 (2012).
Canton, J. et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 22, 140–153 (2021).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008).
Sistigu, A. et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014).
Harding, S. M. et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017).
Yamazaki, T. et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 21, 1160–1171 (2020).
Zitvogel, L., Galluzzi, L., Kepp, O., Smyth, M. J. & Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015).
Yatim, N. et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 (2015).
Aaes, T. L. et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 15, 274–287 (2016).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Lauber, K. et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730 (2003).
Suzuki, J., Denning, D. P., Imanishi, E., Horvitz, H. R. & Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013).
Segawa, K. et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 (2014).
Hangai, S. et al. Orchestration of myeloid-derived suppressor cells in the tumor microenvironment by ubiquitous cellular protein TCTP released by tumor cells. Nat. Immunol. 22, 947–957 (2021).
Ma, Y. et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013).
Minute, L. et al. Cellular cytotoxicity is a form of immunogenic cell death. J. Immunother. Cancer 8, e000325 (2020).
Jaime-Sanchez, P. et al. Cell death induced by cytotoxic CD8+ T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J. Immunother. Cancer 8, e000528 (2020).
Linkermann, A., Stockwell, B. R., Krautwald, S. & Anders, H. J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 14, 759–767 (2014).
Kraehenbuehl, L., Weng, C. H., Eghbali, S., Wolchok, J. D. & Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 19, 37–50 (2021).
Togashi, Y., Shitara, K. & Nishikawa, H. Regulatory T cells in cancer immunosuppression — implications for anticancer therapy. Nat. Rev. Clin. Oncol. 16, 356–371 (2019).
Sautès-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Galluzzi, L., Chan, T. A., Kroemer, G., Wolchok, J. D. & López-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 10, eaat7807 (2018).
Bezu, L. et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 25, 1375–1393 (2018).
Humeau, J. et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol. Med. 12, e11622 (2020).
Senovilla, L. et al. Immunosurveillance against cancer-associated hyperploidy. Oncotarget 3, 1270–1271 (2012).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Costa-Mattioli, M. & Walter, P. The integrated stress response: from mechanism to disease. Science 368, eaat5314 (2020).
Humeau, J. et al. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) in autophagy. Cell Death Dis. 11, 433 (2020).
Liu, Y. et al. The role of host eIF2α in viral infection. Virol. J. 17, 112 (2020).
Fucikova, J., Spisek, R., Kroemer, G. & Galluzzi, L. Calreticulin and cancer. Cell Res. 31, 5–16 (2021).
Panaretakis, T. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 28, 578–590 (2009).
Bagchi, P. Endoplasmic reticulum in viral infection. Int. Rev. Cell Mol. Biol. 350, 265–284 (2020).
Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438 (2020).
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
Yamamoto, K. et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020).
Pietrocola, F. et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell 30, 147–160 (2016).
Ladoire, S. et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 12, 864–875 (2016).
Ferrere, G. et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 6, e145207 (2021).
Clarke, A. J. & Simon, A. K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 19, 170–183 (2019).
Zhao, R. Y. Yeast for virus research. Microb. Cell 4, 311–330 (2017).
Knowles, A., Campbell, S., Cross, N. & Stafford, P. Bacterial manipulation of the integrated stress response: a new perspective on infection. Front. Microbiol. 12, 645161 (2021).
Mizushima, N. & Levine, B. Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576 (2020).
Hopfner, K. P. & Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521 (2020).
Vanpouille-Box, C., Demaria, S., Formenti, S. C. & Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34, 361–378 (2018).
Lind, N. A., Rael, V. E., Pestal, K., Liu, B. & Barton, G. M. Regulation of the nucleic acid-sensing Toll-like receptors. https://doi.org/10.1038/s41577-021-00577-0 (2021).
Büttner, S. et al. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J. Cell Biol. 175, 521–525 (2006).
Barreda, D. R., Neely, H. R. & Flajnik, M. F. Evolution of myeloid cells. https://doi.org/10.1128/microbiolspec.MCHD-0007-2015 (2016).
Mandujano-Tinoco, E. A., Sultan, E., Ottolenghi, A., Gershoni-Yahalom, O. & Rosental, B. Evolution of cellular immunity effector cells; perspective on cytotoxic and phagocytic cellular lineages. Cells 10, 1853 (2021).
Brown, G. C. & Neher, J. J. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem. Sci. 37, 325–332 (2012).
Boada-Romero, E., Martinez, J., Heckmann, B. L. & Green, D. R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 21, 398–414 (2020).
Zitvogel, L., Kepp, O. & Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 140, 798–804 (2010).
Rothlin, C. V., Hille, T. D. & Ghosh, S. Determining the effector response to cell death. Nat. Rev. Immunol. 21, 292–304 (2021).
Ranta, A. & Kumar, S. Recent advancements in role of TAM receptors on efferocytosis, viral infection, autoimmunity, and tissue repair. Int. Rev. Cell Mol. Biol. 357, 1–19 (2020).
López-Otín, C. & Kroemer, G. Hallmarks of health. Cell 184, 33–63 (2021).
Herr, R. A., Wang, X., Loh, J., Virgin, H. W. & Hansen, T. H. Newly discovered viral E3 ligase pK3 induces endoplasmic reticulum-associated degradation of class I major histocompatibility proteins and their membrane-bound chaperones. J. Biol. Chem. 287, 14467–14479 (2012).
Harvey, I. B., Wang, X. & Fremont, D. H. Molluscum contagiosum virus MC80 sabotages MHC-I antigen presentation by targeting tapasin for ER-associated degradation. PLoS Pathog. 15, e1007711 (2019).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
McGranahan, N. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271 (2017).
Garrido, F. HLA class-I expression and cancer immunotherapy. Adv. Exp. Med. Biol. 1151, 79–90 (2019).
Larson, R. C. & Maus, M. V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 21, 145–161 (2021).
Pauleau, A. L. et al. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene 26, 7067–7080 (2007).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012).
Dufour, F. et al. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 16, 256–271 (2011).
Galluzzi, L., Brenner, C., Morselli, E., Touat, Z. & Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 4, e1000018 (2008).
Imre, G. The involvement of regulated cell death forms in modulating the bacterial and viral pathogenesis. Int. Rev. Cell Mol. Biol. 353, 211–253 (2020).
Kvansakul, M., Caria, S. & Hinds, M. G. The Bcl-2 family in host–virus interactions. Viruses 9, 290 (2017).
Saito, K. et al. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 80, 4372–4379 (2006).
Garnett, T. O., Filippova, M. & Duerksen-Hughes, P. J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 13, 1915–1926 (2006).
Nie, Z. et al. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J. Virol. 81, 6947–6956 (2007).
Liu, Z. et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54, 247–258.e247 (2021).
Singh, R., Letai, A. & Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193 (2019).
Rasmussen, M. L. & Gama, V. A connection in life and death: the BCL-2 family coordinates mitochondrial network dynamics and stem cell fate. Int. Rev. Cell Mol. Biol. 353, 255–284 (2020).
Safa, A. R., Kamocki, K., Saadatzadeh, M. R. & Bijangi-Vishehsaraei, K. c-FLIP, a novel biomarker for cancer prognosis, immunosuppression, Alzheimer’s disease, chronic obstructive pulmonary disease (COPD), and a rationale therapeutic target. Biomark J. 5, 4 (2019).
Stoll, G. et al. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology 6, e1299302 (2017).
Broz, P., Pelegrin, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020).
Luchetti, G. et al. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 29, 521–1530 (2021).
Ruggieri, A. et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12, 71–85 (2012).
Gao, B. et al. Inhibition of anti-viral stress granule formation by coronavirus endoribonuclease nsp15 ensures efficient virus replication. PLoS Pathog. 17, e1008690 (2021).
Toroney, R., Nallagatla, S. R., Boyer, J. A., Cameron, C. E. & Bevilacqua, P. C. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J. Mol. Biol. 400, 393–412 (2010).
Rabouw, H. H. et al. Inhibition of the integrated stress response by viral proteins that block p-eIF2–eIF2B association. Nat. Microbiol. 5, 1361–1373 (2020).
Cameron, C. M., Barrett, J. W., Mann, M., Lucas, A. & McFadden, G. Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation in vivo. Virology 337, 55–67 (2005).
Tal, M. C. et al. Upregulation of CD47 is a host checkpoint response to pathogen recognition. mBio 11, e01293-20 (2020).
Song, X. et al. Pharmacologic suppression of B7-H4 glycosylation restores antitumor immunity in immune-cold breast cancers. Cancer Discov. 10, 1872–1893 (2020).
Lin, H. et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell 39, 480–493.e486 (2021).
Feng, M. et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat. Commun. 9, 3194 (2018).
Liu, P. et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol. Cell 77, 748–760.e749 (2020).
Fucikova, J. et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 128, 3113–3124 (2016).
Truxova, I. et al. Calreticulin exposure on malignant blasts correlates with improved natural killer cell-mediated cytotoxicity in acute myeloid leukemia patients. Haematologica 105, 1868–1878 (2020).
Fucikova, J. et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 76, 1746–1756 (2016).
Kasikova, L. et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 7, 312 (2019).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009).
Willingham, S. B. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 109, 6662–6667 (2012).
Fucikova, J. et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front. Immunol. 6, 402 (2015).
Rubio, R. M. & Mohr, I. Inhibition of ULK1 and Beclin1 by an α-herpesvirus Akt-like Ser/Thr kinase limits autophagy to stimulate virus replication. Proc. Natl Acad. Sci. USA 116, 26941–26950 (2019).
Ylä-Anttila, P., Gupta, S. & Masucci, M. G. The Epstein–Barr virus deubiquitinase BPLF1 targets SQSTM1/p62 to inhibit selective autophagy. Autophagy 17, 3461–3474 (2021).
Shiode, Y. et al. Hepatitis C virus enhances Rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci. Rep. 10, 15290 (2020).
Rybstein, M. D., Bravo-San Pedro, J. M., Kroemer, G. & Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 20, 243–251 (2018).
Allard, B., Allard, D., Buisseret, L. & Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 611–629 (2020).
Moesta, A. K., Li, X. Y. & Smyth, M. J. Targeting CD39 in cancer. Nat. Rev. Immunol. 20, 739–755 (2020).
Baracco, E. E. et al. Contribution of annexin A1 to anticancer immunosurveillance. Oncoimmunology 8, e1647760 (2019).
Giampazolias, E. et al. Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell 184, 4016–4031.e4022 (2021).
Chan, Y. K. & Gack, M. U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373 (2016).
Swedan, S., Andrews, J., Majumdar, T., Musiyenko, A. & Barik, S. Multiple functional domains and complexes of the two nonstructural proteins of human respiratory syncytial virus contribute to interferon suppression and cellular location. J. Virol. 85, 10090–10100 (2011).
Wen, W. et al. Seneca Valley virus 2C and 3C inhibit type I interferon production by inducing the degradation of RIG-I. Virology 535, 122–129 (2019).
Ekanayaka, P. et al. Foot-and-mouth disease virus VP1 target the MAVS to inhibit type-I interferon signaling and VP1 E83K mutation results in virus attenuation. PLoS Pathog. 16, e1009057 (2020).
Wang, Z. et al. PCV2 targets cGAS to inhibit type I interferon induction to promote other DNA virus infection. PLoS Pathog. 17, e1009940 (2021).
Christensen, M. H. et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35, 1385–1399 (2016).
Dalrymple, N. A., Cimica, V. & Mackow, E. R. Dengue virus NS proteins inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation: dengue virus serotype 1 NS4A is a unique interferon-regulating virulence determinant. mBio 6, e00553–00515 (2015).
Vazquez, C. et al. SARS-CoV-2 viral proteins NSP1 and NSP13 inhibit interferon activation through distinct mechanisms. PLoS ONE 16, e0253089 (2021).
Liang, Y. et al. Hepatitis C virus NS4B induces the degradation of TRIF to inhibit TLR3-mediated interferon signaling pathway. PLoS Pathog. 14, e1007075 (2018).
Yuen, C. K. et al. Suppression of type I interferon production by human T-cell leukemia virus type 1 oncoprotein tax through inhibition of IRF3 phosphorylation. J. Virol. 90, 3902–3912 (2016).
Rieder, M. et al. Genetic dissection of interferon-antagonistic functions of rabies virus phosphoprotein: inhibition of interferon regulatory factor 3 activation is important for pathogenicity. J. Virol. 85, 842–852 (2011).
Chapon, M., Parvatiyar, K., Aliyari, S. R., Zhao, J. S. & Cheng, G. Comprehensive mutagenesis of herpes simplex virus 1 genome identifies UL42 as an inhibitor of type I interferon induction. J. Virol. 93, e01446-19 (2019).
Aslam, B. et al. Structural modeling and analysis of dengue-mediated inhibition of interferon signaling pathway. Genet. Mol. Res. 14, 4215–4237 (2015).
Vanpouille-Box, C. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618 (2017).
Kim, H. et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 12, eaaz568 (2020).
Chen, Y. J. et al. Interferon regulatory factor family influences tumor immunity and prognosis of patients with colorectal cancer. J. Transl. Med. 19, 379 (2021).
Chon, H. J. et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 10, 4932–4938 (2019).
Mukherjee, S., Huda, S. & Sinha Babu, S. P. Toll-like receptor polymorphism in host immune response to infectious diseases: a review. Scand. J. Immunol. 90, e12771 (2019).
Cordova, M. J., Riba, M. B. & Spiegel, D. Post-traumatic stress disorder and cancer. Lancet Psychiatry 4, 330–338 (2017).
Obradović, M. M. S. et al. Glucocorticoids promote breast cancer metastasis. Nature 567, 540–544 (2019).
Yang, H. et al. Stress–glucocorticoid–TSC22D3 axis compromises therapy-induced antitumor immunity. Nat. Med. 25, 1428–1441 (2019).
Yonekura, S. et al. Cancer-induced ileopathy and dysbiosis contribute to carcinogenesis. Cancer Discov. (in the press).
Derosa, L. et al. Microbiota-centered interventions: the next breakthrough in immuno-oncology. Cancer Discov. 11, 2396–2412 (2021).
Hagan, T. et al. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell 178, 1313–1328 (2019).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Tonnus, W. et al. The role of regulated necrosis in endocrine diseases. Nat. Rev. Endocrinol. 17, 497–510 (2021).
Shalapour, S. & Karin, M. Cruel to be kind: epithelial, microbial, and immune cell interactions in gastrointestinal cancers. Annu. Rev. Immunol. 38, 649–671 (2020).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Di Maio, M. et al. Chemotherapy-induced neutropenia and treatment efficacy in advanced non-small-cell lung cancer: a pooled analysis of three randomised trials. Lancet Oncol. 6, 669–677 (2005).
Ménétrier-Caux, C., Ray-Coquard, I., Blay, J. Y. & Caux, C. Lymphopenia in cancer patients and its effects on response to immunotherapy: an opportunity for combination with cytokines? J. Immunother. Cancer 7, 85 (2019).
Robertson-Tessi, M., El-Kareh, A. & Goriely, A. A mathematical model of tumor–immune interactions. J. Theor. Biol. 294, 56–73 (2012).
Vacchelli, E., Enot, D. P., Pietrocola, F., Zitvogel, L. & Kroemer, G. Impact of pattern recognition receptors on the prognosis of breast cancer patients undergoing adjuvant chemotherapy. Cancer Res. 76, 3122–3126 (2016).
Li, Z. et al. Formyl peptide receptor 1 signaling potentiates inflammatory brain injury. Sci. Transl. Med. 13, eabe9890 (2021).
Le Naour, J. et al. A TLR3 ligand reestablishes chemotherapeutic responses in the context of FPR1 deficiency. Cancer Discov. 11, 408–423 (2021).
Sztupinszki, Z. et al. A major genetic accelerator of cancer diagnosis: rs867228 in FPR1. Oncoimmunology 10, 1859064 (2021).
Osei-Owusu, P., Charlton, T. M., Kim, H. K., Missiakas, D. & Schneewind, O. FPR1 is the plague receptor on host immune cells. Nature 574, 57–62 (2019).
Dorward, D. A. et al. Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax 72, 928–936 (2017).
Kwon, W. Y. et al. Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock. Proc. Natl Acad. Sci. USA 118, e2018538118 (2021).
Martins, I. et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 30, 1147–1158 (2011).
Kepp, O. et al. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle 8, 3971–3977 (2009).
Le Naour, J., Galluzzi, L., Zitvogel, L., Kroemer, G. & Vacchelli, E. Trial watch: TLR3 agonists in cancer therapy. Oncoimmunology 9, 1771143 (2020).
Le Naour, J., Zitvogel, L., Galluzzi, L., Vacchelli, E. & Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 9, 1777624 (2020).
Rodriguez-Ruiz, M. E. et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019).
Yamazaki, T. & Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 9, 1797292 (2020).
Hayashi, K. et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat. Commun. 11, 6299 (2020).
Sikic, B. I. et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 37, 946–953 (2019).
Kepp, O., Marabelle, A., Zitvogel, L. & Kroemer, G. Oncolysis without viruses — inducing systemic anticancer immune responses with local therapies. Nat. Rev. Clin. Oncol. 17, 49–64 (2020).
Banstola, A., Poudel, K., Kim, J. O., Jeong, J. H. & Yook, S. Recent progress in stimuli-responsive nanosystems for inducing immunogenic cell death. J. Control. Release 337, 505–520 (2021).
Ngwa, W. et al. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322 (2018).
Nooraei, S. et al. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnology 19, 59 (2021).
Fabian, K. P., Wolfson, B. & Hodge, J. W. From immunogenic cell death to immunogenic modulation: select chemotherapy regimens induce a spectrum of immune-enhancing activities in the tumor microenvironment. Front. Oncol. 11, 728018 (2021).
Meric-Bernstam, F., Larkin, J., Tabernero, J. & Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 397, 1010–1022 (2021).
Salas-Benito, D. et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discov. 11, 1353–1367 (2021).
Suresh, S. et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat. Cancer 1, 533–545 (2020).
Suresh, S. & O’Donnell, K. A. Translational control of immune evasion in cancer. Trends Cancer 7, 580–582 (2021).
Buqué, A. et al. Immunoprophylactic and immunotherapeutic control of hormone receptor-positive breast cancer. Nat. Commun. 11, 3819 (2020).
Roberti, M. P. et al. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat. Med. 26, 919–931 (2020).
Paillet, J. et al. Autoimmunity affecting the biliary tract fuels the immunosurveillance of cholangiocarcinoma. J. Exp. Med. 218, e20200853 (2021).
Medina, C. B. et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 580, 130–135 (2020).
Saelens, X. et al. Protein synthesis persists during necrotic cell death. J. Cell Biol. 168, 545–551 (2005).
Szekanecz, Z. et al. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 17, 585–595 (2021).
Raffin, C., Vo, L. T. & Bluestone, J. A. Treg cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Roda, G. et al. Crohn’s disease. Nat. Rev. Dis. Primers 6, 22 (2020).
Lochhead, R. B., Strle, K., Arvikar, S. L., Weis, J. J. & Steere, A. C. Lyme arthritis: linking infection, inflammation and autoimmunity. Nat. Rev. Rheumatol. 17, 449–461 (2021).
Picardo, M. et al. Vitiligo. Nat. Rev. Dis. Primers 1, 15011 (2015).
Kallwellis-Opara, A. et al. Autoimmunological features in inflammatory cardiomyopathy. Clin. Res. Cardiol. 96, 469–480 (2007).
Anderton, H., Wicks, I. P. & Silke, J. Cell death in chronic inflammation: breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 16, 496–513 (2020).
Crow, Y. J. & Manel, N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440 (2015).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Patel, P. & Karch, J. Regulation of cell death in the cardiovascular system. Int. Rev. Cell Mol. Biol. 353, 153–209 (2020).
Pandolfi, J. B. et al. ATP-induced inflammation drives tissue-resident TH17 cells in metabolically unhealthy obesity. J. Immunol. 196, 3287–3296 (2016).
Melani, A. et al. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 26, 974–982 (2006).
Sánchez, D. et al. Anti-calreticulin antibodies and calreticulin in sera of patients diagnosed with dilated or hypertrophic cardiomyopathy. Autoimmunity 49, 554–562 (2016).
Komurasaki, R. et al. LKM-1 sera from autoimmune hepatitis patients that recognize ERp57, carboxylesterase 1 and CYP2D6. Drug Metab. Pharmacokinet. 25, 84–92 (2010).
Caorsi, C. et al. Protein disulfide isomerase A3-specific TH1 effector cells infiltrate colon cancer tissue of patients with circulating anti-protein disulfide isomerase A3 autoantibodies. Transl. Res. 171, 17–28 (2016).
Régent, A. et al. Identification of target antigens of anti-endothelial cell antibodies in patients with anti-neutrophil cytoplasmic antibody-associated vasculitides: a proteomic approach. Clin. Immunol. 153, 123–135 (2014).
Yang, W. et al. The role of protein disulphide-isomerase A3 as autoantigen in the pathogenesis of autoimmune thyroiditis and related brain damage in adult mice. Clin. Immunol. 212, 108350 (2020).
Bruschi, M. et al. Annexin A1 and autoimmunity: from basic science to clinical applications. Int. J. Mol. Sci. 19, 1348 (2018).
Mihaylova, N. et al. Suppression of autoreactive T and B lymphocytes by anti-annexin A1 antibody in a humanized NSG murine model of systemic lupus erythematosus. Clin. Exp. Immunol. 199, 278–293 (2020).
Manganelli, V. et al. Elevated serum level of HMGB1 in patients with the antiphospholipid syndrome. J. Immunol. Res. 2017, 4570715 (2017).
Schaper, F. et al. Autoantibodies to box A of high mobility group box 1 in systemic lupus erythematosus. Clin. Exp. Immunol. 188, 412–419 (2017).
Acknowledgements
We are indebted to G. Petroni (Weill Cornell Medical College) for help with figure preparation. G. K. is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Ruban Rose”; Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; the European Joint Programme on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); the Leducq Foundation; a Cancer Research ASPIRE Award from the Mark Foundation; the RHU Torino Lumière; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. L. G.’s lab is supported by a Breakthrough Level 2 grant from the US DoD BRCP (#BC180476P1), by the 2019 Laura Ziskin Prize in Translational Research (no. ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a startup grant from the Department of Radiation Oncology at Weill Cornell Medicine, by a Rapid Response Grant from the Functional Genomics Initiative, by industrial collaborations with Lytix Biopharma and Phosplatin, and by donations from Phosplatin, the Luke Heller TECPR2 Foundation, Sotio a.s., Onxeo and Noxopharm (Chatswood, Australia).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
G. K. has held research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Samsara, Sanofi, Sotio, Vascage and Vasculox/Tiom, is on the Board of Directors of the Bristol Myers Squibb Foundation France, and is a scientific co-founder of everImmune, Samsara Therapeutics and Therafast Bio. L. Z. has held research contracts with GlaxoSmythKline, Incyte, Lytix, Kaleido, Innovate Pharma, Daiichi Sankyo, Merus, Transgene, Tusk and Roche, is on the board of directors of Transgene and is a co-founder of everImmune. L. G. has held research contracts with Lytix Biopharma and Phosplatin, and has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, Sotio, The Longevity Labs, Inzen and the Luke Heller TECPR2 Foundation. All other authors have no conflicts to declare.
Peer review
Peer review information
Nature Immunology thanks Andreas Linkermann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Table 1
Supplementary Table 1
Rights and permissions
About this article

Cite this article
Kroemer, G., Galassi, C., Zitvogel, L. et al. Immunogenic cell stress and death. Nat Immunol 23, 487–500 (2022). https://doi.org/10.1038/s41590-022-01132-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-022-01132-2
This article is cited by
-
Targeting of focal adhesion kinase enhances the immunogenic cell death of PEGylated liposome doxorubicin to optimize therapeutic responses of immune checkpoint blockade
Journal of Experimental & Clinical Cancer Research (2024)
-
Classification and clinical significance of immunogenic cell death-related genes in Plasmodium falciparum infection determined by integrated bioinformatics analysis and machine learning
Malaria Journal (2024)
-
Cancer immunometabolism: advent, challenges, and perspective
Molecular Cancer (2024)
-
In situ chemoimmunotherapy hydrogel elicits immunogenic cell death and evokes efficient antitumor immune response
Journal of Translational Medicine (2024)
-
Shifting the paradigm: engaging multicellular networks for cancer therapy
Journal of Translational Medicine (2024)
