
Abstract
Cellular senescence is a state of irreversible growth arrest; however, the metabolic processes of senescent cells remain active. Our previous studies have shown that radiation induces senescence of human breast cancer cells that display low expression of securin, a protein involved in control of the metaphase–anaphase transition and anaphase onset. In this study, the protein expression profile of senescent cells was resolved by two-dimensional gel electrophoresis to investigate associated metabolic alterations. We found that radiation induced the expression and activation of glyceraldehyde-3-phosphate dehydrogenase that has an important role in glycolysis. The activity of lactate dehydrogenase A, which is involved in the conversion of pyruvate to lactate, the release of lactate and the acidification of the extracellular environment, was also induced. Inhibition of glycolysis by dichloroacetate attenuated radiation-induced senescence. In addition, radiation also induced activation of the 5′-adenosine monophosphate-activated protein kinase (AMPK) and nuclear factor kappa B (NF-κB) pathways to promote senescence. We also found that radiation increased the expression of monocarboxylate transporter 1 (MCT1) that facilitates the export of lactate into the extracellular environment. Inhibition of glycolysis or the AMPK/NF-κB signalling pathways reduced MCT1 expression and rescued the acidification of the extracellular environment. Interestingly, these metabolic-altering signalling pathways were also involved in radiation-induced invasion of the surrounding, non-irradiated breast cancer and normal endothelial cells. Taken together, radiation can induce the senescence of human breast cancer cells through metabolic alterations.
Similar content being viewed by others


Main
Cellular senescence is a state of irreversible cell growth arrest that occurs when cells experience potentially oncogenic stress.1 In addition, senescence can be stimulated by diverse factors, such as telomere dysfunction and oxidative damage, as well as the DNA damage response caused by ionising radiation and several chemotherapeutic drugs.2 Senescence is considered a tumour-suppressive program that halts the proliferation of damaged cells. However, senescent cells remain metabolically active and undergo a plethora of changes in protein expression and the secretion of cytokines and other factors involved in the senescence-associated secretory phenotype (SASP).3 It is well established that SASP is associated with the development of a pro-oncogenic environment.4 In the 1920s, Otto Warburg discovered that cancer cells convert glucose to lactate via a high rate of glycolysis despite oxygen availability. This phenomenon of aerobic glycolysis is known as the ‘Warburg effect’.5 Today, aerobic glycolysis is considered a hallmark of cancer.6 Excess lactate produced by aerobic glycolysis is released into tumour microenvironment, conferring advantages of tumour survival and growth.7 In numerous tumour types, glycolysis genes are overexpressed, leading to altered cancer metabolism.8, 9, 10 Although the Warburg effect is a well-studied phenomenon, its interplay with senescence is largely unclear.
The protein 5′-adenosine monophosphate-activated protein kinase (AMPK) is Ser/Thr protein kinase that is a crucial sensor of energy status that has an important role in the cellular response to metabolic stress.11 During periods of energetic stress, AMPK is activated in response to an increased AMP/ATP ratio and inhibits cell proliferation.12, 13 In addition to the AMP/ATP ratio, AMPK can also be activated in response to DNA-damaging agents, such as etoposide, doxorubicin and ionising radiation.14, 15 Several oncogenic mutations and signalling pathways can suppress AMPK signalling, which uncouples the fuel signals from the growth signals and allow tumour cells to divide under abnormal nutrient conditions. This uncoupling permits tumour cells to respond to inappropriate growth signalling pathways that are activated by oncogenes and the loss of tumour suppressors.11
The nuclear factor kappa B (NF-κB) transcription factors control the expression of genes involved in many critical physiological responses, including the immune and acute phase inflammatory response, cell adhesion, differentiation, oxidative stress responses and apoptosis. Immunity and tumourigenesis also involve a rapid rate of cell division. These conditions demand substantial bioenergetic and biosynthetic requirements, which the cells meet by increasing glucose metabolism.16 The activation of NF-κB signalling enhances the rate of aerobic glycolysis and upregulates glucose transporter 3. In addition, glycolysis drives NF-κB activation, thus forming a positive-feedback loop.17
In this study, we investigated the metabolic alterations associated with radiation-induced senescence. We found that the rate of glycolysis was enhanced in the senescent cells through the AMPK and NF-κB signalling pathways. These metabolic alterations were required for radiation-induced senescence. In addition, the alteration also acted as bystander signalling to promote the invasion of non-irradiated, surrounding cells. Our results provide a novel view of cancer metabolism during radiation-induced senescence.
Results
Radiation induces senescence through the enhancement of glycolysis
Although cellular senescence is a state of growth arrest, senescent cells are metabolically active.3 To gain further insight into the regulation of senescence, a proteomic study using two-dimensional (2D) gel electrophoresis was conducted. According to our previous studies,18, 19 radiation induces senescence in securin-knockdown MDA-MB-231-2A human breast cancer cells but has no effect in parental MDA-MB-231 cells (Figure 1a). Securin gene knockdown may switch radiation-induced apoptosis to growth arrest and senescence, and thus only minimal apoptosis is observed in irradiated MDA-MB-231-2A cells.18, 19 Therefore, MDA-MB-231-2A cells could potentially serve as a suitable model for cellular senescence. For proteomic study, MDA-MB-231-2A cells were irradiated and then allowed to recover for 4 h. Cell lysates were resolved by 2D gel electrophoresis (Supplementary Figure S1A). Among the 69 protein spots that displayed twofold differences, 10 protein spots (designated by arrows in Supplementary Figure S1A) were selected for further mass spectrometric analyses (Table 1). The mRNA expression of these identified proteins was not altered after 4 h of post-irradiation time (Supplementary Figure S1B), suggesting that the expression may be altered immediately after irradiation. Interestingly, the energy-producing enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), involved in glycolysis, was upregulated (Table 1). This finding was further validated by western blot analysis (Figure 1b, upper part) and GAPDH enzymatic activity assay (Supplementary Figure S2A). In contrast, radiation did not alter the expression level of GAPDH in parental MDA-MB-231 cells (Supplementary Figure S3). GAPDH has long been recognised as an important enzyme in ATP production through the anaerobic glycolysis of a monosaccharide (generally glucose) to pyruvate in the cytosol.20 Therefore, we hypothesise that glycolysis is enhanced in radiation-induced senescent cells.

Radiation enhances senescence and glycolysis in securin-knockdown MDA-MB-231-2A cells. (a) Left: Total extracts of MDA-MB-231 and MDA-MB-231-2A cells were prepared for western blot analyses. Right: both cells were exposed to 6 Gy radiation. After 5 days of recovery, cellular senescence was examined by senescence-associated β-galactosidase (SA-β-gal) staining. Senescent cells (blue staining) were observed by bright-field microscopy. (b–d) MDA-MB-231-2A cells were exposed to 6 Gy radiation followed by various recovery periods. The protein expression was examined by western blot analyses (b). The lactate concentration (c) and pH (d) of the culture medium were examined by protocols described in the Materials and Methods section. *P<0.05 or **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicates significant differences compared with time zero. (e) MDA-MB-231-2A cells were pretreated with 5 or 10 mM DCA for 2 h and then exposed to 6 Gy radiation. After 2 days of recovery, cellular senescence was examined by SA-β-gal staining. Senescent cells (blue staining) were observed by bright-field microscopy, and the percentage of SA-β-gal-positive cells was quantified. **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells
Lactate dehydrogenase A (LDHA) catalyses the conversion of pyruvate to lactate in cancer cells.21 Tyrosine phosphorylation of LDHA enhances its enzyme activity.22 Radiation induces transient phosphorylation of LDHA and an increase of LDH enzymatic activity in MDA-MB-231-2A cells (Figure 1b, lower part, and Supplementary Figure S2B). However, only residual induction of LDHA phosphorylation was observed in MDA-MB-231 cells (Supplementary Figure S3). Consistently, the rate of glycolysis was enhanced in irradiated MDA-MB-231-2A cells, as indicated by increased extracellular lactate and decreased cell culture medium pH (Figures 1c and d). To exclude the possibility that radiation-induced metabolic alterations were resulted from the genetic manipulation of MDA-MB-231-2A cells, another human breast cancer cells with low-securin expression (MCF-7) were used. Our previous study has shown that the securin expression level in MCF-7 cells is lower than that in MDA-MB-231 cells (also shown in Supplementary Figure S4A), and radiation also induces senescence in MCF-7 cells.19 Consistent with the results in MDA-MB-231-2A cells, radiation also increased the extracellular level of lactate (Supplementary Figure S4B).
To investigate whether enhanced glycolysis is involved in radiation-induced senescence, MDA-MB-231-2A cells were treated with the glycolytic inhibitor dichloroacetate (DCA) before irradiation. DCA is an inhibitor of pyruvate dehydrogenase kinase (PDK). Inhibition of PDK shifts pyruvate metabolism from glycolysis and lactate production to glucose oxidation in the mitochondria.23 Radiation-induced senescence was inhibited by DCA (Figure 1e), suggesting that enhanced glycolysis promotes radiation-induced senescence.
AMPK is required for radiation-induced glycolytic enhancement and senescence
AMPK, an energy sensor monitoring the cellular energy status, has been reported to regulate metabolism and survival in response to ionising radiation.11, 24 To study the role of AMPK in radiation-induced senescence, the activity of AMPK in irradiated MDA-MB-231-2A and MCF-7 cells was examined via analysis of Thr-172 phosphorylation. As shown in Figure 2a and Supplementary Figure S4C, radiation induced sustained AMPK phosphorylation. Compound C is a reversible and ATP-competitive inhibitor of AMPK.25 Radiation-induced AMPK phosphorylation was inhibited by compound C (Figure 2b and Supplementary Figure S4D). In addition, compound C inhibited radiation-induced senescence, LDH enzymatic activity and lactate release (Figures 2c–e). These results suggest that radiation activates AMPK to enhance glycolysis and induce senescence.

Radiation induced AMPK-dependent senescence and glycolytic enhancement in securin-knockdown MDA-MB-231-2A cells. (a) MDA-MB-231-2A cells were exposed to 6 Gy radiation followed by various recovery periods. The protein expression was examined by western blot analyses. (b) MDA-MB-231-2A cells were pretreated with 5–20 μM compound C for 2 h and then exposed to 6 Gy radiation. The protein expression was examined by western blot analyses after an 8-h recovery period. (c) MDA-MB-231-2A cells were pretreated with 5 or 10 μM compound C for 2 h and then exposed to 6 Gy radiation. After 2 days of recovery, cellular senescence was examined by senescence-associated β-galactosidase (SA-β-gal) staining. Senescent cells (blue staining) were observed by bright-field microscopy, and the percentage of SA-β-gal-positive cells was quantified. **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicate significant differences between inhibitor-treated and untreated cells. (d) MDA-MB-231-2A cells were pretreated with 5 or 20 μM compound C for 2 h, and then exposed to 6 Gy radiation. After a 24-h recovery period, the LDH enzymatic activity was examined by protocols described in the Materials and Methods section. **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells. (e) MDA-MB-231-2A cells were pretreated with 10 μM compound C for 2 h, and then exposed to 6 Gy radiation. After a 24-h recovery period, the lactate concentration in the culture medium was examined by protocols described in the Materials and Methods section. **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells
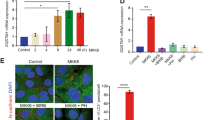
Radiation induces monocarboxylate transporter 1 expression through the AMPK/glycolysis pathway
Monocarboxylate transporters (MCTs) function as proton symporters and are stereoselective for L-lactate.26 To investigate whether the MCT upregulation is responsible for the release of lactate, MCT1 expression was examined because it is the most ubiquitous MCT.27 Radiation induced MCT1 expression in MDA-MB-231-2A cells (Figure 3a), and the increase was inhibited by compound C although compound C alone induced MCT1 expression (Figure 3b, left part and Supplementary Figure S5). To confirm these results, immunofluorescence staining was performed. As shown in Figure 3c, MCT1 protein expression (green fluorescence) localised to the membrane and cytoplasm was induced after irradiation. Consistently, compound C inhibited radiation-induced MCT1 expression (Figure 3c). These results suggest that radiation induces MCT1 expression through the activation of AMPK. Interestingly, the inhibition of glycolysis by DCA also reduced radiation-induced MCT1 expression (Figure 3b, right part and Figure 3c), suggesting that the enhanced glycolysis also promotes MCT1 expression, which is potentially required for the accelerated exportation of intracellular lactate.

Radiation induced MCT1 expression through AMPK/glycolysis-dependent pathways in securin-knockdown MDA-MB-231-2A cells. (a) MDA-MB-231-2A cells were exposed to 6 Gy radiation followed by various periods of recovery. The protein expression was examined by western blot analyses. (b) MDA-MB-231-2A cells were pretreated with 5–20 μM compound C (left part) or 5–20 mM DCA (right part) for 2 h and then exposed to 6 Gy radiation. After an 8-h recovery period, the protein expression was examined by western blot analyses. (c) MDA-MB-231-2A cells were pretreated with 10 μM compound C or 10 mM DCA for 2 h and then exposed to 6 Gy radiation. After a 12-h recovery period, MCT1 protein expression was examined by fluorescent microscopy analysis. MCT1 displayed green fluorescence using the anti-rabbit-conjugated FITC antibody. The nuclei were stained with Hoechst 33258
Radiation induces glycolytic enhancement and MCT1 expression through the AMPK/NF-κB pathway
Glycolysis activates NF-κB signalling, and p53 restricts NF-κB activity through the suppression of glycolysis. In addition, the activation of NF-κB enhances glycolysis, thus forming a positive-feedback loop.17 Because p53 is mutated in MDA-MB-231 cells,28 radiation-induced glycolytic enhancement may further increase NF-κB activity. Indeed, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκB-α) and NF-κB were phosphorylated in MDA-MB-231-2A cells (Figure 4a), suggesting that NF-κB signalling is activated by radiation. In contrast, radiation slightly upregulated NF-κB activity in MCF-7 cells (Supplementary Figure S6A) that carries wild-type p53.28 Radiation-induced NF-κB activation was dependent on AMPK as the IκB-α and NF-κB phosphorylation in response to radiation was attenuated by compound C in both MDA-MB-231-2A and MCF-7 cells (Figure 4b and Supplementary Figure S6B). However, inhibition of glycolysis by DCA augmented radiation-induced NF-κB activation (Figure 4c), suggesting that NF-κB may signal upstream of glycolysis and therefore compensate for the inhibition of glycolysis. To investigate whether NF-κB activation was required for radiation-induced glycolytic enhancement, the NF-κB inhibitor BMS-345541 was used to inhibit radiation-induced NF-κB and IκB-α phosphorylation (Figure 4d). NF-κB inhibition decreased the radiation-induced LDH activation, lactate release and MCT1 expression in MDA-MB-231-2A cells (Figures 4e and f, and Supplementary Figure S7). Therefore, these results indicate that radiation induces the AMPK and NF-κB signalling pathways, leading to the enhancement of glycolysis and MCT1 expression.

The role of NF-κB in radiation-induced metabolic alterations in securin-knockdown MDA-MB-231-2A cells. (a) MDA-MB-231-2A cells were exposed to 6 Gy radiation followed by various recovery periods. The protein expression was examined by western blot analyses. (b–d) MDA-MB-231-2A cells were pretreated with 5–20 μM compound C (b), 5–20 mM DCA (c) or 1–10 μM BMS-345511 (d) for 2 h and then exposed to 6 Gy radiation. After an 8-h recovery period, the protein expression was examined by western blot analyses. (e) MDA-MB-231-2A cells were pretreated with 5 or 10 μM BMS-345511 for 2 h and then exposed to 6 Gy radiation. After a 24-h recovery period, the lactate concentration in the culture medium was examined by protocols described in the Materials and Methods section. **P<0.01 indicates significant differences between control and irradiated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells. (f) MDA-MB-231-2A cells were pretreated with 1–10 μM BMS-345511 for 2 h, and then exposed to 6 Gy radiation. After an 8-h recovery period, the protein expression was examined by western blot analyses
Radiation promotes the invasion of non-irradiated cells through AMPK- and NF-κB-dependent bystander signalling pathways
It is well established that radiation induces damage in cells that are not directly irradiated in a phenomenon called the non-targeted bystander effect.29 Our previous study showed that senescent MDA-MB-231-2A cells promote the invasion of non-irradiated cells.19 In this study, the invasive behaviour of the non-irradiated MDA-MB-231 cells was also enhanced after treatment with the conditioned medium (CM) from the irradiated MDA-MB-231-2A cells (Figure 5a). Interestingly, CM induced AMPK activation in the non-irradiated MDA-MB-231 cells (Figure 5b). AMPK inhibition by compound C attenuated CM-induced invasion of the non-irradiated MDA-MB-231 cells as well as human umbilical vein endothelial cells (HUVECs) (Figure 5c). In addition, DCA also inhibited CM-induced invasion of the non-irradiated MDA-MB-231 cells and HUVECs (Figure 5d). Furthermore, CM induced NF-κB activation in the non-irradiated MDA-MB-231 cells and HUVECs, and NF-κB activity inhibition by BMS-345541 attenuated cell invasion (Figures 6a and b). These results suggest that radiation promotes the invasion of the non-irradiated, surrounding cells through AMPK- and NF-κB-dependent bystander signalling pathways.

Radiation induced a bystander effect through AMPK/glycolysis-dependent pathways. (a) MDA-MB-231-2A cells were exposed to 6 Gy radiation followed by various recovery periods. The conditioned media (2A-CM) was collected and used to treat MDA-MB-231 cells. Serum-free and complete medium (10% FBS) were used as negative and positive controls, respectively. After 16 h, cell invasion was examined using the Boyden chamber assay. The numbers of invading cells were observed by microscopy and quantified. **P<0.01 indicates significant differences between 2A-CM- and 10% FBS-treated cells. (b) MDA-MB-231 cells were treated with 2A-CM for various time intervals. The protein expression was examined by western blot analyses. (c and d) MDA-MB-231 cells or HUVECs were pretreated with compound C (c) or DCA (d) for 2 h and then exposed to 2A-CM for 16 h. Cell invasion was examined using the Boyden chamber assay. The number of invading cells was quantified. **P<0.01 indicates significant differences between 2A-CM- and 10% FBS-treated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells

Radiation induced a bystander effect through NF-κB-dependent pathways. (a) MDA-MB-231 cells were pretreated with BMS-345541 for 2 h, and then exposed to 2A-CM for 4 h. The protein expression was examined by western blot analyses. (b) MDA-MB-231 cells or HUVECs were pretreated with BMS-345541 for 2 h and then exposed to 2A-CM for 16 h. Cell invasion was examined using the Boyden chamber assay. The number of invading cells was quantified. **P<0.01 indicates significant differences between 2A-CM- and 10% FBS-treated cells. ##P<0.01 indicates significant differences between inhibitor-treated and untreated cells
Discussion
Although metabolic deregulation has been linked to cancer8, 9, 10 and senescence cells are thought to remain metabolically active,3 the relationship between cellular metabolism and senescence remains unclear. Our previous studies have shown that ionising radiation induces senescence in the securin-knockdown MDA-MB-231-2A and low-securin-expressing MCF-7 human breast cancer cells, but parental MDA-MB-231 cells are not affected. In addition, only minimal apoptosis is induced in irradiated MDA-MB-231-2A and MCF-7 cells.18, 19 These findings support that MDA-MB-231-2A cells serve as a suitable cellular model for investigating the relationship between senescence and metabolism alternation without the disturbance of cell death. In this study, we found that metabolic alterations occur in senescent MDA-MB-231-2A cells, including the enhancement of glycolysis, accelerated release of lactate and sustained activation of AMPK. Interestingly, these metabolic alterations were also detected in non-irradiated, surrounding cells and promoted their invasiveness. Therefore, changes in metabolism are crucial for both radiation-induced senescence and the bystander effect.
The direct role of securin in cellular senescence is current unclear. It has been reported that overexpression of securin activates DNA damage pathway to induce senescence in normal human fibroblasts.30 In contrast, we and others31 have shown that securin attenuates drug- and radiation-induced senescence in human colorectal and breast cancer cells. These findings suggest a controversial role of securin in cellular senescence. Possibly, securin per se seems not to be a prerequisite for senescence. Securin is involved in controlling the metaphase–anaphase transition and anaphase onset, and fine-tuning of its expression is required for normal cell cycle progression.32 Inhibition or overexpression of securin blocks sister chromatid separation and results in cell cycle dysregulation.32 Therefore, the downstream events, such as DNA damage responses and cell cycle arrest, may be more important to induce cellular senescence.
Using proteomic approach, we hypothesise that increased GAPDH expression has a role in radiation-induced glycolytic enhancement. However, the role of GAPDH expression in radiation-induced senescence remains unclear. In addition to its essential role in glycolysis, GAPDH is also a key mediator of many oxidative stress responses through its nuclear translocation and regulation of cell fate.33 In addition, nuclear translocation of GAPDH also stimulates autophagy by inducing the autophagy protein Atg12.34 The elevation in glycolysis and enhanced autophagy by GAPDH cooperate to protect cells from caspase-independent cell death.34 Because autophagy has been reported to facilitate senescence, and activation of AMPK is known to stimulate GAPDH nuclear translocation,35 we propose that radiation upregulates GAPDH expression to enhance glycolysis and promote autophagy, leading to senescence. However, it has also been reported that S-nitrosylation of GAPDH elicited by nitric oxide augments its binding to Siah1 (an E3 ubiquitin ligase), then promoting nuclear translocation of GAPDH and triggering apoptosis.36 Apoptosis induced by GAPDH-Siah1 cascade is independent of glycolytic impairment because S-nitrosylation of GAPDH also abrogates its catalytic activity.36 Our results found that GAPDH enzymatic activity was reduced after 24 h of post-irradiation time (Supplementary Figure S2A), which raises the possibility that GAPDH-Siah1 cascade may act as a negative feedback mechanism to impair GAPDH activity.
Our results indicated that the enhancement of glycolysis was associated with radiation-induced senescence, indicating increased energy demands during senescence. At the cellular level, AMPK senses increases in the AMP to ATP ratio and then phosphorylates substrates to enhance energy production and reduce energy-consuming processes.37 For example, AMPK phosphorylates and inhibits acetyl-CoA carboxylase that consumes ATP and produces malonyl-CoA for fatty acid synthesis. In addition, AMPK-mediated phosphorylation of ULK-1 triggers autophagy that recycles cellular components for energy production.38, 39 Sustained AMPK activation was observed during radiation-induced senescence. In addition, AMPK is required for radiation-induced glycolytic enhancement and senescence. Consistently, AMP/ATP ratios and AMPK activity increases during cellular senescence in fibroblasts.40 A recent report also demonstrates significant increase in AMPK phosphorylation in H2O2-induced senescent cells.41 Therefore, AMPK could be a general activator of cellular senescence.
The movement of lactate in and out of the cell requires a membrane-bound MCT, such as MCT1.26 In this study, we found that radiation induced MCT1 expression in the securin-depleted MDA-MB-231-2A cells, potentially leading to lactate efflux. Studies have shown that excess lactate acidifies the tumour microenvironment, leading to cancer cell invasion.42 MCT1 is also the main facilitator of lactate uptake in cancer and endothelial cells.43 After entering cells, lactate can trigger the phosphorylation/degradation of IκB-α and subsequently stimulate the NF-κB pathway.44 Our results indicate that CM induced NF-κB activation in the non-irradiated cancer cells and HUVECs, possibly due to lactate uptake.
It is well known that senescent cells exhibit pro-tumourigenic effects on neighbouring cells through the secretion of cytokines and factors involved in the SASP.4 Recently, SASP factors, including TGF-β family ligands, VEGF, CCL2 (MCP-1) and CCL20 (MIP-3α), were shown to mediate paracrine senescence.45 Our previous study also demonstrated that the senescent MDA-MB-231-2A cells release VEGF, MCP-1 and MIP-3α.19 In this study, we observed metabolic alterations not only in irradiated senescent cells but also in neighbouring cells, which may also occur in a paracrine manner through these SASP factors. Whether paracrine senescence was induced in neighbouring cells requires future investigation.
In conclusion, our results revealed a correlation between metabolism and senescence in human breast cancer cells. Radiation induced the activation of metabolic pathways such as AMPK/NF-κB and glycolysis in the securin-depleted MDA-MB-231-2A cells, which are involved in radiation-induced senescence. In addition, a radiation-induced bystander effect appears to be induced by similar alterations in metabolic signalling pathways. Therefore, metabolic alterations are coupled to both senescence and a bystander effect in response to radiation.
Materials and Methods
Chemicals and antibodies
Sodium DCA, compound C and BMS-345541 were purchased from Sigma Chemical (St. Louis, MO, USA). The anti-phospho-LDHA (Tyr-10), anti-LDHA, anti-phospho-AMPK (Thr-172), anti-AMPK, anti-phospho-NF-κB p65 (Ser-468), anti-NF-κB, anti-phospho-IκB-α (Ser-32) anti-IκB-α and antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). The anti-MCT1 was purchased from Merck Millipore (Temecula, CA, USA). The anti-actin and GAPDH antibodies were purchased from Chemicon International (Temecula, CA, USA).
Cell culture
The MDA-MB-231 and human securin shRNA-transfected MDA-MB-231-2A human breast cancer cell lines were kindly provided by Dr. Ji-Hshiung Chen (Department of Molecular Biology and Human Genetics, Tzu Chi University, Taiwan). The cells were grown in DMEM medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and 1 mM sodium pyruvate solution. The MDA-MB-231-2A cells were selected using DMEM containing 0.5 mg/ml puromycin dihydrochloride. HUVECs purchased from ScienCell Research Laboratories (Carlsbad, CA, USA), were grown in endothelial cell medium supplemented with 5% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. The cells were cultured at 37 °C and 5% CO2 in a humidified incubator (310/Thermo, Forma Scientific, Inc., Marietta, OH, USA).
X-ray irradiation
Radiation was generated using an X-ray machine (RS2000; RAD Source Technologies, Coral Springs, FL, USA) operating at 160 kVp (peak kilovoltage) and 25 mA. The cells were replenished with fresh medium and immediately subjected to X-ray irradiation. A 6-Gy radiation dosage was chosen for this study on the basis of our previous study.19
Senescence-associated β-galactosidase staining
Senescent cells were analysed using a β-gal staining kit (Cell Signaling Technology) in accordance with the manufacturer’s instructions. The percentage of senescence-associated β-galactosidase-positive cells was calculated by counting the cells in 10 random fields (at least 100 cells) using bright-field microscopy at a magnification of × 40. The experiments were performed in triplicate.
Western blot analyses
Cellular protein extracts were collected as described previously.46 In brief, equivalent amounts of proteins were subjected to electrophoresis using 10% sodium dodecyl sulfate-polyacrylamide gels. After the electrophoretic transfer of proteins onto polyvinylidene difluoride membranes, the membranes were sequentially hybridised with primary antibody followed by a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The protein bands were visualised using the enhanced chemiluminescence detection system (Immobilon Western Chemiluminescent HRP Substrate, WBKLS0500, Millipore, Bedford, MA, USA). The gel-digitising software Un-Scan-It gel (Version 5.1; Silk Scientific, Inc., Orem, UT, USA) was used to quantify the relative intensity of the bands on the X-ray films.
Immunofluorescence staining and confocal microscopy
To view MCT1 protein expression, the cells were subjected to immunofluorescence staining and confocal microscopy as described.46 At the end of the treatment, the cells were fixed with 4% paraformaldehyde solution in phosphate-buffered saline (PBS) overnight at 4 °C. Thereafter, the cells were washed three times with PBS, and the non-specific binding sites were blocked with PBS containing 10% FBS and 0.3% Triton X-100 for 1 h. The cells were incubated overnight at 4 °C with the rabbit anti-MCT1 (1:100) in PBS containing 10% FBS. Subsequently, the cells were incubated with the anti-rabbit-conjugated FITC antibody (1 : 50) in PBS for 2–3 h at 37 °C. Finally, the nuclei were stained with 2.5 μg/ml Hoechst 33258 for 30 min at room temperature. The samples were examined using a Olympus confocal laser scanning microscope (FluoView FV10i, Olympus, Tokyo, Japan).
Extracellular lactate measurement
For this study, 3 × 105 cells were plated in 35 mm dishes for 24 h, following different experimental treatments. CM from two independent experiments was collected and assayed using a lactate assay kit (BioVision, Milpitas, CA, USA) following the manufacturer’s instructions.
pH measurement
For this study, 3 × 105 cells were seeded in 35 mm dishes for 24 h. CM was collected from plates. The results represent two independent experiments, and pH was measured using a pH metre at approximately 25 °C (Denver Instrument UltraBasic).
GAPDH enzymatic activity assay
Protein extracts were used in the GAPDH assay using a colorimetric assay kit (ScienCell Research Laboratories) according to the manufacturer’s protocol. This method is based on the oxidisation of β-NADH to β-NAD in the presence of 3-phosphoglyceric acid (3-PGA), adenosine 5′-triphosphate (ATP) and GAPDH. The GAPDH activity is determined by assaying the rate of NADH oxidation, which is proportional to the reduction in absorbance at 340 nm over time. In brief, 5 μl of each sample or standard was transferred to the 96-well plate containing 145 μl of GAPDH assay mixture in each well and then mixed immediately. The absorbance at 340 nm was recorded over a 12 min interval. Enzymatic activity was calculated as units/ml according to the absorbance of GAPDH standard.
LDH enzymatic activity assay
The LDH activity assay in cell lysates was measured by a colorimetric assay kit (BioVision, Mountain View, CA, USA). In this assay, LDH reduced NAD to NADH, which then interacts with a specific probe to produce colour (absorbance at 450 nm). In brief, cells were lysed in ice-cold assay buffer. Five microlitres of each sample or standard was mixed with 50 μl of Reaction Mix. The absorbance at 450 nm was recorded over a 30-min interval. Enzymatic activity was calculated as nmol/ml according to the absorbance of NADH standard.
Conditioned media
MDA-MB-231-2A cells were grown in DMEM for 24 h. The culture media was replaced with fresh medium, and the cells were exposed to 6 Gy radiation. The media was collected 12 h after radiation and contained increased lactate levels compared with control. The time point of 12 h post radiation was chosen to exclude the possibility that an increased number of cells could affect the lactate concentration. Subsequently, the collected media was centrifuged at 2000 r.p.m. for 5 min at 4 °C to remove the cells and cell debris.
Boyden chamber assay
An invasion assay was performed using gelatine (Sigma)-coated membranes (pore size, 8 mm; BD Biosciences, San Jose, CA, USA). The membrane was soaked in 0.5 M acetic acid for 24 h to enlarge the pores; subsequently, the membrane was rinsed twice with double deionised water then being soaked in 100 mg/ml gelatine for 16 h. Approximately 8 × 103 cells were suspended in 1% (MDA-MB-231) and 5% (HUVECs) serum medium and plated in the top chamber (Neuro Probe, Inc., Cabin John, MD, USA). Medium supplemented with 10% serum DMEM or 2A-CM was used as a chemoattractant in the lower chamber. After incubation for 16 h, the cells remaining on the bottom side of the membrane were stained with Giemsa (Merck, Darmstadt, Germany), imaged and counted.
Statistical analyses
All data represented the mean±S.E. of at least three independent experiments. Statistical analyses were performed using one-way analyses of variance, and additional post hoc testing was performed using the statistical software GraphPad Prism 4 (GraphPad Software, Inc. San Diego, CA, USA). A P value <0.05 was considered significant.
Abbreviations
- 2D gel electrophoresis:
-
two-dimensional gel electrophoresis
- AMPK:
-
5′-adenosine monophosphate-activated protein kinase
- CM:
-
conditioned medium
- DCA:
-
dichloroacetate
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- HUVECs:
-
human umbilical vein endothelial cells
- IκB-α:
-
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- LDHA:
-
lactate dehydrogenase A
- MCT:
-
monocarboxylate transporter
- MDA-MB-231-2A:
-
securin-knockdown MDA-MB-231 cells
- NF-κB:
-
nuclear factor kappa B
- PDK:
-
pyruvate dehydrogenase kinase
- SASP:
-
senescence-associated secretory phenotype
References
Campisi J, d'Adda di Fagagna F . Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740.
Collado M, Serrano M . The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer 2006; 6: 472–476.
Campisi J . Aging, cellular senescence, and cancer. Annu Rev Physiol 2013; 75: 685–705.
Coppe JP, Desprez PY, Krtolica A, Campisi J . The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5: 99–118.
Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P . The Warburg effect: insights from the past decade. Pharmacol Ther 2013; 137: 318–330.
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Hirschhaeuser F, Sattler UG, Mueller-Klieser W . Lactate: a metabolic key player in cancer. Cancer Res 2011; 71: 6921–6925.
Cairns RA, Harris IS, Mak TW . Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11: 85–95.
Hsu PP, Sabatini DM . Cancer cell metabolism: Warburg and beyond. Cell 2008; 134: 703–707.
Seyfried TN, Shelton LM . Cancer as a metabolic disease. Nutr Metab (Lond) 2010; 7: 7.
Hardie DG, Ross FA, Hawley SA . AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13: 251–262.
Kuhajda FP . AMP-activated protein kinase and human cancer: cancer metabolism revisited. Int J Obes (Lond) 2008; 32 (Suppl 4): S36–S41.
Shackelford DB, Shaw RJ . The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9: 563–575.
Fu X, Wan S, Lyu YL, Liu LF, Qi H . Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One 2008; 3: e2009.
Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys 2010; 78: 221–229.
Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 2011; 13: 1272–1279.
Kawauchi K, Araki K, Tobiume K, Tanaka N . p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 2008; 10: 611–618.
Chen WS, Yu YC, Lee YJ, Chen JH, Hsu HY, Chiu SJ . Depletion of securin induces senescence after irradiation and enhances radiosensitivity in human cancer cells regardless of functional p53 expression. Int J Radiat Oncol Biol Phys 2010; 77: 566–574.
Yu YC, Yang PM, Chuah QY, Huang YH, Peng CW, Lee YJ et al. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci Rep 2013; 3: 1675.
Bruns GA, Gerald PS . Human glyceraldehyde-3-phosphate dehydrogenase in man-rodent somatic cell hybrids. Science 1976; 192: 54–56.
Fantin VR, St-Pierre J, Leder P . Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006; 9: 425–434.
Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu TL et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol 2011; 31: 4938–4950.
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37–51.
Zannella VE, Cojocari D, Hilgendorf S, Vellanki RN, Chung S, Wouters BG et al. AMPK regulates metabolism and survival in response to ionizing radiation. Radiother Oncol 2011; 99: 293–299.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–1174.
Kennedy KM, Dewhirst MW . Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol 2010; 6: 127–148.
Halestrap AP, Meredith D . The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 2004; 447: 619–628.
Negrini M, Sabbioni S, Haldar S, Possati L, Castagnoli A, Corallini A et al. Tumor and growth suppression of breast cancer cells by chromosome 17-associated functions. Cancer Res 1994; 54: 1818–1824.
Morgan WF, Sowa MB . Non-targeted bystander effects induced by ionizing radiation. Mutat Res 2007; 616: 159–164.
Hsu YH, Liao LJ, Yu CH, Chiang CP, Jhan JR, Chang LC et al. Overexpression of the pituitary tumor transforming gene induces p53-dependent senescence through activating DNA damage response pathway in normal human fibroblasts. J Biol Chem 2010; 285: 22630–22638.
Tong Y, Zhao W, Zhou C, Wawrowsky K, Melmed S . PTTG1 attenuates drug-induced cellular senescence. PLoS One 2011; 6: e23754.
Peters JM . The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell 2002; 9: 931–943.
Sirover MA . New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta 1999; 1432: 159–184.
Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007; 129: 983–997.
Narita M, Young AR, Narita M . Autophagy facilitates oncogene-induced senescence. Autophagy 2009; 5: 1046–1047.
Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 2005; 7: 665–674.
Mihaylova MM, Shaw RJ . The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13: 1016–1023.
Rabinowitz JD, White E . Autophagy and metabolism. Science 2010; 330: 1344–1348.
Singh R, Cuervo AM . Autophagy in the cellular energetic balance. Cell Metab 2011; 13: 495–504.
Wang W, Yang X, Lopez de Silanes I, Carling D, Gorospe M . Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem 2003; 278: 27016–27023.
Guo L, Xie B, Mao Z . Autophagy in Premature Senescent Cells Is Activated via AMPK Pathway. Int J Mol Sci 2012; 13: 3563–3582.
Gillies RJ, Gatenby RA . Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 2007; 39: 251–257.
Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 2012; 7: e33418.
Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O . Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 2011; 71: 2550–2560.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15: 978–990.
Kuo PC, Liu HF, Chao JI . Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J Biol Chem 2004; 279: 55875–55885.
Acknowledgements
This work was supported by research grants from the National Science Council, Taiwan to S-J Chiu (NSC99-2314-B-320-004-MY3 and NSC102-2628-B-320-002-MY3), and by a research grant from Taipei Medical University, Taiwan to P-M Yang (TMU101-AE1-B25). We also thank the National Cheng-Kung University Proteomics Research Core Laboratory for their assistance with the two-dimensional gel electrophoresis.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Liao, EC., Hsu, YT., Chuah, QY. et al. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis 5, e1255 (2014). https://doi.org/10.1038/cddis.2014.220
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.220
Keywords
This article is cited by
-
Targeting PDK2 rescues stress-induced impaired brain energy metabolism
Molecular Psychiatry (2023)
-
Comparable radiation sensitivity in p53 wild-type and p53 deficient tumor cells associated with different cell death modalities
Cell Death Discovery (2021)
-
iTRAQ®-based quantitative proteomics reveals the proteomic profiling of methicillin-resistant Staphylococcus aureus-derived extracellular vesicles after exposure to imipenem
Folia Microbiologica (2021)
-
Low doses of ionizing radiation enhance angiogenesis and consequently accelerate post-embryonic development but not regeneration in zebrafish
Scientific Reports (2020)
-
Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche
Cell Death & Disease (2020)
