
Abstract
Oncogenic stress-induced senescence (OIS) prevents the ability of oncogenic signals to induce tumorigenesis. It is now largely admitted that the mitogenic effect of oncogenes requires metabolic adaptations to respond to new energetic and bio constituent needs. Yet, whether glucose metabolism affects OIS response is largely unknown. This is largely because of the fact that most of the OIS cellular models are cultivated in glucose excess. In this study, we used human epithelial cells, cultivated without glucose excess, to study alteration and functional role of glucose metabolism during OIS. We report a slowdown of glucose uptake and metabolism during OIS. Increasing glucose metabolism by expressing hexokinase2 (HK2), which converts glucose to glucose-6-phosphate (G6P), favors escape from OIS. Inversely, expressing a glucose-6-phosphatase, pharmacological inhibition of HK2, or adding nonmetabolizable glucose induced a premature senescence. Manipulations of various metabolites covering G6P downstream pathways (hexosamine, glycolysis, and pentose phosphate pathways) suggest an unexpected role of the hexosamine pathway in controlling OIS. Altogether, our results show that decreased glucose metabolism occurs during and participates to OIS.
Similar content being viewed by others


Main
Otto Warburg was the first to describe a metabolic switch occurring in cancer tissues. In the presence of oxygen, instead of producing adenosine triphosphate (ATP) through oxidative phosphorylation, cancer cells exhibit high rates of glycolysis. Cancer cells divide rapidly and need favorable energy production rates. In these cells, glucose is rendered more bioavailable and metabolizable through upregulation of glucose transporters and metabolic enzymes. Tumor imaging exploits this fact to detect the presence of tumors throughout the body: cancer cells are labeled with the glucose analog 18fluorine-fluorodeoxyglucose. Mechanistically, oncogenes products such as MYC, NF-κB, AKT, HIF, and E2F, and the tumor-suppressor genes products such as p53 and PTEN, which are respectively activated or inhibited in cancer, can act on either glucose transporters, glycolytic enzymes, or both, and this suggests that in cancer cells, the regulation of growth is coupled with that of metabolism.1, 2, 3, 4, 5, 6, 7, 8
Mounting evidence suggesting that increased glycolysis plays a role in maintaining the malignant behavior of tumor cells has raised interest in targeting the metabolism of cancer cells in cancer therapy,9, 10, 11 but the importance of glucose metabolism alterations early in tumorigenesis is poorly known. Failsafe programs can protect cells from transformation, and escape from them is necessary to allow early tumorigenesis. Senescence, because it involves stable cell cycle arrest and activates immune surveillance,12 constitutes such a program. Various studies have demonstrated that activation of oncogenes such as RAS, RAF, MEK, and others induces senescence in vitro. More recently, work on in vivo models has shown that premalignant lesions exhibit high senescence levels, whereas senescence is absent from malignant tumors, thus confirming a tumor-suppressor role for oncogene-induced senescence (OIS).13
Little is known about the features of glucose metabolism in cells undergoing OIS. It is also not known whether the features described in malignant tumors are acquired early or late in tumorigenesis. As far as we know, most of the papers studies senescence response in a context of glucose excess (25 mM). Nevertheless, it is difficult to get a precise idea of glucose concentrations used as in most of the papers they are not mentioned, although they influence cell growth and senescence.14, 15
Alterations in enzyme activities, especially in glycolytic and in tricarboxylic acid pathways, have been reported to modulate senescence response. Indeed, increased glycolytic enzyme activities favor senescence escape in mouse embryonic fiblroblasts.5 Decreased tricarboxylic acid malic enzymes seem to participate in p53-induced senescence,16 whereas the use of pyruvate dehydrogenase to fuel tricarboxylic acid cycle promotes senescence,17 provoking a debate on the role of tricarboxylic acid cycle on senescence. Here, we used human epithelial cells cultivated at 8 mM glucose, rather close to its physiological level, to examine the role, if any, of glucose metabolism during OIS. Surprisingly, we found that glucose uptake and metabolization is altered after oncogenic stress and this alteration participates in senescence.
Results
OIS impairs glucose metabolism
To study glucose metabolism during OIS, we focused on human epithelial cells cultivated without glucose excess. We first immortalized human epithelial cells by expressing hTert to overcome replicative senescence.18 Next, cells were infected with a retroviral vector coding a fusion protein (MEK/ER or RAF/ER) between a constitutively activated form of MEK1 or delta-BRAF and the hormone-binding domain of the human estrogen receptor (hbER).19, 20 In response to 4-hydroxytamoxifen (4-OHT) and, as expected, MEK/ER-expressing cells showed phosphorylation of the MEK substrate ERK. The MEK induction resulted in a strong decrease of the Phospho-S10-Histone3 mitotic marker (Figure 1a). Accordingly, MEK activation blocked cell growth (Figure 1b), induced the appearance of senescence-associated β-galactosidase activity (SA-β-Gal) (Figure 1c), and increased expression of a set of senescence markers: Sprouty homolog 2 (SPRY2),21 the interleukin-8 (IL-8),22 and the Deleted In Esophageal Cancer 1 (DEC1)23 (Figure 1d). Similar results were obtained using the RAF/ER-expressing cells, RAF being the upstream kinase of MEK (Supplementary Figure 1), showing that RAF or MEK are equivalent systems to induce OIS.

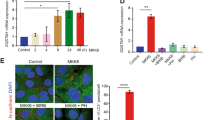
Glucose metabolism decreases during oncogenic stress-induced senescence. Immortalized human epithelial cells expressing the inducible MEK/ER oncogene were treated or not with 4-OHT. (a) Cell extracts were prepared after 0, 3, or 4 days of 4-OHT treatment and analyzed by immunoblotting with the indicated antibodies. (b) Cells were seeded at the same density and treated or not for 3 days with 4-OHT. After 5 days, they were PFA fixed and crystal violet stained. (c) After 3 days with or without 4-OHT treatment, cells were fixed and stained for detection of SA-β-Gal activity. Percentages of stained cells were calculated and representative pictures are shown. (d) After 3 days with or without 4-OHT treatment, RNA was prepared and the expression of the indicated senescence markers was analyzed by RT-qPCR and normalized with respect to actin expression. (e and f) Cells were treated or not for 2 days with 4-OHT, counted, seeded back, and subjected or not to 4-OHT treatment. After 24 h, glucose uptake (e) and lactate production (f) were determined. (g) Cells were treated with or without 4-OHT for 3 days. ATP concentration was determined and normalized with respect to the protein content
To see whether glucose metabolism was altered during OIS, we first compared glucose consumption by MEK/ER-activated and -inactivated human epithelial cells. Uptake of extracellular glucose measurement revealed markedly decreased glucose consumption by senescent cells (Figure 1e). In agreement with decreased glucose consumption, senescent cells displayed a decreased lactate production (Figure 1f) as well as lower ATP levels (Figure 1g). Adding 4-OHT on immortalized cells (without MEK/ER) did not induce any senescence and glucose metabolism changes excluding a MEK-independent effect of the 4-OHT (Supplementary Figure 2). Altogether, these results show a decrease in glucose metabolism during OIS in human epithelial cells.
Inhibition of the first step of glucose metabolism results in premature senescence
Glucose metabolism is thus regulated during OIS. We next wanted to examine whether cellular glucose metabolization shortage is involved in oncogenic stress-mediated senescence. We then blocked glucose-6-phosphate (G6P) accumulation to see if this would have an impact on senescence of human epithelial cells. This was done by constitutively expressing glucose-6-phosphatase (G6PC3) that catalyzes the conversion of G6P into glucose (Figures 2a and b). As expected, G6PC3 constitutive expression correlated with the decrease of glucose consumption (Figure 2c), lactate production (Figure 2d), and ATP level (Figure 2e). Interestingly, this glucose metabolism drop induced by constitutive G6PC3 expression blocked cell growth (Figure 2f), decreased levels of proliferation markers (Figure 2g), and induced both SA-β-Gal activity (Figure 2h) and the expression of various senescence markers (Figure 2i). Pharmacological inhibition of hexokinases with Lonidamine24, 25 (Supplementary Figure 3) or the use of 2-deoxy-D-glucose (2DG), a non-metabolizable form of glucose (Supplementary Figure 4), also blocked glucose metabolism and induced a premature senescence. Blocking glucose metabolism in human epithelial cells thus results in premature senescence supporting its functional role in OIS.

Glucose-6-phosphatase blocks glucose metabolism and causes premature senescence. (a) Schematic representation of actors involved in the first glucose metabolism step. (b–i) Immortalized human epithelial cells were infected with a control or G6PC3-encoding vector and puromycin selected. (b) RNAs were extracted and analyzed by RT-qPCR to check for constitutive G6PC3 expression. (c–e) Equal numbers of cells were seeded and glucose uptake (c) or lactate production (d) was analyzed after 24 h. (e) ATP concentrations were determined and normalized with respect to protein content. (f) Cells were seeded at the same density. At 6 days after seeding, they were fixed with PFA and stained with crystal violet. (g) Cell lysates were prepared and protein expression analyzed by immunoblotting with the indicated antibodies. (h) Cells were PFA fixed and tested for SA-β-Gal activity. (i) RNAs were extracted and analyzed by RT-qPCR for expression of the indicated senescence markers
Hexokinase expression sustains glucose metabolism and favors OIS escape
We next wanted to examine whether increasing glucose metabolism might promote escape of human epithelial cells from OIS by sustaining glucose metabolism. Hexokinases play a major role in this metabolism, catalyzing its first step (conversion of glucose to G6P; Figure 2a) and thus determining both the glucose content of the cell and the level of glucose metabolization.11, 26 We therefore investigated OIS escape and glucose metabolism in human epithelial cells constitutively expressing hexokinase 2 (HK2), the main hexokinase involved in increasing glycolysis in cancer cells.6, 11 After retroviral transduction of HK2, we checked its constitutive expression by immunofluorescence (Figure 3a). As expected, constitutive HK2 expression sustained glucose consumption (Figure 3b), lactate production (Figure 3c), and ATP production (Figure 3d) during oncogenic stress. The sustained glucose metabolism induced by HK2 expression favors escape from OIS, as measured by the ability to grow (Figure 3e), to maintain proliferation markers expression (Figure 3f), and by the absence of SA-β-Gal activity (Figure 3g) and various senescence markers (as determined by RT-qPCR, Figure 3h). Altogether, these results support the view that sustaining glucose metabolism by expressing HK2 allows escape from OIS.

HK2 expression restores glucose metabolism and allows escape from OIS. (a–h) Immortalized human epithelial cells expressing the inducible RAF/ER oncogene were infected with a control or HK2-encoding retroviral vector and neomycin selected. (a) Immunofluorescence against the Flag tag was performed. Nuclei were counterstained with Hoechst. (b–d) Cells were treated or not for 2 days with 4-OHT, counted, and seeded back with or without 4-OHT. After 24 h, glucose uptake (b) and lactate production (c) were determined. (d) Cells were treated with or without 4-OHT for 3 days as indicated. ATP concentrations were determined and normalized with respect to the protein content. (e) Cells were seeded and treated or not for 3 days with 4-OHT. After 5 days, they were PFA fixed and crystal violet stained. (f) Cells were seeded and treated or not for 3 days with 4-OHT. After 2 days, cell extracts were prepared and analyzed by immunoblotting with the indicated antibodies. (g) Cells were PFA fixed and stained for SA-β-Gal detection. Percentages of stained cells were calculated and representative photographs are displayed. (h) RNAs corresponding to the indicated genes were prepared and analyzed by RT-qPCR. Expression levels were normalized with respect to actin expression
Hexosamine pathway is a major pathway controlling OIS
In order to confirm the above described results, we next investigated the ability of glucose metabolites to allow an OIS bypass. We first began by treating the cells with G6P, the upstream glucose metabolite (Figure 4a). Interestingly, daily treatment with G6P permits the cells to escape oncogenic stress-induced growth arrest (Figure 4b) as well as the appearance of SA-β-Gal marker (Figure 4d). Thus, genetic (HK2 and G6PC ectopic expression), pharmacological (HK inhibition or 2DG), as well as metabolite manipulation through G6P all support the view that sustaining glucose metabolization favors OIS escape.

The hexosamine pathway favors OIS escape. (a) Schematic representation of key pathways and metabolites involved in glucose metabolism. (b) Human epithelial cells expressing MEK/ER were seeded at the same density and treated or not for 3 days with 4-OHT, and every 2 days by G6P at 5 mM. After 5 days, they were PFA fixed and crystal violet stained. (c) Human epithelial cells expressing MEK/ER were seeded at the same density and treated every day as indicated with or without NAcGluc at 40 mM, pyruvate at 5 mM, and NADPH at 100 μM, and with 4-OHT for the first 3 days. (d) After treatment as indicated in (b) and (c), cells were PFA fixed and stained for SA-β-Gal detection. Percentages of stained cells were calculated and representative photographs are displayed
The upstream glucose metabolite, the G6P, fuels three main pathways: the glycolytic pathway, the hexosamine pathway, and the pentose phosphate pathway (PPP) (Figure 4a). These pathways participate in producing basic cell components, energy, and in controlling the cell redox state. We then decided to investigate these three pathways. A decreased glycolytic pathway has already been proposed to participate in senescence5 and is thus an obvious candidate pathway. As expected, blocking the glycolysis by the bromopyruvate, a GAPDH inhibitor, induced premature senescence and resulted in strong activation of the energy sensor AMPK (Supplementary Figure 5). Nevertheless, pyruvate glycolysis metabolite was unable to induce an OIS escape (Figures 4c and d) and AMPK was found only slightly activated during OIS (Supplementary Figure 5), suggesting that the glycolysis, even if directly inhibited can induce a premature senescence, was not directly involved in the OIS we observed.
We next investigated a putative role of the PPP in OIS. The knockdown of the G6P dehydrogenase (G6PDH), the rate-limiting enzyme of the PPP, did not modify cell growth (Supplementary Figure 5) and the NADPH PPP metabolite was unable to induce an OIS escape (Figures 4c and d), suggesting that PPP was not involved in OIS in our model.
In conclusion, we investigated the potential role of the hexosamine pathway in OIS. Inhibition by azaserine of the glutamine fructose-6-phosphate amidotransferase (GFAT), the first enzyme of the hexosamine pathway, induced a premature senescence (Supplementary Figure 5). Importantly, N-acetylGlucosamine (NAcGluc) was found to allow cells to bypass growth arrest and SA-β-Gal senescence marker appearance (Figures 4c and d) without modifying glucose consumption or lactate production (Supplementary Figure 6). In addition and as during OIS, and by contrast to the effect of the glycolysis pathway inhibition, inhibition of the hexosamine pathway only slightly activated AMPK (Supplementary Figure 5). Together, these results point out the importance of the hexosamine pathway in the OIS response.
Discussion
The interest in glucose metabolism in cancer research field has recently raised and it is now largely recognized that metabolic alterations are part of the oncogenic program.1, 2, 3, 4, 5, 6, 7, 8, 16, 17 Oncogenic signal, in normal cells, provokes senescence in order to avoid tumorigenesis;13 the role of glucose metabolism in this process is rather unknown. Although it has been reported that glucose levels affect senescence, most of the experiments performed to decipher the events involved in OIS are performed with excess glucose (generally 25 mM). The major problem is that most of the cellular model used (IMR-90, WI38, MEF cells, and so on) are adapted to grow with this high level of glucose, and decreasing this level can affect cell growth.14 Here, we take advantage of primary human epithelial cells that grow at 8 mM glucose, nearly the normal level of glucose, to investigate the function of glucose metabolism during OIS. Our results show that glucose metabolism is impaired during OIS, in part due to a decreased glucose uptake. This decrease in glucose uptake did not seem to be due to a decrease in Glut1 or Glut3 expression, the expressed Glut in our cells (Supplementary Figure 7). Further work looking at glucose transporter activity and glucose metabolism enzyme activity will be necessary to understand the molecular mechanisms inducing these glucose metabolism changes during OIS. Restoring glucose metabolism by constitutively expressing the HK2 and by treating the cells with G6P allows to bypass OIS, whereas inhibiting glucose metabolization by blocking the HK activity, by constitutively expressing the glucose-6-phsphatase, or by treating cells with 2DG induces a premature senescence. At first sight, a recent paper from Dorr et al.,27 showing that human and mouse fibroblasts upon RAS-induced senescence display increased glucose consumption and lactate production, might appear opposite to our results. Nevertheless, we indeed observed that MEF cells (in classical culture condition, and hence in 25 mM glucose) upon constitutive RAS expression enter senescence with increased glucose consumption and lactate production (Supplementary Figure 8). This apparently opposite results might be because of the high glucose concentration used in MEF and/or intrinsic differences between fibroblasts and epithelial cells and/or intrinsic differences between RAS and RAF/MEK. Indeed, RAS activates the RAF/MEK pathway and also activates the PI3K pathway that can activate glucose transport and metabolism,28 eventually explaining the differences between glucose metabolism observed in these different models of OIS. Interestingly, the PI3K pathway has been reported in some models to inhibit RAS/RAF/MEK pathway-induced senescence,29, 30 suggesting, with our results, that the anti-OIS effect of the PI3K pathway might, in part, be mediated by its ability to increase glucose uptake and metabolism.
Examining the pathways downstream of G6P involved in OIS surprisingly suggest that the hexosamine pathway is a key pathway in the response to oncogenic stress. This pathway controls the N- and O-glycosylation of the proteins and is involved in numerous biological processes such as ER stress, gene expression, signaling, and trafficking, and thus perturbation in that pathway may have pathophysiological consequences.31, 32, 33, 34 This does not exclude that for other inducers of senescence other pathways might be involved. For example, ectopic expression of some glycolytic enzymes has been shown to enable MEFs to bypass senescence by enhancing the glycolytic flux, whereas inhibition of these enzymes results in senescence induction.5
Enhancement of glucose metabolism, extensively studied in malignant cells, has been shown to participate in malignant behavior.2 HK2, the most upstream glucose-metabolizing kinase, seems to play an important role in malignant cell growth, as the oncogenic transcription factors Myc and HIF can increase HK2 transcription, and as HK2 expression seems necessary to elicit a malignant cell phenotype.6, 11, 35, 36 The increased glucose uptake and metabolism observed in malignant cells might thus be, at least in part, acquired during evasion to the oncogenic stress-induced senescence. It might not be surprising taking into account the fact that OIS escape is often considered as a step participating in the malignant conversion of benign tumor.13, 37 Supporting this assumption, HK2 expression is significantly increased in 32% (15.9-fold, P=0.00018) of the malignant melanomas tested as compared with melanocytic nevi, their benign, senescent counterparts (Supplementary Figure 9).38 In addition and in agreement with our results, a recent paper showed that HK2 is required for tumor initiation induced by KRas or ErbB2.39
Altogether, our results support the view that the level of glucose metabolization and the hexosamine pathway are key determinants of the OIS response in human cells. This study naturally opens new avenues of research into relationships between OIS and glucose metabolism.
Materials and Methods
Cell culture
Human mammary epithelial cells (Lonza, Barcelona, Spain) were cultured in mammary epithelial cell growth medium containing 8 mM glucose (Promocell, Heidelberg, Germany) and 100 U/ml penicillin/streptomycin (Life Technologies, Saint Aubin, France). Virus-packaging GP293 cells (Clontech, Saint-Germain-en-Laye, France) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) with 10% FBS (Life Technologies) and penicillin/streptomycin. The cells were maintained at 37°C under a 5% CO2 atmosphere.
Retroviral infection
GP293 cells were transfected with PEI reagent (Euromedex, Souffelweyersheim, France) according to the manufacturer’s recommendations. At 2 days after transfection, the viral supernatant was mixed with fresh medium (1/2) and polybrene at 8 μg/ml (Sigma, Lyon, France) to infect target cells. Human epithelial cells were selected with G418 (Life Technologies) at 100 μg/ml or/and puromycin (InvivoGen, Toulouse, France) at 500 ng/ml.
Plasmids and reagents
The plasmids were pNLCΔMEK1 (ΔN3, S218E, S222D)/ER (Neo R) and pBabe-puro-BRAF/ER.20 pWZL Neo Myr Flag HK2 was obtained from Addgene (Cambridge, MA, USA) (Plasmid 20501).40 Human G6PC3 cDNA (Human MGC Verified FL cDNA, Clone ID 3050476; Thermo Scientific, Waltham, MA, USA) was excised with EcoRI–XhoI and inserted into the pLPC-puro vector.
The 4-OHT (H7904, Sigma) was used daily for 3 days at 500 nM. The hexokinase inhibitor Lonidamine (L4900, Sigma) was used daily at 25 μM. G6P (G7772, Sigma) was used every other day at 5 mM, NADPH (N5130, Sigma) was used daily at 100 μM, sodium pyruvate (11360039, Life Technologies) was used at 5 mM daily, and NAcGluc (A3286, Sigma) was used daily at 40 mM.
Immunoblotting and immunofluorescence
Immunoblot and immunofluorescence analyses were performed as described in Bernard et al.41 The primary antibodies used were: anti-phospho-ERK (9101, Cell Signaling, Danvers, MA, USA), anti-Flag (200472, Stratagene, Agilent Technologies, Les Ulis, France), anti-phospho-histone3Ser10 (ab14955, Abcam, Paris, France), anti-cyclin A (H432, sc-751, Santa Cruz Technology, Heidelberg, Germany), and anti-tubulin (T6199, Sigma). Quantification of immunoblots signals were performed using ImageJ software (National Institute of Health, Bethesda, MD, USA) and normalized to normalizator signal (tubulin or actin).
Reverse transcription and real-time quantitative PCR
TriReagent (Sigma-Aldrich) and PhaseLockGel tubes (Eppendorf, Hamburg, Germany) were used for total RNA preparation. The First-Strand cDNA Synthesis Kit (GE Healthcare, Chalfont St. Giles, UK) was used to synthesize cDNA from 2 μg total RNA. The RT reaction mixture was diluted 1/60 and used as cDNA template for quantitative PCR. TaqMan quantitative PCR was carried out on a LightCycler 2.0 System (Roche Applied Science, Meylan, France). The PCR mixture contained 1.33 μl LightCycler TaqMan mix (Roche Applied Science), 0.201 μl of a pre-mix of primers and the UPL probe, and 1.67 μl cDNA template in a 6.67-μl reaction volume. The relative amount of mRNA was calculated by the comparative Cp method after normalization against β-actin. The primers used were: actin (UPL probe #11) forward 5′-ATTGGCAATGAGCGGTTC-3′ and reverse 5′-GGATGCCACAGGACTCCAT-3′, IL8 (UPL probe #72) forward 5′-ATGGTTCCTT CCGGTGGT-3′ and reverse 5′-AGACAGCAGAGCACACAAGC-3′, Sprouty2 (UPL probe #40) forward 5′- TCAGGTCTTGGAAGTGTGGTC-3′ and reverse 5′-TTTGCACATCGCAGAAAGAA-3′, DEC-1 (UPL probe #84) forward 5′-TTTCTCCCTGACAGCTCACC-3′ and reverse 5′-TGAAAGCACTAACAAACCTAATTGA-3′, and G6PC3 (UPL probe #19) forward 5′-TGGCTCAACCTCATCTTCAA and reverse 5′-AGAAGAGGGGAACTGGTGAAC-3′.
SA-β-Gal analysis
At 6–10 days after initial treatment, the cells were fixed with 4% PFA and processed as described in Augert et al.42
ATP-level measurement
Human epithelial cells were seeded into 10-cm dishes at 2.105 cells/dish and treated with 500 nM 4-OHT daily for 3 days. ATP levels were measured 1 day after the third 4-OHT treatment for human epithelial cells. The cells were washed with iced-cold 1 × PBS and extracted in an ATP-releasing buffer containing 100 nM potassium phosphate buffer at pH 7.8, 2 mM EDTA, 1 mM dithiothreitol, and 1% Triton X-100.43 Then, 2 μl of lysate was used for protein determination by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). This involves adding an acidic dye to the protein-containing lysate and measuring the absorbance at 595 nm with a spectrophotometer. Comparison with a standard curve of bovine serum albumin (BSA, Sigma) provides a relative measurement of protein concentration. Samples were diluted in order to assess the ATP content per 10 μg protein in each condition. ATP was determined according to the manufacturer’s instructions (ATP determination kit, Life Technologies). ATP levels were first expressed in nM ATP per 10 μg protein and then normalized with respect to the level in control samples.
Glucose and lactate assays
After senescence induction by 2 days of 500 nM 4-OHT treatment, 9.104 cells were seeded into six-well plates and treated or not with 4-OHT. After 24 h, the supernatants were collected and filtered and then glucose and acid lactic were assayed. Assays for glucose and lactic acid were performed with an automated analyzer, Architect C16000 (Abbott Laboratories, Abbott Park, IL, USA).
Lactic acid was quantified on the basis of its lactate oxidase-catalyzed conversion to pyruvate and hydrogen peroxide (H2O2), followed by peroxidase-catalyzed conversion of the chromogen precursor ABTS (2,2′-azino-di-(3-ethylbenzthiazoline sulfonate)) to its chromogen in the presence of H2O2. The chromogen was quantified spectrophotometrically at 548 nm, and its appearance was strictly proportional to the lactic acid concentration. This assay is linear between 0.020 and 13.32 mmol/l. Manual dilution was performed before quantification in the case of the highest concentrations. Lactate production was calculated as the lactate concentration measured in the cell supernatants minus the lactate concentration measured in the cell medium.
Glucose was quantified enzymatically by coupling of the actions of hexokinase (HK) and G6P dehydrogenase. In the presence of ATP and Mg2+, glucose is phosphorylated to G6P by hexokinase, and ADP is produced. The G6P is converted to 6-phosphogluconate by G6P dehydrogenase. This reaction requires conversion nicotinamide adenine dinucleotide from its oxidized form (NAD+) to its reduced form (NADH). The glucose concentration is directly proportional to the generated NADH concentration that is spectrophotometrically measured at 340 nm. This assay is linear between 0.28 and 44.40 mmol/l. Samples containing higher concentrations were diluted manually before quantification. Glucose uptake was calculated as the glucose concentration of the medium minus the glucose concentration measured in the cell supernatant.
Statistical analysis
The values are presented as mean±S.D. unless stated otherwise. Statistical analyses were performed using Student’s-t-test (mean *P<0.05, mean **P<0.01, mean ***P<0.001). Each experience was at least replicated.
Change history
14 August 2014
This article has been corrected since Online Publication and a corrigendum has also been published
Abbreviations
- 2DG:
-
2-deoxy-D-glucose
- 4-OHT:
-
4-hydroxytamoxifen
- ATP:
-
adenosine triphosphate
- HK2:
-
hexokinase-2
- G6P:
-
glucose-6-phosphate
- OIS:
-
oncogene-induced senescence
- PPP:
-
pentose phosphate pathway
- SA-β-Gal:
-
senescence-associated β-galactosidase
References
Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012; 149: 49–62.
Ward PS, Thompson CB . Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 2012; 21: 297–308.
Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol 2011; 13: 1146–1152.
Jones RG, Thompson CB . Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009; 23: 537–548.
Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G et al. Glycolytic enzymes can modulate cellular life span. Cancer Res 2005; 65: 177–185.
Mathupala SP, Rempel A, Pedersen PL . Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 2001; 276: 43407–43412.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 2010; 107: 7461–7466.
Kim JW, Gao P, Liu YC, Semenza GL, Dang CV . Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol 2007; 27: 7381–7393.
Levine AJ, Puzio-Kuter AM . The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010; 330: 1340–1344.
Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux SP, Wilson GL et al. Emerging metabolic targets in cancer therapy. Front Biosci 2011; 16: 1844–1860.
Mathupala SP, Ko YH, Pedersen PL . Hexokinase-2 bound to mitochondria: cancer’s stygian link to the ‘Warburg Effect’ and a pivotal target for effective therapy. Semin Cancer Biol 2009; 19: 17–24.
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479: 547–551.
Collado M, Serrano M . Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10: 51–57.
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18: 283–293.
Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P . Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J 2003; 376: 403–411.
Jiang P, Du W, Mancuso A, Wellen KE, Yang X . Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013; 493: 689–693.
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013; 498: 109–112.
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ . Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998; 396: 84–88.
Blalock WL, Pearce M, Steelman LS, Franklin RA, McCarthy SA, Cherwinski H et al. A conditionally-active form of MEK1 results in autocrine tranformation of human and mouse hematopoietic cells. Oncogene 2000; 19: 526–536.
Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M . Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 1997; 17: 5598–5611.
Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006; 10: 459–472.
Acosta JC, O'Loghlen A, Banito A, Raguz S, Gil J . Control of senescence by CXCR2 and its ligands. Cell Cycle 2008; 7: 2956–2959.
Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M et al. Tumour biology: senescence in premalignant tumours. Nature 2005; 436: 642.
Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, C De Martino . Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst 1981; 66: 497–499.
Floridi A, Paggi MG, D'Atri S, De Martino C, Marcante ML, Silvestrini B et al. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res 1981; 41: 4661–4666.
Wilson JE . Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 2003; 206: 2049–2057.
Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013; 501: 421–425.
Riley JK, Carayannopoulos MO, Wyman AH, Chi M, Moley KH . Phosphatidylinositol 3-kinase activity is critical for glucose metabolism and embryo survival in murine blastocysts. J Biol Chem 2006; 281: 6010–6019.
Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell 2011; 42: 36–49.
Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012; 26: 1055–1069.
Dennis JW, Lau KS, Demetriou M, Nabi IR . Adaptive regulation at the cell surface by N-glycosylation. Traffic 2009; 10: 1569–1578.
Love DC, Krause MW, Hanover JA . O-GlcNAc cycling: emerging roles in development and epigenetics. Semin Cell Dev Biol 2010; 21: 646–654.
Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 2010; 24: 2784–2799.
Huber AL, Lebeau J, Guillaumot P, Petrilli V, Malek M, Chilloux J et al. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol Cell 2013; 49: 1049–1059.
Smith TA . Mammalian hexokinases and their abnormal expression in cancer. Br J Biomed Sci 2000; 57: 170–178.
Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 2011; 208: 313–326.
Braig M, Schmitt CA . Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res 2006; 66: 2881–2884.
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436: 720–724.
Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013; 24: 213–228.
Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007; 129: 1065–1079.
Bernard D, Quatannens B, Begue A, Vandenbunder B, Abbadie C . Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res 2001; 61: 2656–2664.
Augert A, Payre C, de Launoit Y, Gil J, Lambeau G, Bernard D . The M-type receptor PLA2R regulates senescence through the p53 pathway. EMBO Rep 2009; 10: 271–277.
Yang SH, Sarkar SN, Liu R, Perez EJ, Wang X, Wen Y et al. Estrogen receptor beta as a mitochondrial vulnerability factor. J Biol Chem 2009; 284: 9540–9548.
Acknowledgements
We thank M Ferrand, M Samyn, M Manchon, and D Collin Chavagnac for help and materials, and other laboratory members for helpful suggestions and discussions. This work was carried out with the support of the ‘Fondation de France’, the ‘Comités de l’Ardèche et de la Drome de la Ligue nationale contre le Cancer’, the ‘Institut National du Cancer’, and the ‘RTRS Fondation Synergie Lyon Cancer’. CW is supported by the ‘Ligue contre le Cancer’ and the ‘Fondation pour la Recherche Médicale’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Gitenay, D., Wiel, C., Lallet-Daher, H. et al. Glucose metabolism and hexosamine pathway regulate oncogene-induced senescence. Cell Death Dis 5, e1089 (2014). https://doi.org/10.1038/cddis.2014.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.63
Keywords
This article is cited by
-
RNA sequencing profiles reveal dynamic signaling and glucose metabolic features during bone marrow mesenchymal stem cell senescence
Cell & Bioscience (2022)
-
Multidrug resistance protein 3 loss promotes tumor formation by inducing senescence escape
Oncogene (2016)
-
Dysregulated glycolysis as an oncogenic event
Cellular and Molecular Life Sciences (2015)
-
Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence
Nature Communications (2014)
