
Abstract
Smac mimetics (SMs) comprise a class of small molecules that target members of the inhibitor of apoptosis family of pro-survival proteins, whose expression in cancer cells hinders the action of conventional chemotherapeutics. Herein, we describe the activity of SM83, a newly synthesised dimeric SM, in two cancer ascites models: athymic nude mice injected intraperitoneally with IGROV-1 human ovarian carcinoma cells and immunocompetent BALB/c mice injected with murine Meth A sarcoma cells. SM83 rapidly killed ascitic IGROV-1 and Meth A cells in vivo (prolonging mouse survival), but was ineffective against the same cells in vitro. IGROV-1 cells in nude mice were killed within the ascites by a non-apoptotic, tumour necrosis factor (TNF)-dependent mechanism. SM83 administration triggered a rapid inflammatory event characterised by host secretion of TNF, interleukin-1β and interferon-γ. This inflammatory response was associated with the reversion of the phenotype of tumour-associated macrophages from a pro-tumoural M2- to a pro-inflammatory M1-like state. SM83 treatment was also associated with a massive recruitment of neutrophils that, however, was not essential for the antitumoural activity of this compound. In BALB/c mice bearing Meth A ascites, SM83 treatment was in some cases curative, and these mice became resistant to a second injection of cancer cells, suggesting that they had developed an adaptive immune response. Altogether, these results indicate that, in vivo, SM83 modulates the immune system within the tumour microenvironment and, through its pro-inflammatory action, leads cancer cells to die by necrosis with the release of high-mobility group box-1. In conclusion, our work provides evidence that SMs could be more therapeutically active than expected by stimulating the immune system.
Similar content being viewed by others


Main
Resistance to death is a hallmark of cancer cells1 that renders them unresponsive to chemotherapy. Chemoresistance is often attributed to the inhibitor of apoptosis (IAP) proteins that control many aspects of cellular life, including the response to environmental stimuli, the regulation of cell death2 and cellular motility.3, 4 X-linked IAP (XIAP) is the only IAP to directly interact with caspases, which occurs through conserved domains named baculoviral IAP repeats.5 This interaction hinders the activity of both initiator caspase-96 and effector caspases-3 and -7.7 XIAP activity is antagonised by second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (Smac/DIABLO), which binds to its baculoviral IAP repeats, thereby releasing the caspases6 and favouring the apoptotic cascade. Starting from this observation, many groups have designed small peptidomimetics that mimic the structure of the N-terminal tetrapeptide (AVPI) of Smac/DIABLO in order to prevent XIAP from inhibiting caspases.8, 9, 10 These compounds, referred to as Smac mimetics (SMs), enhanced the antitumour activity of radiation or chemotherapeutic treatments in combination experiments.11 Furthermore, they have been shown to potentiate the cytotoxic effects of ‘death ligands’, in particular, than that of tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL).9, 12, 13, 14 Therefore, these compounds have been proposed as novel cancer treatments and are currently being tested, as part of combination therapies, in clinical trials.2
Despite the ability of SMs to enhance the cytotoxicity of other compounds, they are often ineffective in monotherapy. Still, some cancer cell lines undergo a TNF-dependent apoptotic process in response to the administration of a SM alone. In fact, although these compounds were designed to target XIAP, they also bind other IAPs, such as cellular IAP1 (cIAP1) and cIAP2, leading to their self-ubiquitination15, 16 and rapid proteasomal degradation.17, 18 As cIAPs are negative regulators of NF-κB-inducing kinase, their SM-triggered depletion results in the stabilisation of NF-κB-inducing kinase19 with consequent induction of the non-canonical NF-κB pathway.17 In cell lines sensitive to these compounds, this event is sufficient to trigger the secretion of high levels of TNF, thus killing the cells through an autocrine or paracrine mechanism.17, 18 Despite the clear in vitro assessment of this process, the role of TNF in SM-induced cell death in vivo is still controversial. In fact, the employment of these compounds in pre-clinical models, either as monotherapy or in combination with other drugs, has resulted in conflicting evidence,11, 20, 21 indicating a need to clarify the mechanism of action of SMs in vivo.
One way to study the mechanisms of action of antitumoural agents involves the use of animal models of cancer ascites. The formation of ascites characterises the advanced-stage of various types of tumours, especially ovarian carcinomas. This condition depends on a mechanical constriction exerted by the tumour and is determined by a peritoneal increase of fluid production associated with decreased lymphatic absorption. Ascites is often the cause of death for ovarian cancer patients, while rarely there is metastatic spread of tumour cells outside the peritoneum.22 In peritoneal ovarian carcinomatosis, the ascitic fluid represents the microenvironment in which soluble factors are interchanged between cancer and stromal and inflammatory cells, in order to support tumour growth and invasion. This microenvironment also promotes immunosuppression favouring tumour escape from immune control. Thus, although the ascitic fluid is normally enriched in immune cells, they are often anergic and unable to elicit an antitumour immune response. Animal models of cancer ascites facilitate the study of immune cells such as macrophages in response to antitumoural treatments.
Macrophages are plastic cells characterised by different states of activation.23 Classically activated macrophages (M1) and ‘alternatively’ activated macrophages (M2) represent the two extremes in the spectrum of the macrophage phenotype. Tumour-associated macrophages are classified as M2 macrophages because of their cytokine expression profile;24 they produce high amounts of immunosuppressive cytokines such as interleukin-10 (IL-10) and, mostly in solid tumours, share some tissue repair functions with fibroblasts.23, 25 In contrast, M1 macrophages produce large amounts of pro-inflammatory cytokines including interferon-γ (IFNγ) and IL-12, and can be involved in the elicitation of an effective antitumour immune response. The plasticity of macrophages has suggested new therapeutic approaches aiming to revert the M2 phenotype26 especially in tumours that strictly relies on the macrophage infiltration such as ovarian carcinomas. Accordingly, macrophages have been reprogrammed in a mouse ovarian ascites model by modulating NF-κB signalling.27
We recently described the synthesis of novel dimeric molecules that target XIAP, cIAP1 and cIAP2.28, 29 One of these molecules, SM83, inhibited the growth of SM-sensitive human mammary adenocarcinoma MDA-MB231 and rhabdomyosarcoma Kym-1, but not of other cell lines. Here, we use two murine xenograft models of cancer ascites to show that SM83, when administered in monotherapy, increases the survival of these mice by targeting tumour-associated macrophages. Through TNF, SM83 rapidly induces necrosis of the intraperitoneally (i.p.) injected cancer cells, which are otherwise completely resistant in vitro to the antitumoural effects of SM83. Therefore, our work shows that SM83 displays different mechanisms of action in vitro and in vivo, and that in vivo it exerts its antitumoural activity by stimulating the immune system.
Results
SM83 sensitises the IGROV-1 ovarian carcinoma cell line to the apoptotic effects of TRAIL
SM83 (Figure 1a) is a novel inhibitor of XIAP, cIAP1 and cIAP2. When administered to human IGROV-1 ovarian carcinoma cells, SM83 in monotherapy at two doses had no inhibitory effect on cell growth (Figure 1b). Instead, when administered together with TRAIL, cell growth was substantially reduced to about 50 (2 ng/ml TRAIL) and 28% (10 ng/ml TRAIL) of that of untreated cells, without a dose-dependent effect for SM83. TRAIL treatment alone had a negligible effect at this concentration, whereas SM83 monotherapy was ineffective on a panel of other human cancer cell lines (A2780, H460, SW48, HCT-116 and DLD-1 cells; data not shown). The apoptotic effects of these treatments on IGROV-1 cells at 3 and 24 h were assessed by western blotting (Figure 1c). Treatment with SM83 alone decreased cIAP1 and cIAP2 to almost undetectable levels already at 3 h. Treatment with SM83 and TRAIL, at 24 h, strongly increased cleaved poly (ADP-ribose) polymerase (PARP), a marker of activated apoptosis. Similar results were obtained when cells were treated with SM59 (Figure 1d). These results suggest that SMs sensitise IGROV-1 cells to TRAIL-induced cell death without causing death themselves.

SM83 induces apoptosis in vitro when combined with TRAIL. (a) Chemical structure of the dimeric SM SM83. (b) IGROV-1 cells were treated with 0.1 or 1.0 μM SM83 alone or in combination with 2 or 10 ng/ml TRAIL. Cell growth is expressed as a percentage relative to cells mock-treated with vehicle. Values are mean and S.D. from one experiment representative experiment of three performed. (c and d) Western blot analysis of XIAP, cIAP1, cIAP2 and cleaved PARP (Cl PARP) in IGROV-1 cells treated 3 or 24 h with SM83 (c) or SM59 (SM-164) (d) in the absence or presence of 10 ng/ml TRAIL. Actin is shown as loading control. Arrow, specific XIAP band
SMs in monotherapy increase the survival of mice bearing cancer ascites
SM83 and SM59 were then tested in vivo using a murine xenograft model in which IGROV-1 cells are injected i.p. into athymic nude mice, leading to ascites and death. Treatment with both SM83 (Figure 2a) and SM59 (Figure 2b) increased mouse survival (P<0.05 versus control mice), but SM83 was slightly more effective than SM59 (T/C% 180 versus 164). Furthermore, SM83 administration significantly reduced the formation of the ascites (Figure 2c). Treatment with TRAIL alone did not increase mouse survival, and the combination of TRAIL plus SM83 had no additive effect (Figure 2a). These findings, which are contrary to the in vitro results, suggest that SMs alone slow the progression of ovarian ascites but are not curative in these mice, whereas TRAIL alone is ineffective at the concentration used.

Treatment with SM83 in monotherapy increases the survival of mice bearing cancer ascites. (a) Nude mice were injected i.p. with IGROV-1 cells and left untreated (○) or treated 5 times a week, for 2 consecutive weeks starting the day after injection, with 5 mg/kg SM83 (▴), 2.5 mg/kg TRAIL (∇) or with the same doses of SM83 and TRAIL together (▪). One experiment representative of two performed is shown. Each treatment group contained seven mice. Survival curve for SM83-treated mice and controls. (b) Survival curve for SM59-treated and control mice. Untreated (○) or treated with SM SM59 (∇). (c) The formation of ascites was checked by monitoring body weight on day 17. (d and e) BALB/c mice were injected with Meth A cells and, starting on day 7, were treated daily with 5 mg/kg SM83. (d) The formation of ascites was checked by monitoring body weight on day 13. The horizontal line represents the mean. (e) Survival curve for untreated (●) or SM83-treated mice (○) starting from day 7
To test whether the in vivo activity of SM83 was cell line-specific or limited to immunodeficient mice, we used another ascites model in which murine Meth A sarcoma cells are injected i.p. into immunocompetent syngeneic BALB/c mice. Similar to IGROV-1 cells, Meth A cells were not growth-inhibited by SM83 in vitro (data not shown). In mice, SM83 reduced the progression of the ascites, seen as a smaller increase in body weight (Figure 2d), and augmented the median survival time from 15 days for untreated animals to 26 days (P=0.0721; Figure 2e). Also in this model, combination with TRAIL was not beneficial (data not shown). Moreover, when mice cured by SM83 (4 of 14 mice tested) were challenged with a new injection of 4 × 105 Meth A cells (twice that of the first administration) none formed new ascites (data not shown). These results strongly suggest that these animals developed an adaptive immunity.
SM83 rapidly kills cancer cells floating in the ascites by a non-apoptotic mechanism
To investigate the mechanism of SM83 activity in vivo, the ascitic fluid from mice injected with IGROV-1 cells and treated or not with SM83 for 3, 6 or 24 h was collected and tumour cells were counted. The results showed a striking decrease (P<0.001) in the total number of ascites tumour cells in SM83-treated mice at 24 h compared with control untreated mice, without appreciable changes at 3 or 6 h (Figure 3a). Autopsy revealed no adhesion of single cancer cells to the peritoneal wall nor migration outside the peritoneum. Furthermore, we detected a significant increase of human cytokeratin-18 (P<0.0001), a protein released from dying human epithelial cells, in the serum of mice treated for 24 h (Figure 3b), suggesting that SM83 triggered tumour cell death. Corroborating these in vivo findings, western blot analysis of proteins from floating tumour cells within the ascites confirmed that SM83 induced the degradation of cIAP1 and cIAP2, but not of XIAP (Figure 3c).

SM83 kills ascites tumour cells through a rapid, non-apoptotic event. Ascitic fluids were collected from nude mice injected with IGROV-1 cells and left untreated or treated with a single injection of SM83 (5 mg/kg). (a) Number of tumour cells within the ascites of untreated animals (n=11) or of animals treated for 3 (n=4), 6 (n=4) and 24 h (n=9). Data are mean and S.D. (b) Levels of human cytokeratin-18 in the serum collected 24 h after a single dose of SM83 (5 mg/kg) or no treatment. (c) Western blot of apoptosis markers in IGROV-1 cells cultured in vitro or recovered from the ascites of control mice (n=2) or mice treated with SM83 (n=2) or TRAIL for 24 h. (d) Western blot to detect activated apoptosis markers cleaved PARP (p89), caspase-8 (precursor p55 and cleaved form p43/41) and cleaved caspase-3 (p19/p17) in IGROV-1 cells cultured in vitro or recovered from the ascites (24 h) of control mice (n=2) and of mice treated with 5 mg/kg SM83 (n=2) or 10 mg/kg TRAIL. (e) Western blot of apoptotic markers in tumour cells collected from the ascites of untreated mice (n=2) or from mice treated for 3 or 6 h with SM83 (n=2 for both) or with TRAIL for 24 h. Arrow, specific band for XIAP
To understand the mechanism of cell death accounting for the decrease in the number of floating tumour cells within ascites, we examined the expression of mediators of apoptosis in these cells at different time points. At 24 h, SM83 treatment led to only a faint increase of cleaved PARP, no evidence of cleaved, active caspase-8 (which was expected because these cells are killed in vivo by SM monotherapy30) and only a modest effect on cleavage of caspase-3 (Figure 3d). In contrast, cells treated with TRAIL showed a greater activation of these apoptotic markers even though TRAIL was ineffective in reducing the ascitic cell number (Supplementary Figure S1). In an attempt to detect the apoptotic events, western blotting was done on the cells collected after 3 and 6 h of treatment (Figure 3e). Also in this case, SM83 caused the complete degradation of cIAP1 and cIAP2, but there was only a faint accumulation of the cleaved forms of PARP, caspase-8 and caspase-3. This low activation of the apoptotic cascade suggested that the cause of death of the ascites tumour cells was not primarily apoptotic.
To assess the possibility that autophagy – another mechanism that can cause cell death when abnormally and continuously activated – was responsible for the loss of ascites tumour cells, we measured the levels of beclin-1 and cleaved LC3B. Treatment with SM83 did not increase the levels of these proteins (Supplementary Figure S2), suggesting that autophagy is not involved in SM83-induced tumour cell death.
SM83 triggers an inflammatory event in vivo
Having excluded apoptosis and autophagy in SM83-mediated cell death, we investigated the involvement of necrosis by measuring the levels of murine inflammatory cytokines in the ascites. There was a basal level of murine TNF in the ascites of untreated mice, whereas in mice treated with SM83, the level of murine TNF was significantly higher at 3 and 6 h but not at 24 h (Figure 4a). On the contrary, human TNF was not affected by SM83 treatment (Supplementary Figure S3). We tested the relevance of TNF in this model by pretreating mice with two inhibitors of TNF, etanercept and infliximab. Etanercept at the higher dose tested completely blocked the cytotoxic effects of SM83 (Figure 4b), without preventing SM83-triggered degradation of cIAPs (Figure 4c). Pretreatment of mice with infliximab, instead, did not block the cytotoxic effects of SM83 on ascites tumour cells (data not shown), probably owing to its lower affinity for mouse TNF or to an insufficient concentration. Overall, these data suggest that SMs are effective in vivo by stimulating the release of murine TNF that activates the TNF receptor leading to the creation of a pro-inflammatory microenvironment. Accordingly, the murine inflammatory cytokines IL-1β (Figure 4d) and IFN-γ (Figure 4e) were upregulated in the peritoneal ascites in response to SM83 administration. In contrast, levels of the immunosuppressive cytokines IL-10 (Figure 4f), transforming growth factor-β (Figure 4g) and IL-4 (Figure 4h) remained mostly unchanged, with the exception of IL-4 that increased significantly at 24 h; this increase could be explained by the anti-inflammatory role of this cytokine, secreted after 24 h to restrain the strong inflammation in process.

SM83 treatment induces the expression of inflammatory cytokines in vivo. Nude mice were injected i.p. with IGROV-1 cells and treated or not with a single dose of 5 mg/kg SM83; ascites was collected 3, 6 and 24 h after SM83 administration. (a) SM83 transiently increased the level of murine TNF measured by ELISA. (b) SM83 treatment reduced ascites cell counts but this action was blocked when TNF was sequestered with the higher dose of etanercept (P=0.0118 relative to treated with SM83 alone). (c) Western blots of ascites tumour cell protein from untreated mice and mice treated with SM83 alone or in combination with 150 mg/kg etanercept. Arrow, specific band for XIAP. (d–h) ELISA results for IL-1β (d), IFN-γ (e), IL-10 (f), transforming growth factor-β (TGF-β) (g) and IL-4 (h) in the ascitic fluid of mice treated as above. Results for IL-10 are shown as fold induction owing to the lack of recombinant protein standard
SM83 promotes macrophage activation towards an M1-like phenotype
To evaluate whether the pro-inflammatory effect of SM83 depends on the activation of tumour-associated macrophages, bone marrow (BM)-derived macrophages (BMDM) from BALB/c mice were treated in vitro with SM83, and the cell supernatants were assayed for TNF and IL-1β. SM83 induced the secretion of TNF starting at 6 h (Figure 5a) and of IL-1β at 24 h (Figure 5b). Moreover, as macrophage activation is regulated by NF-κB, we checked whether SM83 activated this pathway using a macrophage cell line stably expressing a luciferase reporter gene under control of the NF-κB-response element. NF-κB activation was detectable after 3 h of treatment with SM83 but returned to basal levels at 24 h (Figure 5c). In agreement with an earlier study,31 SM83 treatment also promoted the death of BMDM through necroptosis, which was seen 24 h after treatment both morphologically (Figure 5d, second panel) and with the CellTiter-Glo assay (Figure 5e, first bar). This cytotoxicity was prevented by pretreatment with necrostatin-1, a specific inhibitor of receptor-interacting protein-1 (a crucial mediator of necroptosis), but not by the pan-caspase inhibitor z-vad-fmk, which actually increased BMDM death (Figures 5d and e). Overall, these data suggest that macrophages could be the first target of SM83 activity that fosters their production of pro-inflammatory M1-like molecules such as TNF and IL-1β.

SM treatment activates macrophage and sensitises them to necroptotic death. (a and b) BALB/c BMDM (5 × 105) were seeded in 96-well plates, left to adhere for 16 h and treated or not with 1 μM SM83 for up to 24 h. SM83 treatment promoted the secretion of the pro-inflammatory cytokines TNF (a) and IL-1β (b). (c) NF-κB activation by SM83 evaluated in the macrophage cell line RAW stably transfected with a NF-κB luciferase reporter gene. One representative experiment of two performed is shown. Values are mean and S.D.; n=3. (d and e) BALB/c BMDM were seeded in six-well plates, left to adhere for 16 h and treated with serial dilutions of SM83 in the absence or presence of necrostatin-1 or z-vad-fmk to inhibit necroptosis or apoptosis, respectively. (d) Representative images showing cell morphology after 24 h of treatment. (e) Cell viability after treatments evaluated using the CellTiter-Glo assay
To exclude the possibility that the SM83-induced IL-1β secretion was due to contamination with lipopolysaccharides (LPS), by the stimulation of toll-like receptor-4 (TLR432), we prepared BMDM from TLR4-knockout (KO) mice and found that IL-1β was secreted to a similar extent as it was secreted from BALB/c cells (Supplementary Figure S4). Furthermore, SM83 preparations for injection were found to not contain detectable levels of LPS on the Endosafe-PTS (Charles River Laboratories, Calco, Italy) test (data not shown). These results indicate that the SM83-induced IL-1β production was not due to LPS contamination.
Bystander accumulation of neutrophils in the ascites
Having demonstrated a pro-inflammatory effect of SM83 in the tumour microenvironment, we next evaluated whether SM83 treatment affected the innate immune cells infiltrating the ascites. To this end, we used immunofluorescence to examine the expression of leucocyte antigens on cells harvested from the ascites of nude mice. This assay allowed us to identify both macrophages (CD11b+ Gr-1−) and neutrophils (CD11b+ Gr-1+) in mice that were either untreated or treated for 3 and 6 h before harvesting (Figure 6a), without noticeable differences in cell density. However, after 24 h of treatment, neutrophils (but not macrophages) had massively accumulated in the ascitic fluid (Figure 6a, Supplementary Figure S5). Neutrophils had also infiltrated the tumour nodules (Supplementary Figure S6). This massive neutrophil recruitment was likely because of the presence, in the ascites, of high-mobility group box-1 (HMGB-1; Figures 6b and c) and TNF (Figure 4a). HMGB-1, which is released during necrosis and immunogenic cell death, behaves as an ‘alarmin’ that, together with TNF, attracts and activates neutrophils.33 In our IGROV-1 model, HMGB-1 was detected in the ascitic fluid starting 6 h after SM83 treatment (Figure 6b) and was located within the cytoplasm of dying tumour cells (Figure 6c).

SM83 treatment promotes peritoneal neutrophil recruitment and activation. Cells were harvested from the ascitic fluid of mice treated with 5 mg/kg SM83. (a) Immunofluorescence for CD11b (green) and Gr-1 (red) was performed on Cytospin cell preparations. SM83 treatment induced a massive recruitment of neutrophils (CD11b+ Gr-1+) at 24 h but not at earlier time points. (b) Dot blot of HMGB-1 (left panel) was performed on cleared ascitic fluids collected from untreated (un) or treated mice at 3, 6 and 24 h after a single injection of 5 mg/kg SM83. Right panel, loading control. (c) Infiltration of neutrophils (Gr-1; red) and HMGB-1 expression in dying tumour cells (green) in ascites untreated (upper row) and treated for 24 h (lower row) with a single injection of SM83. Left and right panels show two different magnifications ( × 10 and × 40). (d) PMN migration assessed by Transwell assay. Wild-type PMN from BALB/c mice (WT) or TNF-R1-deficient PMN from TNF-R1-KO mice (KO) were seeded into the upper chamber in the absence or presence of the HMGB-1 inhibitor glycyrrhizin (GLZ), whereas the ascitic fluid of mice untreated or treated for 6 h (1:20 in medium) was added to the lower chamber of the Transwell insert. PMN were left to migrate overnight, harvested and counted. (e) Activation of wild-type PMNs was colorimetrically evaluated on the basis of the ability of cells to oxidise the cytochrome c in presence of the ascitic fluid collected from mice 6 h after treating with a single injection of SM83. The experiment was also performed in the presence of GLZ and the TNF inhibitor etanercept. (f) Tumour cell counts in the ascites of untreated mice (UN), mice treated with a single injection of SM83 alone or mice treated with SM83 24 h after depletion of neutrophils by injection with the 1A8 mAb. (g) BALB/c mice were injected i.p. with Meth A cells and treated with 1A8 alone (▪) or in combination with 5 mg/kg SM83 (□), starting from day 7
Next, neutrophils were isolated from spleens of BALB/c (wild-type) and TNF-receptor-1 (TNF-R1)-deficient mice to investigate in vitro the role of neutrophils in the antitumoural activity of SM83. In Transwell assays, the migration of wild-type neutrophils towards ascites from SM83-treated mice was greater than that from untreated mice (Figure 6d). The HMGB-1 inhibitor partially reduced the migration, whereas neutrophils from TNF-R1-deficient mice did not migrate in response to ascites from SM83-treated mice. Ascites from mice treated with SM83 activated wild-type neutrophils to produce superoxide even in the presence of the TNF blocker etanercept, whereas glycyrrhizin completely blocked such activity (Figure 6e). These findings suggest that neutrophil migration in the presence of ascites from SM83-treated mice is stimulated by TNF while activation is attributable to HMGB-1. The importance of TNF in neutrophil migration was also shown in vivo, where etanercept pretreatment blocked the recruitment of these cells into the ascites (Supplementary Figure S5).
Considering that, in certain conditions, neutrophils can be responsible for tumour cell killing,34, 35 we depleted ascites-bearing mice of neutrophils before SM83 treatment. In nude mice bearing IGROV-1 ascites, neutrophil depletion did not impede the ability of SM83 to reduce the number of floating tumour cells in the ascites (Figure 6f). Furthermore, in immunocompetent BALB/c mice bearing Meth A ascites, depletion of neutrophils (Supplementary Figure S7) did not prevent SM83 from prolonging survival (Figure 6g), even though these mice have CD8+ T cells that are known to support the activity of neutrophils.36 On the contrary, neutrophil depletion improved the efficacy of SM83 treatment by significantly augmenting the overall survival of mice treated with SM83 (P=0.0458 versus 1A8 alone). These results support the notion that, in our models, neutrophil recruitment is a bystander effect due to SM83-induced TNF secretion. The accumulation of neutrophils has no role in SM-dependent cancer cell killing but rather, at least in the Meth A model, could be in some way detrimental to the host.
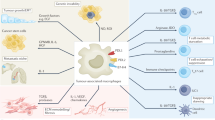
In conclusion, our work shows that SM83 acts in vivo in cancer ascites by reverting macrophages from an M2-like phenotype, supporting the tumour, to an M1-like phenotype, endowed with antitumour activity. M1 macrophages secrete cytokines such as TNF, IL-1β and IFNγ that cause a rapid TNF-dependent necrotic death of the ascites cancer cells (Figure 7); subsequently, the dying cells release HMGB-1 that, together with TNF, stimulates a massive infiltration of neutrophils.

Proposed mechanism of action of SM83 in cancer ascites. SM83 stimulates the reversal of macrophages from M2 to M1 phenotype. TNF secreted by M1 macrophages triggers necrotic death of the cancer cells within the ascitic fluid; the dying cells release HMGB-1 that, together with TNF, recruits neutrophils
Discussion
In this study, we describe the in vivo activity of newly synthesised SM SM83 in two murine xenograft models of cancer ascites. We show that SM83 is active as monotherapy in vivo on human and murine cancer cells that are refractory to SMs in vitro, and demonstrate that these cells die through a non-apoptotic, TNF-dependent mechanism. We also provide evidence that SM83 exerts its activity by inducing inflammation and immune cell activation, resulting in immunogenic death of cancer cells. Moreover, our study shows that the activity of SM83 (and possibly other SMs) in the complex in vivo environment is different from that already observed in vitro.
The activity of SMs in vitro has been widely shown to depend on the secretion of TNF from tumour cells leading to apoptosis in an autocrine manner.17, 18 Our in vivo work confirms that the antitumoural activity of SM83 depends on TNF production. However, in our xenograft models, TNF originated primarily from the host rather than from the tumour cells. TNF was secreted by the immune cells, and by macrophages in particular, and this effect was preceded temporally by activation of the NF-κB-response element. Following SM83 treatment, macrophages acquired an M1-like phenotype characterised by the release of pro-inflammatory cytokines such as TNF and IL-1β. Both cytokines have been associated with the cytotoxic activity of macrophages on tumour cells.23, 37 Our experiments show that SM83 treatment increased the concentration of these cytokines in the ascites without a major influx of macrophages. Thus, SM83 treatment may revert existing M2 macrophages to the M1 phenotype rather than recruit new macrophages to the ascites. A mechanistic interpretation of these results (Figure 7) is that the cytokines secreted by M1 macrophages cause necrotic death of the ascites cancer cells; the dying cells release HMGB-1 that, together with TNF, recruits neutrophils as a side effect rather than a needed step in the antitumoural activity of SM83.
In tumour microenvironment, including ovarian cancer, macrophages acquire an M2-like phenotype.38, 39 Nevertheless, some authors have proposed various strategies23, 24 that are able to interchange the status of macrophages towards an efficient antitumour response. On this line, it has also been shown the possibility of increasing efficacy of standard therapies interfering with M2 macrophages,40 particularly in tumours whose progression strictly relies on their support such as ovarian carcinomas. These tumours establish a complex relationship with the associated immune cells within the ascites,41 developing an immunosuppressive microenvironment elicited by macrophages. Thus, in ovarian cancer, the re-polarisation of macrophages or the inhibition of their recruitment could be a new effective therapeutic strategy.
Our results suggest that SMs regulate the immune response to cancer ascites by polarising macrophages and thus have therapeutic potential. In fact, Smac/DIABLO42 and its mimetic SM83, as shown here, trigger necrotic or necroptotic cell death. Necrotic cells, different from apoptotic ones, release a non-oxidised, immunogenic form of the alarmin HMGB-143 that activates dendritic cells by engaging receptor for advanced glycation endproducts, TLR4, TLR7 and TLR9 receptors.44 Furthermore, necrotic cells can prime CD4+ T cells, essential for the development of an adaptive immune response.45 The possibility that SM83-induced necrosis primed an adaptive immune response is raised by our results using BALB/c-immunocompetent mice bearing Meth A ascites. Some of these mice were cured by SM83 treatment and, when challenged with a new, double dose of Meth A cells, they did not form another ascites, suggesting that they were immune to Meth A sarcoma cells.
In conclusion, our data demonstrate that the SM SM83 is active in monotherapy by promoting inflammation and immunogenic cell death. These observations provide an explanation for why SMs increase the effectiveness of standard therapies, namely by stimulating the immune system. Finally, the evidence that SMs can induce an inflammatory response and activate the immune system provides an interpretation of why SMs can be more effective in vivo than in vitro,11, 46 and our results in cancer cell killing.
Materials and Methods
SMs, cell lines and mice strains
The compounds used in this study were SM83 (called 9a elsewhere29, 47), SM59 (also called SM-16448) and TRAIL. Recombinant TRAIL was purchased from Enzo Life Sciences (Farmingdale, NY, USA), whereas SM83 and SM59 were synthesised by CISI scrl (purity>99%). Synthesis and chemical structure of these SMs have been described elsewhere.28, 29
Human ovarian carcinoma IGROV-1 and A2780, lung cancer H460, colon cancer SW48, HCT-116 and DLD-1, and murine sarcoma Meth A cell lines were cultured in vitro with RPMI plus 10% foetal calf serum. The murine macrophage cell line RAW 264.7, stably transfected with an NF-κB luciferase reporter plasmid, was cultured in DMEM plus 10% foetal calf serum.
KO C57/BL6 mice strains deficient in TNF-R1 (TNF-R1-KO49) or in TLR4-KO50 and female athymic Swiss nude mice, all 8–10 weeks of age (Charles River Laboratories), were maintained in laminar flow rooms keeping temperature and humidity constant. Female BALB/c mice 7–8 weeks of age (Charles River Laboratories) were housed in filter-top cages. Room sentinels were checked for pathogens every 6 months by Charles River Laboratories staff. Experiments were approved by the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale dei Tumori of Milan according to institutional guidelines.
Cell treatments and assays
Cells were plated at a density of about 60% and grown overnight before being treated with SM83 or SM59 in the absence or presence of 2 or 10 ng/ml TRAIL. Cell growth was determined after 3 days by cell counting, whereas viability was tested using the CellTiter-Glo (Promega Italia srl, Milan, Italy) assay for ATP after 24 h of treatment. For apoptosis assays, cells were harvested after 3 or 24 h of treatment and used in western blotting.
Western blotting
Cells were harvested at the end of the treatment and lysed in 125 mM Tris HCl pH 6.8, 5% SDS by boiling; protein concentration was quantified. Lysates (50 μg) were fractionated by SDS-PAGE and proteins were blotted onto PVDF membranes. Free protein-binding sites were blocked with 5% non-fat dry milk in phosphate-buffered saline (PBS) and the membranes were incubated overnight with primary antibodies diluted in non-fat dry milk-PBS. The primary antibodies used were directed against: cleaved caspase-3, beclin-1, cleaved PARP and LC3B (Cell Signaling Technology, Danvers, MA, USA); caspase-8 (Enzo Life Sciences); XIAP and cIAP2 (BD Biosciences, Milan, Italy), β-actin (Sigma, Rome, Italy); and cIAP1 (R&D Systems, Minneapolis, MN, USA). After washing, the membranes were exposed to horseradish peroxidase-linked secondary antibody (GE Healthcare, Milan, Italy) for 1 h and the bound antibodies were detected using ECL (Thermo Scientific, Rockford, IL, USA).
Animal models of cancer ascites
The in vivo effects of SMs were tested using two murine xenograft models of ovarian cancer. In one model, human ovarian carcinoma IGROV-1 cells are injected i.p. into athymic nude mice.51 The cells adapt to grow i.p. and are maintained by i.p. serial passages of ascitic cells. Mice develop haemorrhagic ascites and diffuse carcinomatosis and eventually die.51 In order to assess the effects of SMs on survival, mice were inoculated i.p. with 2.5 × 106 cells in 0.2 ml saline and, starting the day after cell injection, injected i.p. with the compounds (dissolved in saline) at 5 mg/kg body weight, once daily for 5 days per week for 2 weeks (qdx5/wx2w). The animals were inspected and weighed daily. The progression of ascites was assessed from an increase in body weight (measured on day 17). For ethical reasons, they were killed before impending death, recognised by their suffering status and loss of reactivity to external stimuli. The day of killing was taken as the day of death for calculating the median survival time. The antitumour activity of treatments was expressed as the ratio of median survival time in treated mice to that of untreated control mice × 100 (T/C%).
In the other ascites model, murine Meth A cells are injected i.p. into immunocompetent syngeneic BALB/c mice.52 Here, 2 × 105 Meth A cells were injected i.p., and treatment, starting 7 days later, consisted of five consecutive daily i.p. injections of SM83 at a dose of 5 mg/kg body weight. The progression of ascites was assessed from both an increase in body weight (measured on day 13) and overall survival, as calculated above. Mice in whom the ascites resolved and did not relapse within 60 days were considered to be cured, and were challenged with a new injection of 4 × 105 Meth A cells.
To assess the effects of SMs on ascites tumour cells, athymic nude mice were injected with IGROV-1 cells as above. Ten days after IGROV-1 inoculation, ascites-bearing mice were treated with a single injection of SM83 (5 mg/kg) or left untreated; in some experiments, mice were pretreated 1 h before SM83 administration by i.p. injection with one of two TNF inhibitors: 5 or 150 mg/kg etanercept (Wyeth Pharmaceuticals, Collegeville, PA, USA)53 or 5 mg/kg infliximab (Schering-Plough, Milan, Italy). Mice were killed 3, 6 or 24 h after SM83 treatment, and ascitic fluid was collected with a heparinised syringe, transferred to a centrifuge tube on ice and centrifuged to pellet the cells for blotting. The supernatant was removed and stored at −80 °C until it was assayed for cytokines (human and murine TNF, and murine IL-1β, IL-4, IL-10, IFNγ and transforming growth factor-β enzyme-linked immunosorbent assay (ELISA) kits (Affymetrix, San Diego, CA, USA) or used in blotting and in neutrophil assays. The pellet was suspended in 0.17 M ammonium chloride for 10 min at 41 °C to lyse red blood cells; after washing in saline, the remaining tumour cells were counted using the trypan blue exclusion assay and used in immunofluorescence. Death of IGROV-1 cells was also checked by quantifying human cytokeratin-18, a cytokeratin released by dying cells, in the serum collected from killed ascites-bearing mice using M65 ELISA (Enzo Life Sciences).
Animals were subjected to autopsy to search for adherent tumour cells or extraperitoneal migration. As IGROV-1 cells injected i.p. in nude mice also form solid nodules, these were collected and fixed in formalin. After 24 h, tumours were washed in PBS and kept in 70% ethanol until they were embedded in paraffin and prepared for haematoxylin and eosin staining.
To deplete neutrophils, nude mice bearing IGROV-1 ascites were injected i.p. with the anti-Ly-6G antibody (clone 1A8; Affymetrix) 24 h before SM83 administration. In the Meth A model, mice were injected with 20 μg 1A8 antibody every second day, for 14 consecutive days, starting one day before SM83 treatment (day 7). The efficiency of polymorphonuclear leucocytes (PMNs) depletion in nude mice was tested by sampling the bone marrow (BM) 24 h after injection of the anti-Ly-6G antibody. Cells were double-stained with anti-CD11b/Ly-6G and cell populations were quantified by flow cytometry.54
Macrophage primary cultures and reporter gene assays
BM precursors were obtained from BALB/c and TLR4-KO mice and used to isolate macrophages as previously described.26 Briefly, macrophages were isolated from the precursors by cultivation in RPMI medium 10% foetal calf serum containing 5 ng/ml macrophage colony-stimulating factor. On day 5, the medium was replaced with fresh medium containing macrophage colony-stimulating factor. On day 7, adherent cells were harvested and phenotypically characterised by immunofluorescence using mAb to the macrophage markers CD11b and F4/80 (BD Biosciences). Up to 90% of the adherent population consisted of BMDM.
For in vitro experiments, 5 × 105 BMDM were seeded in six-well plates, allowed to attach for 1 h at 37 °C and then treated or not with 1 μM SM83. The release of TNF and IL-1β to the culture medium, after 3, 6 and 24 h, was evaluated using ELISA kits (Affymetrix). In viability experiments, 1 × 104 cells were seeded in 96-well plates and exposed to serial dilutions of SM83 in the absence or presence of either 50 μM necrostatin-1 (Enzo Life Sciences) or 20 μM z-vad-fmk (Enzo Life Sciences). After 24 h, cells were examined morphologically by microscopy and tested for viability using the CellTiter-Glo (Thermo Scientific) assay.
In other experiments, 104 RAW 264.7 macrophages were plated in 96-well plates, grown for 16 h and then treated or not with 1 μM SM83 for up to 24 h. The activation of the NF-κB promoter was assayed using the Dual-Luciferase Reporter Assay System (Thermo Scientific).
Immunofluorescence and dot blotting
Floating cells harvested from the ascites were prepared for double-marker immunofluorescence by centrifugation on slides using a Cytospin cytocentrifuge. Briefly, acetone-fixed cells were re-hydrated in PBS and incubated 1 h with a primary antibody. Cells were then washed in PBS and incubated for 30 min with the appropriate Alexa Fluor 488-conjugated secondary antibody. After washing, slides were incubated for 30 min with another primary antibody, washed and incubated for 30 min with an Alexa Fluor 546-conjugated secondary antibody. The primary antibodies used were: rat anti-mouse CD11b mAb (1:200; BD Biosciences); rat anti-mouse Ly-6G mAb (1:200; BD Biosciences); and rabbit anti-HMGB-1 Ab (1:400; Abcam, Cambridge, UK). In some experiments, nuclei were stained with 4′,6-diamidino-2-phenylindole. Slides were mounted with Prolong medium (Life Technologies, Monza, Italy) and examined under a RADiance-2000 (Bio-Rad, Milan, Italy) Nikon-TE300 laser scanning confocal microscope (Nikon Instruments S.p.A, Florence, Italy).
For dot blotting, ascitic fluid samples cleared of cells were spotted on PDVF membranes and let to dry. After saturation with non-fat dry milk-PBS, filters were hybridised with anti-HMGB-1 Ab (Abcam, 1:2000). The HMGB-1 was then detected as for western blotting.
Spleen neutrophil preparation and assays
Neutrophils were obtained from BALB/c (wild-type) or TNF-R1-KO mice through immunomagnetic cell separation of a spleen cell suspension, using the Anti-Ly-6G MicroBead Kit (Miltenyi Biotec, Bologna, Italy), as already described.54
Migration of neutrophils PMNs was tested using the Transwell system (Corning, Tewksbury, MA, USA). Briefly, 5 × 105 PMNs were placed in serum-free RPMI in the upper chamber of the Transwell insert. The lower chamber was filled with ascites (1:20) or TNF in serum-free DMEM. PMNs were allowed to migrate for 24 h and then collected from the lower chamber. In some cases, experiments were performed by adding 100 μM of the HMGB-1 inhibitor glycyrrhizin (Sigma-Aldrich, St. Louis, MO, USA) to the top chamber.55
Neutrophil activation by cleared ascites was evaluated by measuring the rate of formation of superoxide. Briefly, neutrophils (2 × 106/0.1 ml cells/PBS solution) were incubated with 20 μl of cytochrome c (0.1 mM final, bovine, Sigma), 20 μl of ascites or 10 μg/ml of PMA (as positive control for the neutrophil stimuli) in 1.76 ml PiCM-G buffer (138 mM NaCl, 2.7 mM KCl, 0.6 mM CaCl2, 1.0 mM MgCl2, 5 mM glucose and 10 mM NaH2PO4/Na2HPO4, pH 7.4). The mixture was added to cuvettes for spectrophotometer and absorbance was read at 550 nm every 15 min as described.56
Statistical analyses
For survival analyses, the percent survivorship over time was estimated using the two-sided Kaplan–Meier product method; differences between groups were compared using the log-rank test. In other analyses, differences between groups were tested for significance using a two-tailed unpaired t-test. A value of P<0.05 indicated significance. The analyses were performed using Graphpad Prism v.5.02 (GraphPad Software, La Jolla, CA, USA).
Abbreviations
- SM:
-
smac mimetic
- IAP:
-
inhibitor of apoptosis
- TNF:
-
tumour necrosis factor
- XIAP:
-
X-linked inhibitor of apoptosis
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- PARP:
-
poly (ADP-ribose) polymerase
- LPS:
-
lipopolysaccharides
- HMGB-1:
-
high-mobility group box-1
- TNF-R1:
-
TNF-receptor-1
- TLR:
-
toll-like receptor
- BMDM:
-
bone marrow-derived macrophages
- PMN:
-
polymorphonuclear leucocytes
References
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Gyrd-Hansen M, Meier P . IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 2010; 10: 561–574.
Lopez J, John SW, Tenev T, Rautureau GJ, Hinds MG, Francalanci F et al. CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol Cell 2011; 42: 569–583.
Fingas CD, Blechacz BR, Smoot RL, Guicciardi ME, Mott J, Bronk SF et al. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology 2010; 52: 550–561.
Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS et al. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 1998; 273: 7787–7790.
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410: 112–116.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC . X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997; 388: 300–304.
Oost TK, Sun C, Armstrong RC, Al-Assaad A, Betz SF, Deckwerth TL et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 2004; 47: 4417–4426.
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG . A small molecule smac mimic potentiates TRAIL- and TNF{alpha}-mediated cell death. Science 2004; 305: 1471–1474.
Seneci P, Bianchi A, Battaglia C, Belvisi L, Bolognesi M, Caprini A et al. Rational design, synthesis and characterization of potent, non-peptidic Smac mimics/XIAP inhibitors as proapoptotic agents for cancer therapy. Bioorg Med Chem 2009; 17: 5834–5856.
Probst BL, Liu L, Ramesh V, Li L, Sun H, Minna JD et al. Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner. Cell Death Differ 2010; 17: 1645–1654.
Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res 2009; 69: 2425–2434.
Lecis D, Drago C, Manzoni L, Seneci P, Scolastico C, Mastrangelo E et al. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. Br J Cancer 2010; 102: 1707–1716.
Lu J, McEachern D, Sun H, Bai L, Peng Y, Qiu S et al. Therapeutic potential and molecular mechanism of a novel, potent, nonpeptide, Smac mimetic SM-164 in combination with TRAIL for cancer treatment. Mol Cancer Ther 2011; 10: 902–914.
Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM et al. SMAC-mimetics activate the E3 ligase activity of cIAP1 by promoting RING dimerisation. J Biol Chem 2011; 286: 17015–17028.
Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J et al. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ 2011; 18: 1376–1386.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 2007; 131: 682–693.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007; 131: 669–681.
Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 2012; 31: 1679–1691.
Eschenburg G, Eggert A, Schramm A, Lode HN, Hundsdoerfer P . Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-alpha independent apoptosis. Cancer Res 2012; 72: 2645–2656.
Greer RM, Peyton M, Larsen JE, Girard L, Xie Y, Gazdar AF et al. SMAC mimetic (JP1201) sensitizes non-small cell lung cancers to multiple chemotherapy agents in an IAP-dependent but TNF-alpha-independent manner. Cancer Res 2011; 71: 7640–7648.
Lengyel E . Ovarian cancer development and metastasis. Am J Pathol 2010; 177: 1053–1064.
Sica A, Mantovani A . Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122: 787–795.
Sica A, Schioppa T, Mantovani A, Allavena P . Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 2006; 42: 717–727.
Martinez FO, Helming L, Gordon S . Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009; 27: 451–483.
Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP . Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res 2005; 65: 3437–3446.
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med 2008; 205: 1261–1268.
Manzoni L, Belvisi L, Bianchi A, Conti A, Drago C, de Matteo M et al. Homo- and heterodimeric Smac mimetics/IAP inhibitors as in vivo-active pro-apoptotic agents. Part I: synthesis. Bioorg Med Chem 2012; 20: 6687–6708.
Lecis D, Mastrangelo E, Belvisi L, Bolognesi M, Civera M, Cossu F et al. Dimeric Smac mimetics/IAP inhibitors as in vivo-active pro-apoptotic agents. Part II: Structural and biological characterization. Bioorg Med Chem 2012; 20: 6709–6723.
Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-{alpha} signaling. Cancer Res 2007; 67: 11493–11498.
Muller-Sienerth N, Dietz L, Holtz P, Kapp M, Grigoleit GU, Schmuck C et al. SMAC mimetic BV6 induces cell death in monocytes and maturation of monocyte-derived dendritic cells. PLoS One 2011; 6: e21556.
Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med 1999; 189: 615–625.
Sitia G, Iannacone M, Muller S, Bianchi ME, Guidotti LG . Treatment with HMGB1 inhibitors diminishes CTL-induced liver disease in HBV transgenic mice. J Leukoc Biol 2007; 81: 100–107.
Colombo MP, Modesti A, Parmiani G, Forni G . Local cytokine availability elicits tumor rejection and systemic immunity through granulocyte-T-lymphocyte cross-talk. Cancer Res 1992; 52: 4853–4857.
Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R . Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011; 20: 300–314.
Stoppacciaro A, Melani C, Parenza M, Mastracchio A, Bassi C, Baroni C et al. Regression of an established tumor genetically modified to release granulocyte colony-stimulating factor requires granulocyte-T cell cooperation and T cell-produced interferon gamma. J Exp Med 1993; 178: 151–161.
Bonta IL, Ben-Efraim S . Involvement of inflammatory mediators in macrophage antitumor activity. J Leukoc Biol 1993; 54: 613–626.
Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ et al. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol 2011; 187: 1157–1165.
Said NA, Elmarakby AA, Imig JD, Fulton DJ, Motamed K . SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia 2008; 10: 1092–1104.
Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res 2011; 17: 6083–6096.
Jeon BH, Jang C, Han J, Kataru RP, Piao L, Jung K et al. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res 2008; 68: 1100–1109.
Emeagi PU, Van Lint S, Goyvaerts C, Maenhout S, Cauwels A, McNeish IA et al. Proinflammatory characteristics of SMAC/DIABLO-induced cell death in antitumor therapy. Cancer Res 2012; 72: 1342–1352.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA . Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ . HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010; 28: 367–388.
Bartholomae WC, Rininsland FH, Eisenberg JC, Boehm BO, Lehmann PV, Tary-Lehmann M . T cell immunity induced by live, necrotic, and apoptotic tumor cells. J Immunol 2004; 173: 1012–1022.
Dineen SP, Roland CL, Greer R, Carbon JG, Toombs JE, Gupta P et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res 2010; 70: 2852–2861.
Cossu F, Milani M, Vachette P, Malvezzi F, Grassi S, Lecis D et al. Structural insight into inhibitor of apoptosis proteins recognition by a potent divalent smac-mimetic. PLoS One 2012; 7: e49527.
Sun H, Nikolovska-Coleska Z, Lu J, Meagher JL, Yang CY, Qiu S et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc 2007; 129: 15279–15294.
Sangaletti S, Tripodo C, Ratti C, Piconese S, Porcasi R, Salcedo R et al. Oncogene-driven intrinsic inflammation induces leukocyte production of tumor necrosis factor that critically contributes to mammary carcinogenesis. Cancer Res 2010; 70: 7764–7775.
Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Menard S et al. Critical role of TLR9 in acute graft-versus-host disease. J Immunol 2008; 181: 6132–6139.
De Cesare M, Sfondrini L, Campiglio M, Sommariva M, Bianchi F, Perego P et al. Ascites regression and survival increase in mice bearing advanced-stage human ovarian carcinomas and repeatedly treated intraperitoneally with CpG-ODN. J Immunother 2010; 33: 8–15.
Watanabe N, Niitsu Y, Sone H, Neda H, Urushizaki I, Yamamoto A et al. Therapeutic effect of endogenous tumor necrosis factor on ascites Meth A sarcoma. J Immunopharmacol 1986; 8: 271–283.
Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther 2002; 301: 418–426.
Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012; 120: 3007–3018.
Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M et al. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol 2007; 14: 431–441.
Clark RA, Nauseef WM . Isolation and Functional Analysis of Neutrophils. Current Protocols in Immunology. John Wiley & Sons, Inc, 2001.
Acknowledgements
We thank Vinci-Biochem for the gift of the M65 ELISA kit, Dr. C Tripodo for the evaluation of immunohistochemistry, Dr. A Coliva for testing for LPS in SM preparations, Dr. P Casalini for confocal immunofluorescence analyses, Professor C Carlo-Stella for providing reagents, the Immunohistochemistry Facility of the Fondazione IRCCS Istituto Nazionale dei Tumori for sample preparation and staining, and Professor E Berti for providing infliximab and etanercept. Valerie Matarese provided scientific editing. This work was financially supported by grants of the CARIPLO Foundation (Project 2009-2534, IAPs as anticancer therapeutics) and the Italian Association for Cancer Research (AIRC Special Program Molecular Clinical Oncology – 5 per mille – Project 2010-9998; My First Grant no 12810 to SS).
Author Contributions
Conception and design: DL, MDC, PP, MPC, DD and SS. Development of methodology: DL, MDC, AC, EC and SS. Acquisition of data: DL, MDC, PP and SS. Analysis and interpretation of data: DL, MDC, PP, PS, MPC, DD and SS. Writing, review and/or revision of the manuscript: DL, MDC, PP, PS, HW, DD, SS and MPC. Administrative, technical or material support: CD and PS. Study supervision: PP, PS, HW, MPC, DD.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lecis, D., De Cesare, M., Perego, P. et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis 4, e920 (2013). https://doi.org/10.1038/cddis.2013.449
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.449
Keywords
This article is cited by
-
The Smac mimetic BV6 cooperates with STING to induce necroptosis in apoptosis-resistant pancreatic carcinoma cells
Cell Death & Disease (2021)
-
In vitro analysis reveals necroptotic signaling does not provoke DNA damage or HPRT mutations
Cell Death & Disease (2020)
-
Inhibitor of apoptosis proteins are potential targets for treatment of granulosa cell tumors – implications from studies in KGN
Journal of Ovarian Research (2019)
-
Antigen-specific primed cytotoxic T cells eliminate tumour cells in vivo and prevent tumour development, regardless of the presence of anti-apoptotic mutations conferring drug resistance
Cell Death & Differentiation (2018)
-
cIAP1 regulates the EGFR/Snai2 axis in triple-negative breast cancer cells
Cell Death & Differentiation (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}