
Abstract
Tumour-associated macrophages are an essential component of the tumour microenvironment and have a role in the orchestration of angiogenesis, extracellular matrix remodelling, cancer cell proliferation, metastasis and immunosuppression, as well as in resistance to chemotherapeutic agents and checkpoint blockade immunotherapy. Conversely, when appropriately activated, macrophages can mediate phagocytosis of cancer cells and cytotoxic tumour killing, and engage in effective bidirectional interactions with components of the innate and adaptive immune system. Therefore, they have emerged as therapeutic targets in cancer therapy. Macrophage-targeting strategies include inhibitors of cytokines and chemokines involved in the recruitment and polarization of tumour-promoting myeloid cells as well as activators of their antitumorigenic and immunostimulating functions. Early clinical trials suggest that targeting negative regulators (checkpoints) of myeloid cell function indeed has antitumor potential. Finally, given the continuous recruitment of myelomonocytic cells into tumour tissues, macrophages are candidates for cell therapy with the development of chimeric antigen receptor effector cells. Macrophage-centred therapeutic strategies have the potential to complement, and synergize with, currently available tools in the oncology armamentarium.
Similar content being viewed by others

Introduction
The tumour microenvironment (TME) provides an essential ecological niche for cancer initiation and progression1,2,3,4,5,6,7,8. Inflammatory cells and mediators are key universal components of the TME, and tumour-associated macrophages (TAMs) have served as a paradigm for the connection between inflammation and cancer9.
The construction and orchestration of an inflammatory TME can be driven by genetic events that cause cell transformation and progression (the so-called intrinsic pathway) and by inflammatory conditions that predispose to neoplasia (the extrinsic pathway) such as inflammatory bowel disease2. There is considerable diversity in the inflammatory components of the TME in cancers from different tissues. However, infiltration of myelomonocytic cells, specifically monocytes, macrophages and dendritic cells, represents a common denominator of cancers, irrespective of their origin and localization10.
Among myelomonocytic cells, macrophages are double-edged swords with dual potential in cancer, a reflection of their plasticity in response to environmental cues4,5,7. Macrophages have the potential to kill tumour cells, mediate antibody-dependent cellular cytotoxicity and phagocytosis, elicit vascular damage and tumour necrosis4, and activate innate or adaptive lymphoid cell-mediated mechanisms of tumour resistance. By contrast, in most established tumours, macrophages contribute to cancer progression and metastasis by various mechanisms, including promotion of cancer cell survival and proliferation, angiogenesis, and suppression of innate and adaptive immune responses4,5,7,11 (Fig. 1).

The pro-tumour functions of tumour-associated macrophages (TAMs) are diverse and act at different phases of tumour development. TAMs release nitric oxide (NO) and reactive oxygen intermediates (ROI), which cause DNA damage and genetic instability during the initiation phase. TAMs produce epidermal growth factor (EGF) and several mediators such as IL-6, hepatocyte growth factor (HGF) and GPNMB, which support cancer stem cell expansion. At later stages, TAMs contribute to metastatic spread by releasing IL-1 and transforming growth factor-β (TGFβ), which are also involved — together with several proteases — in extracellular matrix (ECM) remodelling and pathological fibrosis. TAMs are a critical source of angiogenic factors: vascular endothelial growth factor (VEGF) and pro-angiogenic chemokines. TAMs are drivers of immunosuppression in the tumour microenvironment. Secretion of IL-10, TGFβ, prostaglandins and indoleamine 2,3-dioxygenase (IDO) promote the expansion of regulatory T (Treg) cells, inappropriate skewing of dendritic cells towards an immature and tolerogenic state, and T cell metabolic starvation. Immunosuppressive TAMs are characterized by a high expression of immune-checkpoint molecules (PDL1, PDL2, B7-H4) causing T cell exhaustion. EMT, epithelial–mesenchymal transition; ILC3, type 3 innate lymphoid cell; TH17, T helper 17.
Macrophages have an important role in the antitumour activity of chemotherapy, radiotherapy and monoclonal antibodies (mAbs)4,6 by mediating tumoricidal activity and eliciting adaptive immune responses. Moreover, they are an important target of current checkpoint blockade immunotherapy by expressing inhibitory counter-receptors (such as PDL1 and PDL2), thus suppressing adaptive immune responses3,4,5,6,12,13.
Strategies specifically aimed at targeting macrophages, including mononuclear phagocytes engineered for cell therapy such as chimeric antigen receptor macrophages (CAR-M), have entered clinical evaluation14. Therapeutic approaches undergoing clinical assessment cover a broad range of strategies, ranging from targeting recruitment and differentiation to functional reprogramming by engaging activating or inhibitory (checkpoint) receptors, the latter with encouraging results15,16,17.
Here, we will review strategies to exploit macrophages as therapeutic tools and targets in cancer therapy, including the role of TAMs in conventional anticancer therapies and checkpoint blockade, strategies to reshape and activate TAMs, metabolic approaches, and macrophage cell therapies. Previous reviews on the basic immunobiology of TAMs1,2,3,4,5,6,7,9,11 will provide a framework for this essay.
TAM diversity and prognosis
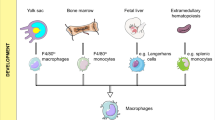
It has long been held that TAMs originate from bone marrow (BM)-derived monocytic precursors, which replenish the tumour compartment4,5 and, in a number of mouse models, the majority of TAMs indeed derive from monocytic precursors18,19 (Box 1). However, the discovery that, under homeostatic conditions, tissue-resident macrophages (TRMs) originate from embryonic precursors that seed organs in embryonic life20 has raised the issue that TRMs, in addition to BM-derived precursors, might sustain TAM levels (Fig. 1). In a mouse model of pancreatic cancer, TRMs proliferated locally and acquired a pro-fibrotic transcriptional profile, promoting a desmoplastic reaction, typical of pancreatic ductal adenocarcinoma21. In addition, in murine ovarian cancer, a population of self-renewing CD163+ Tim4+ macrophages that reside in the omentum promoted metastatic spread by generating a protecting niche for cancer stem cells22.
Brain macrophages, also known as microglia, were the first TRM population shown to derive from embryonic precursors; they expand during development and maintain themselves until adulthood. However, in inflammatory conditions associated with disruption of the blood–brain barrier, BM-derived progenitors can supplement the microglial population23 (Box 1). In murine glioma, although both brain microglia and infiltrating peripheral macrophages can promote glioma cell proliferation and migration24, TAMs mostly derive from resident microglia25. Microglia, and not BM-derived macrophages, promote murine glioblastoma via mTOR-mediated immunosuppression26. Furthermore, immunoediting induced by the immune response, and specifically by macrophage-derived IFNγ, results in stable epigenetic changes in transcriptional circuits in glioblastoma cells. These include the activation of critical myeloid-affiliated transcription factors and other immune-related pathways, which leads to increased recruitment of TAMs and the establishment of a pro-tumorigenic microenvironment27.
Using a rigorous genetic approach, Casanova-Acebes et al. shed new light on the origin and dynamics of TAMs in a mouse model of lung adenocarcinoma28. They used lineage tracing to genetically fate-map monocyte-derived macrophages and self-maintained TRMs combined with single-cell RNA sequencing (scRNA-seq) and found that tumour-infiltrating macrophage populations differ in origin and have a distinct temporal and spatial distribution in the TME. TRMs were the main source of TAMs and provided a nurturing niche in the very early phases of progression. Later, monocyte-derived macrophages were recruited at the tumour site and propelled neoplastic development28. Similar findings were reported in mouse and human glioblastoma29. In apparent contrast to these studies, in lung cancer models, both TRM and monocyte-derived TAMs contributed to generating an immunosuppressive TME in early stages by sustaining regulatory T cells30. Perhaps unexpectedly, macrophages in early-stage human lung cancer did not exert immunosuppressive functions31.
There are many substantial differences between mouse and human mononuclear phagocytes (including their proliferative capacity and their response to classic polarization signals such as those triggered by IFNγ, bacterial lipopolysaccharides and IL-4)32,33. These differences caution against mechanical extrapolations of data in mouse models to human cancers.
Based on available evidence, we propose that TRMs may contribute to the construction of a nurturing niche early in carcinogenesis and to the diversity of TAM populations in established tumours. Local proliferation of TRMs and monocyte-delivered macrophages may contribute to sustaining macrophages in specific domains in the TME, a point that needs to be addressed in human tumours using spatial transcriptomics and proteomics. In general, evidence in mouse and human tumours10,28,34 suggests that, although TRMs may contribute to the diversity of TAM populations in clinical tumours, the bulk of macrophages in established tumours derive from circulating monocytes10. This general view has implications for therapeutic approaches, particularly in adoptive cell therapy as discussed below.
Macrophages are recruited and set in tumour-promoting mode by signals produced by tumour cells, polarized type 2 adaptive immune responses, B cells, stromal fibroblasts and macrophages themselves. Molecules involved in the recruitment and education of TAMs include transforming growth factor-β (TGFβ), macrophage colony-stimulating factor 1 (CSF1), chemokines such as CCL2, cytokines such as IL-4 and IL-1, immune complexes recognized by receptors against the Fc portion of immunoglobulin G (FcγR), and complement (Fig. 1). In addition, histamine released in allergic reactions and from tumour cells activates macrophages, promoting an M2-like immunosuppressive phenotype in mice and humans, leading to the suppression of CD8+ T cell functions through the immune checkpoint V-type immunoglobulin domain-containing suppressor of T cell activation (VISTA) and conferring resistance to immunotherapy35. The function of TAMs can also be affected by the interaction with the microbiota or in response to the dysbiosis associated with cancer development36,37. Bacterial components enriched in tumour tissues can increase the recruitment of suppressive myeloid cells and their pro-tumour polarization38. Similarly, antibiotic treatment can have dual effects on tumour growth and a critical interaction of the microbiome with anticancer strategies has been documented37,39 as discussed in the next section.
Analysis of TAMs at a whole-population level has shown an M2-like phenotype in murine and human tumours4 (see Box 1 for a definition of M1 and M2 phenotypes and their limitations). However, dissection of TAMs, using conventional and scRNA-seq or mass cytometry by time-of-flight (CyTOF) approaches, has revealed the existence of diverse macrophage cell clusters with distinct transcriptomic and proteomic profiles, including macrophages with potential antitumour activity22,40,41,42,43,44,45,46,47,48,49,50,51. In the context of lung cancer, single-cell transcriptomics revealed conserved myeloid (Box 2) populations across patients as well as in mice43. In addition, analyses of scRNA-seq combined with cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), which allow transcriptomic and multiplexed single-cell surface protein dissection, showed that the predominant populations enriched in human non-small-cell lung cancer (NSCLC) were monocyte-derived macrophages, whereas alveolar and interstitial macrophages were respectively depleted and stable34. In colorectal cancer (CRC), scRNA-seq analyses highlighted the presence of conserved subsets of myeloid cells across human and mouse species, which showed distinct functional signatures (inflammatory or angiogenic), and differential sensitivity to myeloid-targeted therapeutic approaches such as treatment with the antibodies anti-CSF1R and CD40 agonists, which target macrophages and conventional dendritic cells, respectively52. Multicellular interaction networks, analysed by spatial profiling, underlie immunological and tumorigenic processes in human CRC. These include a myeloid-rich inflammatory hub at the luminal margin of primary tumours associated with tissue damage, and an immune hub within tumours deficient in mismatch repair pathway, which are enriched with activated T cells and myeloid cells expressing T cell-attracting chemokines53. ScRNA-seq analysis of human brain tumours and metastases revealed disease-specific leukocyte enrichment, with pronounced differences in the abundance of microglia, infiltrating monocyte-derived macrophages and other leukocytes between primary and metastatic tumours, indicating that tumours arising within the brain shape their TME differently compared with cancers that metastasize from extracranial sites44.
Altogether, single-cell-resolution approaches have been instrumental to recognize the heterogeneity of TAMs beyond the classic dichotomous classification as M1-like or M2-like (Box 1) and have shed light on their functional relevance and ontogeny in cancer. The distinction of myeloid clusters based on genes and markers differentially expressed by tumour-supporting TAMs or immunostimulatory myeloid effectors could be important for the development of innovative targeting strategies and predictive prognostic markers.
Consistently with a prevailing pro-tumour orientation of TAMs, high macrophage infiltration is associated with poor prognosis in most human tumours1,2,3,4,5,6,7,9,54 (Box 3). A notable exception is CRC, in which TAMs predict a better prognosis55, in particular for patients with stage III CRC that respond to 5-fluorouracil adjuvant therapy56. In liver metastasis from CRC, TAMs with different morphologies and molecular fingerprints coexist and correlate with clinicopathological variables, whereby large TAMs were associated with a poor survival rate compared with small TAMs50. In breast cancer, crown-like structures, composed of macrophages surrounding dying adipocytes in a crown-like pattern, support tumour progression and have prognostic potential57.
A better understanding of pathways responsible for recruitment and functional skewing of TAMs has provided a basis for the development of macrophage-centred therapeutic strategies. The negative prognostic significance of macrophage infiltration, as assessed by conventional immunohistology, molecular signatures or single-cell analysis, has given impetus for the clinical assessment of macrophage-targeting strategies. However, the redundancy of regulatory pathways and the diversity and plasticity of mononuclear phagocytes in situ represent stumbling blocks and may account for failures as described below.
TAMs in conventional cancer therapies
Macrophages have an important, dual role in the activity of different anticancer modalities, including chemotherapy, radiotherapy, anti-angiogenic and hormonal therapies, and immune-checkpoint blockade (ICB) immunotherapy4,6,58. Some selected chemotherapeutic agents (such as doxorubicin) induce the release of tumour antigens and adjuvant molecules, a process known as immunogenic cell death, engaging macrophages in a fruitful cancer-immunity cycle59. Other cytoreductive therapies target macrophages by depleting them. Trabectedin is a registered compound extracted from a marine organism and approved as a single agent for the treatment of soft tissue sarcomas and ovarian cancer. Trabectedin and its analogue lurbinectedin — recently approved for the treatment of NSCLC — selectively deplete monocytes and TAMs in patients and mice through the tumour necrosis factor receptor superfamily member 10 (TNFRSF10, also known as TRAIL)-dependent apoptosis pathway. TAM depletion is essential for the full antitumour activity of trabectedin60.
Selected anticancer drugs can revert TAM polarization, resulting in increased response to the treatment such as gemcitabine in pancreatic cancer61, 5-fluorouracil in CRC56 and platinum-based neoadjuvant chemotherapy in high-grade ovarian cancer62. For drugs that induce DNA damage via the generation of reactive oxygen species (ROS), such as platinum-based chemotherapies, the gut microbiome can prime intratumour mononuclear phagocytes to produce ROS, positively modulating the efficacy of these agents39,63. In the case of radiotherapy, opposing effects of commensal bacteria and fungi were reported following radiation, with depletion of fungi enhancing responsiveness to radiation in mouse models of breast cancer and melanoma64.
In this context, macrophages act by contributing to the activation of adaptive immune responses by anticancer treatments59,62, by cooperating in tumour cell killing56,61 and by being a target themselves60,65. In a seemingly opposite direction, TAMs limit the effectiveness of selected chemotherapeutic agents, driving detrimental reactive responses to tissue damage cues and rapidly reprogramming towards a pro-remodelling state4,58,66. Glioblastoma-associated microglia can activate the STAT3–MYC signalling axis in tumour cells, promoting resistance to temozolomide67. Antibodies against CSF1 and CSF1R are used to target macrophages by inhibiting their recruitment and depleting and re-educating them (see below, in the Reshaping TAMs section). Accordingly, this strategy may synergize with chemotherapy as shown in breast cancer preclinical models68,69,70,71 and is being evaluated in clinical trials5,72.
Tumour-infiltrating leukocytes are key players in the antitumour activity of selected mAbs that act by triggering immune cells — TAMs in particular — that express FcγR to perform tumour cell killing and phagocytosis73,74. Such therapeutic antibodies are currently in use in the clinical setting and include rituximab (targeting CD20 (ref.75)), trastuzumab (an antibody against HER2), cetuximab (which targets epithelial growth factor receptor (EGFR))74 and daratumumab (which targets CD38 on myeloma cells). Functional polymorphisms in human FcγRIIIA that affect the killing capability of macrophages correlate with rates of response to mAbs in patients with lymphoma treated with rituximab76, patients with breast cancer treated with trastuzumab77 and patients with metastatic CRC treated with cetuximab78.
The density of TAMs frequently correlates with the density of vessels in tumour tissues79 as TAMs both secrete and actively respond to angiogenic growth factors, primarily vascular endothelial growth factor (VEGF)4,6,79. Accordingly, the activity of anti-angiogenic therapy is modulated by TAMs. VEGF antagonists induce vascular normalization80 and concomitantly remodel the TAM phenotype81,82, whereas myeloid cells mediate resistance to anti-angiogenic therapies via compensatory pathways such as cathepsin B and angiopoietin 2 (ANG2). This evidence has provided a basis to test bispecific ANG2–VEGF antibodies, which have shown promising results in preclinical models of glioblastoma81,82.
Similarly, hormonal therapy is influenced by inflammatory pathways orchestrated by myeloid cells. Inflammatory cytokines, such as IL-1 and IL-6, can activate oestrogen receptor signalling on tumour cells83,84, linking inflammation to tumour growth and endocrine resistance. In prostate cancer, TAMs express the androgen receptor, which drives their pro-tumour activation85. Anti-androgen treatment reduced the number of TAMs in human prostate cancer85 whereas, in a preclinical model, blockade of myeloid cell-derived IL-23, another key inflammatory cytokine released by myeloid-derived suppressor cells (MDSCs), improved response to androgen-deprivation therapy86.
The results presented above suggest that myelomonocytic cells have an important influence on the activity of cancer chemotherapy, radiotherapy87, anti-angiogenic agents and hormonal therapy. Their role is complex and dual, serving as amplifiers or inhibitors of antineoplastic activity. Although progress has been made in dissecting the yin yang of macrophages in conventional antitumour treatment modalities, the actual translation of deeper knowledge into more effective treatments remains a challenge.
TAMs and ICB immunotherapy
Unleashing T cell-mediated type 1 immune responses represents the cornerstone of ICB immunotherapy. Myelomonocytic cells, major orchestrators of immunosuppressive circuits, are an important determinant of response to ICB and are critically involved in resistance and eligibility for this treatment4,6.
Myelomonocytic cells are part of tumour-extrinsic pathways of primary and adaptive resistance to ICB88 by expressing several immunosuppressive molecules, including checkpoint ligands, such as PDL1, PDL2, CD80 (also known as B7-1) and CD86 (also known as B7-2), and poliovirus receptor (PVR; also known as CD155 and one of the TIGIT ligands)12,51,89,90 (Fig. 1). PDL1 assessment by immunohistochemistry is approved as a companion diagnostic for anti-PD1 therapy in NSCLC and other tumours91,92, but its predictive capability may vary according to the cell type considered, such as tumour or immune cells93. In preclinical models, PDL1 expression on tumour-infiltrating immune cells but not on tumour cells was associated with response to anti-PD1 or anti-PDL192 antibodies. Unexpectedly, macrophages were reported to express PD1, which negatively correlates with their phagocytic activity against tumour cells94,95,96. Interestingly, myelomonocytic cells express other counter-receptors that interact with negative regulators expressed by T cells and natural killer (NK) cells. VISTA is expressed by macrophages97 and interacts with P-selectin glycoprotein ligand 1 (PSGL1)98, functioning as a T cell checkpoint inhibitory ligand. In human monocytes, mAbs against VISTA elicited transcriptional and functional changes, including increased antigen presentation, activation and migration99.
Tissue contexture is important in dictating the role of macrophages in ICB. For instance, in triple-negative breast cancer, distinct myeloid cell profiles, including neutrophils and macrophages, mediated resistance to ICB13 and hepatic macrophages in liver metastases negatively regulated responsiveness to systemic immunotherapy by eliminating T cells100. The pleural and peritoneal cavity represent sites of tumour progression in an immunosuppressive microenvironment. In mouse models and in patients, TIM4+ macrophages in coelomic cavities suppress CD8+ T cell responses and inhibit ICB101, and blocking TIM4 with antibodies enhanced the efficacy of ICB at these sites. In human renal cell carcinoma (RCC), a predominance of M2-like macrophages was associated with resistance to ICB102,103. Response to immunotherapy can be modulated by the composition of the microbiome as shown for anti-CTLA4104 and anti-PD1105, in which abundance and diversity of the gut bacteria shape tumour myeloid infiltration105.
Depletion of macrophages can potentiate various immunotherapeutic strategies, including vaccination106 and checkpoint inhibitors107,108,109. Several clinical trials combining checkpoint inhibitors and anti-CSF1R antibodies or other TAM-centred therapeutic strategies are ongoing4,5,6 (Table 1).
Reshaping TAMs
Functional reprogramming of TAMs can be achieved with different strategies (Fig. 2).

Cytokines (such as interferons), Toll-like receptors, stimulator of interferon genes (STING) agonists, and monoclonal antibodies activate macrophage-mediated tumour cell killing (red shading). A number of signals, including complement, inflammasome activators, ligands for scavenger receptors MARCO and CD206, and myeloid checkpoints (Fig. 3) can set macrophages in a pro-tumour mode (brown shading). MicroRNAs (miRNAs) and mRNA represent strategies to reprogramme macrophage functions. Macrophage reprogramming induces macrophage-mediated killing of cancer cells, recruitment and activation of innate and adaptive lymphoid cells, and reshaping of the tumour microenvironment. IFNR, interferon receptor; NK, natural killer; TAM, tumour-associated macrophages.
Targeting TAM recruitment and accumulation
It has long been known that chemokines and CSF1 have a major role in the recruitment of monocytes in tumours and in shaping their function in the TME1,2,3,4,5,6,7,9,18,110. Monocyte attractants, such as CCL2 and its cognate receptor CCR2, have been targeted using mAb and receptor antagonists in solid tumours and haematological malignancies4,5,111. However, despite a wealth of data in preclinical models, chemokine targeting strategies have not yielded positive clinical results alone or in combination and have been discontinued (Table 1). Therapeutic strategies targeting CSF1 and CSF1R have also used mAbs or receptor antagonists. Several studies in preclinical models proved that CSF1R inhibition reduced the density of TAMs and tumour growth, and increased the sensitivity to chemotherapy5,110,112,113. Although the small-molecule inhibitor BLZ945 did not deplete TAMs, these underwent a reprogramming towards an antitumour phenotype112,114; upon CSF1R blockade, macrophages showed lower expression of pro-tumorigenic genes and upregulation of genes associated with antigen presentation and lymphocyte activation. BLZ945 reshaped the immune microenvironment inducing positive crosstalk among TAMs, IFNγ-producing NK and T cells, and IL-12-producing dendritic cells, thus sustaining a virtuous network of antitumour responses114. Inhibition of CSF1R in patients with a rare tumour (diffuse-type tenosynovial giant cells) overexpressing CSF1 led to significant clinical benefit in 71% of patients in the study115. In other solid tumours, CSF1R inhibitors as single agent demonstrated very modest or no activity. Several clinical trials have assessed combinations of CSF1R inhibition with chemotherapy, radiotherapy or ICB (Table 1). In those phase II trials for which results are available, no durable clinical responses have been reported, but some patients experienced disease stabilization or even a partial response. Most studies targeting CSF1R are still ongoing; therefore, the jury is still out on the clinical benefit of this approach. In terms of adverse effects, CSF1R blockade by both kinase inhibitors and mAbs caused periorbital oedema in a considerable proportion of treated patients owing to loss of dermal macrophages. which leads to extracellular matrix remodelling, activation of matrix metalloproteinases and hyaluronan deposition, causing increased fluid retention116.
Targeting TAM recruitment strategies also faces major stumbling blocks such as compensatory accumulation of neutrophils, which may play an immunosuppressive part, and the apparent redundancy of the chemokine system with multiple ligands and receptors acting on monocytes.
Rodriguez-Garcia et al. have proposed an original approach to reduce the number of TAMs in tumours by exploiting the cytotoxic activity of CAR T cells against macrophages117. The authors identified a subset of TAMs with immunosuppressive activity that expresses the folate receptor-β (FRβ) in addition to other M2 markers (CD204, CD206 and CD163). Engineered T cells with a CAR directed to FRβ specifically targeted immunosuppressive TAMs; in murine tumour models, these CAR T cells mediated selective elimination of FRβ+ TAMs, which resulted in the recruitment of pro-inflammatory monocytes and tumour-specific CD8+ T cells and restrained tumour growth117.
Another limitation of strategies targeting macrophage recruitment and survival rests in the diversity of the mononuclear phagocyte populations in tumours. In a mouse melanoma model, global targeting of the total TAM population was not as effective as depletion of the CD163+ cells endowed with immunosuppression activity118. Hence, strategies for specific TAM subsets are warranted.
Complement
Complement has emerged as an important component of tumour-promoting inflammation in murine and human cancers119,120. The complement system consists of a cascade of sequential proteolytic reactions activated by three different pathways: the classical pathway, which is initiated by binding of C1q to immune complexes; the lectin pathway, which is initiated by the interaction of mannose-binding lectin or ficolins with pathogen-associated molecular patterns or aberrant carbohydrate structures on damaged cells; and the alternative pathway, which is directly activated by damaged cells or microbes. The activation of the central components C3 and C5 results in the release of their bioactive fragments (anaphylatoxins) and in the assembly of the cytolytic membrane attack complex119,120. Unconventional complement activation may also occur owing to the proteolytic activity of coagulation and fibrinolytic enzymes.
In transplanted tumour models, the release of these anaphylatoxins, C5a in particular, was associated with the recruitment of MDSCs into the tumour, thus promoting immunosuppression121. In mouse tumour models of sarcomagenesis induced by 3-methylcholanthrene and in two transplanted sarcoma models, the lectin pathway-dependent C3a–C3aR axis played a dominant role in TAM recruitment and functional skewing, driving immunosuppression and tumour promotion122. In squamous carcinogenesis, urokinase (uPA)+ macrophages regulated C3-independent release of C5a during premalignant progression, which in turn promoted the pro-tumorigenic properties of C5aR1+ mast cells and macrophages, including suppression of T cell cytotoxicity119,123. Both in mice and humans, factor H deficiency has been recently associated with increased susceptibility to hepatocellular carcinoma; the deficiency of this negative regulator of the alternative pathway was associated with spontaneous deposition of complement activation fragments throughout the sinusoids, chronic activation of inflammatory signalling pathways, and increased hepatic infiltration of macrophages and T lymphocytes124.
In patients, the prognostic potential of complement molecules, in particular C4d, C5a and C5aR1, has been demonstrated in different types of cancer120. For instance, the complement activation fragment C4d served as a diagnostic and prognostic marker in lung cancer and C5–C5aR1 in breast cancer120, whereas C1s and C4d act as biomarkers of poor prognosis in patients with clear cell RCC125. In addition, transcriptomic data available in The Cancer Genome Atlas (TCGA) database showed that high expression of classical and alternative pathway genes correlates with poor prognosis in selected tumours, including uveal melanoma, glioma, lung squamous carcinoma and clear cell RCC126. In clear cell RCC, infiltration by C1q-producing TAMs was associated with an immunosuppressed TME, characterized by high expression of immune-checkpoint molecules (PD1, LAG3, PDL1 and PDL2), suggesting a correlation between complement activation and T cell exhaustion127. In agreement with these data, the soluble pattern recognition molecule pentraxin 3 (PTX3) can act as an extrinsic tumour suppressor gene through the regulation of complement-dependent and macrophage-sustained tumour-promoting inflammation in sarcomas128,129.
Preclinical studies provide evidence for the therapeutic potential of C3a and C5a inhibition, for instance, by targeting immunosuppressive MDSCs in cervical cancer121 and lung metastasis from breast cancer130, or macrophages in sarcomas128. Of particular interest are studies on the combined blockade of complement and the PDL1–PD1 immune-checkpoint axis. In particular, in lung cancer models, the combination of C5a and PD1 blockade markedly reduced tumour growth and metastasis and led to prolonged survival131. This effect was associated with increased frequency of CD8+ T cells and reduced frequency of MDSCs within tumours, suggesting the restoration of antitumour immune responses131 (Fig. 2). Along the same line, the combination of a C3aR antagonist (SB 290157 trifluoroacetate salt) with anti-PD1 treatment resulted in a significant reduction of primary tumour growth and incidence, compared with anti-PD1 or C3aR treatment alone122, indicating that complement inhibition increases the clinical benefit of ICB.
Based on the above preclinical and clinical evidence, complement targeting strategies deserve to undergo (or are undergoing) clinical assessment (Table 1). We surmise that assessment of the actual involvement of complement pathways in different tumours or tumour subsets should guide clinical assessment of anti-complement strategies.
Inflammasome and IL-1
Although IL-1 can trigger protective immune responses, the predominant function of this pathway is to promote carcinogenesis and metastasis132,133,134. IL-1 promotes cancer-associated immunosuppression through complex mechanisms, including driving emergency haematopoiesis and the generation and recruitment of MDSCs as well as promoting TAM immunosuppressive and tumour-promoting functions135. Accordingly, in a model of RCC, the combination treatment with anti-IL-1β and anti-PD1 or the multi-targeted tyrosine kinase inhibitor cabozantinib led to a synergistic antitumour effect, a pronounced decrease of MDSCs and skewing of TAMs136. Incidental findings from CANTOS — a large, phase III randomized, double-blind, placebo-controlled trial of canakinumab, an IL-1β blocking antibody, involving 10,061 patients with a history of myocardial infarction with atherosclerosis — demonstrated that canakinumab was associated with a dramatic (>60%) reduction of incidence and mortality from lung cancer133. Based on preclinical studies, it is conceivable that inhibition of MDSCs and unleashing of innate and adaptive effective antitumour responses contribute to cancer prevention by canakinumab as observed in the CANTOS trial132,133,137.
IL-1 can be targeted directly, for instance, through canakinumab, or upstream through inflammasome inhibition. For instance, in a mouse melanoma model, pharmacological inhibition of tumour-derived NLRP3, a sensor component of the inflammasome, limited MDSC-mediated T cell suppression and tumour progression. The antitumour potential of NLRP3 inhibition was further amplified by the combination with anti-PD1 treatment138. These results call for prevention trials with anti-IL-1β and/or inflammasome inhibitors. A pharmacoprevention trial (Canal, 2020-002773-10), using canakinumab (anti-IL-1β) in patients at high risk of developing lung cancer (such as heavy smokers) has been planned but not activated to date owing to the SARS-CoV-2 pandemic. We feel that bold pharmacoprevention approaches targeting tumour-promoting inflammation pathways should be considered in selected patient populations.
mRNA and microRNA
In vitro-transcribed mRNA has recently come into focus as a potential new drug class to induce the production of effector molecules or reprogramme cells. mRNA formulated into an injectable nanocarrier has been used to genetically reprogramme TAMs into antitumour effectors. In different mouse cancer models, infusion of nanoparticles formulated with mRNAs encoding the transcription factor interferon regulatory factor 5 (IRF5) in combination with its activating kinase, inhibitor of NF-κB kinase subunit-β (IKKβ), reversed the immunosuppressive TME and reprogrammed TAMs, achieving tumour regression139.
MicroRNA (miRNA) are small non-coding RNA molecules with an important role in regulating post-transcriptional gene expression. miRNA have emerged as key modulators of macrophage differentiation and polarization, including TAMs7,140. miRNA-based therapies in the context of cancer have been tested in experimental tumour models with multiple delivery strategies. As an example, miRNA-155 enveloped in lipid-coated phosphonate nanoparticles decorated with mannose can specifically deliver miRNA-155 to TAMs and successfully reprogrammed them towards antitumour effectors (Fig. 2); treated mice showed a significantly reduced tumour burden141. Of interest, conditional deletion in macrophages of Dicer1, encoding miRNA-processing enzyme endoribonuclease DICER, resulted in marked activation of IFNγ–STAT1 signalling that rewired TAM immunosuppression142. So far, no clinical trials have been initiated using miRNA delivery to reprogramme tumour macrophages.
Macrophage activation
The tumoricidal activity of M1-like macrophages (Box 1) is triggered by receptors sensing microbial molecules and cytokines such as IFNγ. In this section, we will focus on three classes of macrophage activators4,143,144 (Fig. 2).
CD40
The CD40 receptor is a member of the TNF receptor family expressed on antigen-presenting cells, including macrophages. When engaged by its specific ligand CD40L, expressed on the surface of activated T helper lymphocytes, it triggers the production of TNF, ROS and reactive nitrogen species. These factors mediate the bactericidal and tumoricidal activity of macrophages. The CD40–CD40L axis is a feedforward loop that activates antigen-presenting cells, which further stimulate T cell-mediated immunity (Fig. 2). Several agonistic CD40 mAbs have been generated that mimic the effect of CD40. In preclinical studies, these CD40 agonistic mAbs successfully re-educated immunosuppressive TAMs by switching them into cytotoxic effectors, eventually resulting in immune surveillance and reduced tumour growth145. Upon administration of CD40 agonists, there was an increased influx of CD4+ T cells into the TME in response to the chemokine CCL5 produced by CD40-stimulated TAMs146. In addition, the treatment caused a transitory modulation of the macrophage phenotype with increased expression of CD86 and MHC class II, promoting antigen-presenting potential. Agonistic CD40 mAbs are currently under clinical evaluation and may be used in combination with other treatments. Transient inhibition of CSF1R combined with CD40 agonists resulted in rapid reprogramming of TAMs before their depletion147. The safety of the CD40 agonistic antibody APX005M was assessed in a phase I study in patients with metastatic pancreatic cancer in combination with chemotherapy, with or without immunotherapy; treatment-related toxicity was clinically manageable and 14 of the 24 evaluated patients had measurable clinical responses148. These encouraging results warrant further investigation in phase II/III clinical trials (Table 1).
Toll-like receptors
Engagement of toll-like receptors (TLRs) activates an immunostimulatory response and this approach has been used to bypass the immunosuppressive activity of macrophages in tumours (Fig. 2). Several agonists of TLRs with immunostimulatory activity have been investigated for the reprogramming of TAMs in different tumour models, showing increased cytotoxic activity against tumour cells and production of immunostimulating cytokines149,150,151. The first TLR-stimulating agent approved by the FDA was BCG (Bacillus Calmette-Guérin), a live attenuated strain of Mycobacterium bovis that stimulates TLR2 and TLR4. BCG is still in use to treat patients with bladder cancer149,152. In mice, monophosphoryl lipid A (MPLA), a TLR4 ligand, injected with IFNγ, reprogrammed TAMs to become tumoricidal153.
TLR3, TLR7, TLR8 and TLR9 are localized in endosomal compartments and serve as nucleic acid sensors. Engagement of these receptors activates NF-kB, the master transcription factor of inflammation, and triggers the secretion of immunostimulatory cytokines, including the antitumour cytokine type I interferon. The TLR7 agonist imiquimod is the only one that has been approved by the FDA for the topical treatment of squamous and basal cell carcinoma. Two derivatives of imiquimod, resiquimod or R848 (TLR7 and TLR8 agonist) and motolimod (TLR8 agonist), showed promising immunostimulatory activity in a variety of preclinical models154. Although other cells of the innate immunity can be activated by TLR ligands, in these models, priming of the antitumour immune response was mediated by TAMs154,155. Poly(I:C), a TLR3 agonist, demonstrated significant antitumour responses in experimental settings and its nanocomplex formulation with polyethyleneimine (BO-112) is now being evaluated in clinical trials156. Another analogue, poly-ICLC, complexed with carboxymethylcellulose (Hiltonol), is under investigation in combination with anti-PD1 therapy157. Most frequently, TLR agonists have been formulated with nanoparticles and administered intratumourally in accessible lesions as their systemic administration may cause inflammatory toxicity156,158,159,160. The TLR9 ligand CpG formulated in a virus-like particle was administered intratumourally together with systemic anti-PD1 mAb pembrolizumab to patients with melanoma who were refractory to PD1 blockade. Promising clinical activity was observed in 25% of patients161.
With the idea to combine the precision of a therapeutic antibody with the stimulation of innate immunity cells into a single agent, Ackerman et al. recently reported the design of BDC-1001, an immune-stimulating antibody conjugate, engaging TLR7 and TLR8 and conjugated to a tumour-targeting antibody against HER2. In mice, systemic administration of this immune-stimulating antibody conjugate successfully reduced tumour growth and stimulated an antitumour immune response mediated by macrophages and other antigen-presenting cells, without eliciting inflammation162. SBT6050 is a TLR8 agonist conjugated to a HER2-directed antibody. Both BDC-1001 and SBT6050 are under evaluation in a first-in-human trial in patients with HER2-positive tumours (Table 1).
Several clinical phase I–III trials involving the administration of TLR agonists to patients with cancer are ongoing, either as monotherapy or, most frequently, in combination with targeted therapy or immunotherapy161,163,164,165,166. TLR agonists have also been used as adjuvants for anticancer vaccines and in combination with checkpoint inhibitors or adoptive T cell therapy to ‘warm’ immunologically cold tumours167,168,169. Overall, these treatments seem well tolerated, and evidence of clinical activity, especially disease stabilization, has been observed also in heavily pretreated patients. Still, none of these novel agents has so far been approved by regulatory agencies for use in patients with cancer170 (Table 1).
STING
Stimulator of interferon genes (STING) protein receives input from different cytoplasmic receptors that sense ectopic nucleotides from pathogens as well as from endogenous damaged DNA, leading to the production of type I interferon. Therefore, STING also has a key role in antitumour immune responses171,172. This property of STING generated attention and prompted the development of synthetic STING agonists173 (Fig. 2). In early times before the identification of STING, DMXAA (5,6-dimethylxanthenone-4-acetic acid), a vascular disrupting agent based on flavone acetic acid, was known to have anticancer activity mediated by interferon and TNF in experimental tumour models174. DMXAA is the best-known STING activator, and stimulation of STING in innate immunity cells (but also in cancer cells) successfully elicited antitumour immunity in several preclinical tumour models and synergized with radio-chemotherapy and immunotherapy172,173. Although compounds targeting the STING pathway are not macrophage specific, preclinical studies have demonstrated that activation of antitumour immunity engages infiltrating macrophages and dendritic cells. Because early pharmacokinetic studies showed that most STING agonists have a short half-life, novel compounds with improved half-life have been developed and are moving ahead for evaluation in patients with cancer173,175,176. Most frequently, STING agonists have been administered intratumourally; however, an available formulation for oral administration has been tested in mice and resulted in significant tumour regression and synergism with checkpoint blockade immunotherapy177. STING is also activated by microbiota-derived agonists, such as c-di-AMP, which regulate interferon production and macrophage polarization178. Interestingly, modulation of the microbiota with a high-fibre diet improved the efficacy of ICB178.
Myeloid checkpoints and regulators
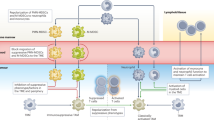
The function of myelomonocytic cells is tightly controlled by a number of negative regulators that directly inhibit or divert their effector functions15,118,179. Some of these molecules (such as members of the signal regulatory protein-α (SIRPα), sialic acid-binding immunoglobulin-like lectin (SIGLEC) and leukocyte immunoglobulin-like receptor B (LILRB) families) have been referred to as myeloid checkpoints, similar to regulatory molecules of T cells15,180 (Fig. 3a). Other relevant molecules are scavenger receptors, which mediate the clearance of debris and dead cells by macrophages, a function that is pivotal to mitigating inflammation (during the resolution phase) and preventing tissue damage. These pathways, physiologically used to protect from unwanted attack of host tissues, result in a state of immunosuppression and activation of the TAM tissue-trophic functions, which eventually can be hijacked by tumour cells to evade recognition from the immune system and proliferate.

a | Overview of myeloid checkpoints and inhibitory receptors expressed by tumour-associated macrophages (TAMs) and their ligands expressed on tumour cells or cell debris. These include the receptor/ligand pairs signal regulatory protein-α (SIRPα)–CD47, LILRB1–HLA1, sialic acid-binding immunoglobulin-like lectin 10 (SIGLEC10)–CD24, and PD1–PDL1, which inhibit phagocytosis, and macrophage receptor with collagenous structure (MARCO), CD169 and mannose receptor scavenger receptors. Clever 1, triggering receptor expressed on myeloid cells 2 (TREM2) and P-selectin glycoprotein ligand 1 (PSGL1) are also depicted. Targeting of Clever 1 and TREM2 does not specifically interfere with phagocytosis but with immunosuppressive activation. b | In strategies featuring the use of therapeutic antibodies, such as anti-CD20 or anti-epithelial growth factor receptor (EGFR), combinatorial use of anti-CD47 enhances antibody-dependent cellular phagocytosis (ADCP) and increases antigen presentation to T cells. c | Bispecific CD47 antibodies are designed to recognize CD47 and tumour-associated antigens (such as CD20 or PDL1), which enhances selective blocking of CD47 on tumour cells, avoiding on-target toxicity due to recognition of CD47 on red blood cells and platelets. ADCC, antibody-dependent cellular cytotoxicity; MHC II, MHC class II.
SIRPα and CD47
CD47 is expressed on normal cells and serves as a ‘don’t eat me’ signal, instructing mononuclear phagocytes and neutrophils expressing SIRPα to spare host cells from removal181,182,183,184. Loss of CD47 is associated with ageing of red blood cells and allows their disposal, an illustration of the importance of this pathway. Tumour cells overexpress CD47 in many cancer types, disguising as healthy cells and avoiding phagocytosis. CD47-targeting approaches include both, antibodies anti-CD47 (Hu5F9-G4 (NCT02216409), SRF231 (NCT035123), IBI188 (NCT03763149)) and anti-SIRPα (BI-765063 (NCT03990233)) (Fig. 3b). Emerging evidence points to combination as the key to success of immune-based strategies. The strength of therapeutics disrupting the CD47–SIRPα axis resides in the possibility to concomitantly render macrophages more phagocytic and increase their antigen load, thus enhancing antigen presentation to T cells185 (Fig. 3b). Therefore, in a stepwise manner, CD47 approaches may be synergistically effective in combination with T cell checkpoint inhibitors, first improving phagocytosis and antigen presentation and second unleashing a response of activated T cells.
In preclinical models, therapeutic combinations with anti-CD47 were particularly effective in those tumours for which targeted therapies against tumour antigens are already available such as CD20 for lymphoma, HER2 for breast cancer or EGFR for CRC (Fig. 3b). The mechanism of action of tumour-opsonizing mAbs, such as rituximab (anti-CD20), trastuzumab (anti-HER2) or cetuximab (anti-EGFR), can be greatly enhanced with a concomitant signal that dampens CD47 and boosts Fc-mediated functions186. The combination of cetuximab with an anti-CD47 antibody markedly increased macrophage antibody-dependent phagocytosis in preclinical models187 and is being tested in clinical trials for CRC (NCT02953782). In a phase Ib study, CD47 blockade combined with rituximab showed activity in patients with lymphoma15,180. So far, promising results have been observed for haematological malignancies15,180, but studies on solid tumours are under way and results are awaited. Preclinical results in cell lines of lung188, breast189 and other types of cancer show that macrophage-mediated phagocytosis can critically contribute to the therapeutic function of the antibody and trigger adaptive immune responses.
The blockade of CD47, a ubiquitously expressed molecule on normal cells, raised concern about potential on-target toxicity, in particular anaemia and thrombocytopenia, owing to the expression of CD47 on platelets and red blood cells. A possible strategy to overcome this problem is blocking SIRPα186 or using anti-CD47 antibodies with weak killing properties, such as antibodies with an IgG4 tail, which can reduce NK cell engagement; for example, TTI-621 (a SIRPα decoy receptor fused to an active IgG1 Fc; NCT03530683) or TTI-622 (identical to TTI-621 but with an IgG4 Fc that has a weaker killing function; NCT02890368). ALX148 (NCT04675333) combines a high-affinity CD47 binding domain with an inactive Fc domain. Off-tumour effects can also be reduced by increasing specificity for the tumour target such as with bispecific agents that recognize CD47 and a tumour-associated antigen (Fig. 3c). Such molecules, so far developed for dual recognition of CD47 and CD20 (RTX-CD47190), PDL1191, EGFR192 and CD19 (TG-1801, NCT03804996), restrain the phagocytic action of the unleashed macrophage to the tumour cells that express the antigen, sparing the host cells that do not express the tumour antigen, such as platelets and red blood cells (Fig. 3c), showing limited toxicity. More recently, delivery of the anti-CD47 antibody with an oncolytic virus has been tested in preclinical models as a strategy to improve drug availability to the tumour site, reducing its toxicity193.
The SIGLEC family
SIGLEC molecules are membrane proteins that bind sialic acid and engage in cell–cell interactions. These proteins contain tyrosine-based inhibitory receptor motifs (ITIMs) in their cytoplasmic tail, which are typically components of those immune receptors that inhibit and suppress activation signals, thus regulating the functions of several immune cells.
SIGLEC1 (also known as sialoadhesin or CD169) is expressed by a fraction of macrophages and is upregulated in human cancer, with its expression correlating with worse prognosis10,143. Depletion of CD169+ TAMs was effective in reducing tumour burden and metastasis in mouse models of breast cancer194. SIGLEC7 and SIGLEC9 were targeted in a humanized murine model with a significant reduction in tumour burden195.
The sialoglycoprotein signal transducer CD24 has been evaluated as an anti-phagocytic molecule expressed by multiple cancer cells and holds promise as an additional checkpoint with therapeutic potential179. Binding of CD24 to SIGLEC10, which is overexpressed by TAMs, inhibited phagocytosis of tumour cells and interference of this axis with a mAb against SIGLEC10 rescued the macrophage capability to limit tumour growth in preclinical models of ovarian cancer179.
The LILRB family
Downregulation of MHC class I molecules is probably one of the best-known mechanisms of evasion used by cancer cells to circumvent recognition by T cells196. However, tumour cells can exploit MHC class I as a mechanism of evasion from phagocytosis by interacting with LILRB family members. LILRB1 is an MHC-binding protein widely expressed on immune cells and enriched on TAMs197; it contains an ITIM motif and transduces an inhibitory signal. LILRB expression was associated with the inhibition of phagocytosis of cancer cells. In fact, its role as a myeloid checkpoint was discovered by analysing cancer cell lines resistant to the anti-CD47 antibody197. The expression of MHC class I by tumour cells correlated with the degree of their resistance to anti-CD47, and phagocytosis induced by the anti-CD47 antibody was restored by LILRB1-blocking antibody.
Macrophages also express LILRB2, whose blocking enhanced their pro-inflammatory activation and phagocytic activity198. Despite the similarity between the two members of the LILRB family, whether the checkpoint activity of LILRB2 is accounted for blocking phagocytosis or for a general modulation of macrophage activation remains to be elucidated. In terms of the development of new macrophage-based therapeutics, efforts are ongoing to identify binding partners to LILRB molecules, which are expected to provide control over the capability of the tumour to evade phagocytosis. MK-4830, a human mAb directed against LILRB2, was tested in advanced-stage solid tumours in a phase I dose-escalating study (NCT03564691) as monotherapy or in combination with pembrolizumab. Initial results showed durable responses, supporting further development16. Immune correlates of response included expression of pro-inflammatory cytokines, such as GM–CSF and TNF, and an enhanced cytotoxic T lymphocyte-mediated antitumour immune response.
LILRB4 was found on a variety of intratumour immune cells in murine tumour models and human cancers, most prominently on TAMs, where it strongly suppressed tumour immunity199. Its blockade reshaped tumour-infiltrating T cells and modulated phenotypes of TAMs towards a less suppressive phenotype199.
Scavenger receptors
Several types of scavenging receptor are abundantly expressed in TAMs, and their targeting is emerging as an option to potentiate a pro-inflammatory switch (Fig. 2).
Clinical evidence points to a significant association between macrophages expressing the scavenging receptor CD163 and tumour progression in several cancers, including pancreatic cancer61 and melanoma200. CD163 enables macrophages to remove damaged erythrocytes by binding to haptoglobin–haemoglobin complexes formed upon intravascular haemolysis201 and its expression is traditionally associated with M2-like macrophages. Notwithstanding, the exact mechanisms of its tumour-promoting functions are unclear. Depletion of CD163+ macrophages, through a genetic and a nanoparticle-based approach resulted in tumour regression in a mouse model of melanoma resistant to anti-PD1 therapy118. This strategy induced a complete re-education of the TME, featuring infiltration of cytotoxic T cells and inflammatory monocytes, ultimately restoring response to anti-PD1 treatment.
Macrophage mannose receptor 1 (MRC1, also known as CD206) is a macrophage scavenger receptor that binds several endogenous ligands in addition to pathogen moieties such as tumour mucins202. Engagement of the mannose receptor (MR) on macrophages, either by mucins or agonist anti-MR mAbs, induced an immunosuppressive phenotype with increased IL-10 production203. This immunoregulatory function of the MRC1 was confirmed in models of intestinal inflammatory conditions, in which MRC1-deficient mice had more-severe colitis204,205. Jaynes et al. identified the RP-182 peptide, which binds to the MRC1 and induces a change in conformation. When administered to tumour-bearing mice, the synthetic peptide RP-182 partially depleted CD206+ macrophages and reprogrammed the remaining TAMs into antitumour M1-like effectors with increased inflammatory cytokine production and the ability to phagocytose cancer cells. In murine cancer models, RP-182 suppressed tumour growth, extended survival and synergized with combined immunotherapy206.
The macrophage receptor with collagenous structure (MARCO) is highly expressed on TAMs. When antibodies blocking this receptor were tested in preclinical tumour models, they did not reduce the number of TAMs but they induced an antitumour immune response through reprogramming of TAMs into pro-inflammatory effectors207. In NSCLC, macrophages polarized to express MARCO showed an immunosuppressive phenotype. Silencing of cancer cell-derived IL-37, an anti-inflammatory cytokine of the IL-1 family, or blocking of its receptor208, which signals through IL-1R8, rescued the MARCO-associated immunosuppressive phenotype. IL-1R8 also negatively regulates IL-18 signalling and acts as a new NK cell checkpoint restraining NK cell antitumour and anti-viral potential209. Activation of NK cell killing through the TRAIL pathway has been recently identified as one of the mechanisms of action of anti-MARCO mAbs. Indeed, treatment with anti-MARCO mAbs in a mouse melanoma model did not engage CD8+ T cells and was mediated only by NK cells210. In glioblastoma, MARCOhigh TAMs significantly accelerated tumour engraftment and growth in vivo211.
The receptor Clever 1, also known as Stabilin 1, was originally described as an adhesion and scavenger receptor expressed by a variety of cells, including circulating monocytes, lymphatic and sinusoid endothelial cells, and immunosuppressive M2-like macrophages212. In homeostatic conditions, Clever 1 binds several ligands, primarily lipoproteins and carbohydrates, mediating endocytosis of scavenged material and its delivery to the endosomal compartment, ultimately resulting in suppression of macrophages and impaired activation of T helper 1 lymphocytes. Antibody blockade of Clever 1 in preclinical studies caused a phenotypic switch in TAMs from immunosuppressive to pro-inflammatory, activation of T cell responses and delayed tumour growth212,213. These results provided a roadmap for a phase I trial to test the safety and preliminary efficacy of FP-1305, a humanized anti-Clever 1 antibody administered to heavily pretreated patients with metastatic solid tumours, with encouraging results17 (Table 1). To avoid potential side effects due to the expression of Clever 1 on lymphatic vessels, the antibody has been optimized to escape Fcγ-mediated cytotoxicity and complement-mediated functions. Notwithstanding, blocking of Clever 1 on the lymphatic endothelium may have an impact on tumour metastases. Mechanistically, inhibition of Clever 1 resulted in a pro-inflammatory switch of CD14+ blood monocytes, enhanced the capability of macrophages to cross-present scavenged antigens, and activated peripheral T cells17.
PD1
PD1 expression by TAMs inhibits phagocytosis and tumour immunity94, twisting the traditional view of the PD1–PDL1 axis as a specific T cell checkpoint. PDL1 expression on cancer cells may thus concomitantly enable evasion from T cell cytotoxicity and macrophage-mediated phagocytosis, suggesting that blockade of this axis might unleash antitumour immunity by both adaptive and innate mechanisms. The mechanism of phagocytosis inhibition triggered by the engagement of PD1 on macrophages has not yet been elucidated nor have the signals inducing PD1 upregulation. SIRPα, LILRB1 and PD1 all contain an ITIM domain, which could be instrumental for the downstream signals inhibiting phagocytosis, but a formal demonstration has not been provided. On this basis, studies aimed at monitoring response in patients with cancer undergoing checkpoint inhibitor treatment should consider the myeloid compartment as a potential target and as a predictive biomarker.
TREM2
Triggering receptor expressed on myeloid cells 2 (TREM2) is expressed by macrophages of several tissues and is upregulated on TAMs in human and mouse tumours214,215, in which targeting of TREM2+ macrophages restricted tumour growth and sensitized the response to anti-PD1 therapy216. TREM2 scavenges large molecules such as phospholipids, lipoproteins and apoptotic cells217. PY314, a humanized mAb targeting TREM2+ macrophages, is currently undergoing evaluation in a phase I clinical trial in advanced solid tumours that are refractory to previous treatments (NCT04691375) (Table 1).
PSGL1
PSGL1 is widely expressed in cells of haematopoietic origin, strongly upregulated by M2 polarizing signals in macrophages and expressed at high levels in TAMs. Its ligands include VISTA and selectins98. An anti-PSGL1 mAb repolarized human M2 macrophages to an M1-like phenotype, induced a hot inflammatory functional pattern in ex vivo human tumour tissue cultures, and had antitumour activity in humanized mouse models218. Thus, PSGL1 can represent a valuable target to re-educate TAMs218.
Targeting TAM metabolism
A network of transcription factors, epigenetic modifications and mRNAs underlies the plasticity and polarized activation of macrophages4,5,7. Downstream rewiring of macrophage function involves profound changes in amino acid, lipid and iron metabolism219,220,221. This complex cascade provides potential targets to reprogramme macrophage function. Epigenetic regulation by inhibition of class IIa histone deacetylases (HDACs) is a promising approach to harness the antitumour potential of macrophages. TMP195 was identified as a selective class IIa HDAC inhibitor that was able to modify the transcription profile of macrophages. In preclinical breast cancer models, TMP195 resulted in a macrophage-mediated reduction of tumour growth222. Tefinostat (CHR-2845) is an HDAC inhibitor that is cleaved to an active acid by non-specific esterase liver carboxylesterase 1 (CES1), the expression of which is limited to cells of monocytoid lineage and some hepatocytes, allowing selective accumulation of active drug within monocytoid cells. Because of this feature, it has been successfully used in a phase I clinical trial in patients with advanced haematological malignancies such as myelodysplastic syndromes and chronic myeloid leukaemia (NCT00820508)223. In principle, CES1 may provide a tool to develop macrophage-selective targeting of drugs.
The extreme metabolic conditions generated in the TME in terms of hypoxia and nutrient deprivation are causatively linked to the recruitment of macrophages that are, most commonly, functionally impaired6,220.
Lactic acid, produced by tumour cells as a by-product of glycolysis, functionally polarizes macrophages towards an M2-like phenotype characterized by higher expression of arginase 1224, suggesting that strategies blocking glycolysis may positively reprogramme TAMs. However, the same glycolytic pathway is fundamental for macrophage activity against tumour cells and glucose supply is needed for ROS production and phagocytosis6,220, raising questions as to the utility of blocking glycolytic pathways. So far, most of the studies aimed at targeting glycolysis to revert macrophage polarization have relied upon glycolytic inhibitors, such as 2-deoxy-d-glucose (2-DG)225, which are far from specific. In this line, the respiratory complex I inhibitor metformin, an antidiabetic agent, is being investigated in several clinical trials enrolling patients with different tumour types. In preclinical models, metformin was able to remodulate the TME, reducing the density of TAMs and increasing their phagocytic function226.
Despite the clear causative association between a hyperglycolitic environment and macrophage impairment, no data is available on the effect of caloric restriction on TAMs. Fasting-mimicking diets can increase response to chemotherapy in preclinical models227,228, an effect mediated by adaptive immune cells, although the myeloid compartment was not studied. Future studies are expected to unravel whether this debated approach could have an impact on TAMs. Among the modulators of macrophage polarization, pharmacological inhibition of retinoic acid impairs the differentiation of monocytes to immunosuppressive macrophages in soft tissue sarcoma229.
M2-like TAMs exhibit elevated consumption of glutamine, whose metabolism is instrumental to accomplish several cellular processes230, opening a possibility to rewire macrophage metabolism as an antitumour strategy. Blocking glutamine metabolism by small molecules or inhibiting its synthase resulted in decreased tumour growth by modulation of suppressive myeloid cells231,232. Elevated expression of the enzyme indoleamine 2,3-dioxygenase 1 (IDO1) by TAMs results in the consumption of tryptophan, an amino acid that is essential for the functional activation of T cells. Thus, tryptophan consumption by TAMs can severely impact the functional status of T cells and favour the generation of regulatory T cells with immunosuppressive activity. Several clinical trials are ongoing testing IDO inhibitors, with contrasting results233. In a randomized phase III study (ECHO-301/KN-252), an IDO inhibitor was tested in combination with pembrolizumab in metastatic melanoma but the result was negative234. Compensatory expression of similar enzymes, such as tryptophan 2,3-dioxygenase (TDO) and IDO2, could be a possible mechanism.
Among metabolic derangements frequently observed in TAMs, lipids take the stage. TAMs are defective in their lipid handling, which is directly linked to the activation of immunosuppressive pathways via the oxysterol receptor LXR transcription factor. Strategies targeting LXR have been developed, such as synthetic LXR agonists, shown to have anti-inflammatory actions in atherosclerosis and affecting the egress of macrophages from established lesions235. Among lipids, prostaglandins act as both pro-inflammatory mediators and orchestrators of mechanisms of tumour evasion. Tumour-derived prostaglandin E2 (PGE2) blocks early activation of NK cells236 and inflammatory activation of myeloid cells, driving their suppressive phenotype237. A TCGA analysis revealed that, in many human cancers, altered prostaglandin pathways have a negative impact on the efficacy of ICB236. In mouse preclinical models, inhibitors of prostaglandin G/H synthase 2 (PTGS2, also known as COX2) or antagonists of the PGE2 receptors EP1/EP2 can reprogramme the antitumour effectors and enhance the efficacy of ICB237,238.
Macrophage cell therapy
As discussed above, the TAM pool is constantly replenished by recruitment of circulating monocytes. CAR T cells are effective in the treatment of haematological malignancies but entry into solid tumours represents a stumbling block for T cell-based cellular therapies. Macrophage-based cell therapies may overcome this limitation, given the constant trafficking of mononuclear phagocytes into tumours. Macrophage-based cell therapy strategies have been based either on the capacity of monocytes to act as Trojan horses, delivering cytokines or nanoparticles to the TME, or on arming mononuclear phagocytes with engineered receptors. In a proof-of-concept study, monocytes replenished with drug-loaded nanoparticles and intravenously injected into tumour-bearing mice were able to reach the tumour site with superior efficiency than free nanoparticles239. The possibility of using macrophages to bring IFNα to the tumour site and consequently activate an immune response was investigated by De Palma et al.240, who transduced the Ifna1 gene in haematopoietic progenitors under the Tie2 promoter. Tie2-expressing monocytes, having a high tumour-homing ability, successfully migrated to tumours and delivered IFNα in the TME, triggering the activation of immune cells and inhibiting tumour growth and angiogenesis240. A clinical trial in glioblastoma patients is ongoing (Table 1). Similarly, soft particles called ‘backpacks’, containing the cytokine IFNα on their internal side, were stuck on the macrophage surface241. The study demonstrated that backpack-loaded macrophages acquire an M1 phenotype and, when injected intratumourally, maintain this phenotype without being affected by the immunosuppressive TME. Tumour growth and metastatic burden were significantly reduced in mice treated with macrophages carrying IFNγ backpacks241. In a murine sarcoma model, premetastatic niches were characterized by an immune suppression gene signature centred on myeloid cells. Myeloid cells were genetically engineered to express IL-12. Upon adoptive transfer, IL-12 expressing bone marrow-derived myeloid cells elicited a type 1 immune response in the lungs and reduced metastasis and primary tumour growth242.
The difficulty in transducing human macrophages has been a hurdle in developing mononuclear phagocyte-based cellular therapy, a stumbling block addressed by the development of different technological platforms243,244. Human CAR-M armed with receptors recognizing carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), CD19, CD22, HER2 and CD514,245,246,247 have been developed to attack haematopoietic and solid tumours (Fig. 4); various signalling platforms have been used (see Related link). CAR-M cells mediate phagocytosis and express M1 functions in a stable way14 and traffic to primary and metastatic tumours. Clinical trials are currently under way or planned to assess the potential of CAR-M in different tumours (Table 1).

a | Schematic of chimeric antigen receptor (CAR) molecule designed to be expressed by macrophages (CAR-M). Antibody specificity is provided by the extracellular module recognizing tumour antigens. Transmembrane and intracellular modules allow downstream activation signalling. Common CAR molecules have a T cell-activating module, which is preserved in one of the CAR-M developed so far because it is shown to retain activator functions on macrophages. b | Upon binding of CAR-M to tumour cells expressing the antigen, phagocytosis is increased and the pro-inflammatory M1 programme is induced, featuring release of interferon and upregulation of antigen-presentation molecules as well as enhanced presentation to T cells.
Conclusion
Macrophages are a universal component of the TME and engage in a complex interaction with cancer cells, stroma and immunocompetent cells. The recent discovery that glioblastoma cells acquire the expression of myeloid genetic programmes speaks to the fundamental role of macrophages in carcinogenesis and cancer progression27.
Macrophages play a dual part in the effectiveness of current treatment modalities from chemotherapy to ICB immunotherapy. In many human tumours, TAMs are the main drivers of checkpoint blockade in T cells and mediate resistance to immunotherapy. Despite their crucial role in the origin, progression and treatment of cancer, direct targeting of myelomonocytic cells has so far failed to have a major therapeutic impact, although ongoing trials targeting TAMs in combination with other treatment modalities may change the overall picture248 (Table 1). Stumbling blocks in the therapeutic translation of TAM immunobiology include the diversity and plasticity of mononuclear phagocytes in tumours. The dissection of TAM diversity at the single-cell level and their ontogenetic relationship may provide new views on strategies to selectively deplete tumour-promoting versus tumour-inhibiting subsets. It is our tenet that spatial and single-cell analysis will require crystallization of working hypotheses amenable to diagnostic and therapeutic testing in a clinical context.
Although macrophage infiltration is a common denominator of different tumours with shared properties, there is evidence for substantial differences in TAM phenotype and role in tumours arising in or disseminating to different organs. For instance, while TAM infiltration is associated with poor prognosis in most tumours, there are notable exceptions such as primary CRC249. Moreover, as discussed above, the tissue-intrinsic properties at the primary site or in metastatic lesions imprint the characteristics of the inflammatory microenvironment to some extent. Therefore, exploitation of TAMs may have to consider the tissue contexture of primary and secondary localization of tumours.
Macrophage function is regulated by surface receptors, some of which inhibit effector function or skew it in an inappropriate direction. These regulators of myelomonocytic cell function belong to different molecular classes and are obvious therapeutic targets. Early clinical trials of mAbs targeting the CD47–SIRP1α axis, LILRB2 and Clever 1 for the first time gave positive signals15,16,17. These positive responses, though still at an early stage, may herald the dawn of myeloid checkpoint immunotherapy. Based on the current understanding of the immunobiology of TAMs, it is our tenet that these approaches are unlikely to represent stand-alone strategies and their potential rests in complementing cytoreductive therapy and ICB immunotherapy.
In most, if not all, human tumours, TAMs are replenished by the circulating myeloid precursor pool. This long-held view provided the basis to develop CAR-M strategies. Poor recruitment at tumour sites has been a limiting factor for the application of CAR T cells to solid tumours. The ‘fatal attraction’ of monocytes to tumour tissues may provide a tool to circumvent this stumbling block in the development of cell therapy for solid tumours. However, several open questions remain for the exploitation of the cancer-homing propensity of monocytes. In addition to the choice of receptor and signalling components to arm monocytes, there is still limited or no understanding of the replenishment rate of the intratumoural pool by circulating precursors, the length of survival in the tumour context, the retention of appropriate functional orientation, or the spatial localization within the tumour tissues. Clinical trials may represent a unique opportunity to address these key questions of paramount importance for the development of CAR-M therapies.
Prevention of cancer using retinoic acid or drugs targeting hormones (pharmacoprevention) has failed. Macrophages and their mediators are companions and propellers of carcinogenesis. This fundamental consideration and the results of the CANTOS trial discussed above raise the issue of revisiting pharmacoprevention from the point of view of tumour-promoting inflammation. A trial using an anti-IL-1β mAb in participants at high risk of developing lung cancer (Canal trial) is being planned. Orally active inflammasome inhibitors, if proven safe and effective in conventional inflammatory conditions, could be ideally suited for pharmacoprevention efforts. The immunobiology of cancer-related inflammation and TAMs calls for giving careful consideration to prevention strategies in selected high-risk patients based on the premises discussed here.
References
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Engblom, C., Pfirschke, C. & Pittet, M. J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Locati, M., Curtale, G. & Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147 (2020).
Jahchan, N. S. et al. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 10, 1611 (2019).
Coussens, L. M., Zitvogel, L. & Palucka, A. K. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339, 286–291 (2013).
Cassetta, L. et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35, 588–602 (2019).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Kryczek, I. et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 203, 871–881 (2006).
Kim, I. S. et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 21, 1113–1126 (2019).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020). This is the first study to use primary human macrophages transduced with a CAR recognizing the HER2 antigen; a phase I clinical trial using CAR-M is under way.
Advani, R. et al. CD47 Blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379, 1711–1721 (2018). This is a phase Ib study in patients with non-Hodgkin lymphoma that showed promising activity of combining rituximab and CD47 blockade, pointing to CD47 as a novel myeloid checkpoint.
Siu, L. L. et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin. Cancer Res. 28, 57–70 (2022). This paper presents promising preliminary results observed with the MK-4830 antibody, targeting the myeloid-specific ILT4 receptor in advanced solid tumours; the mechanism of action includes reprogramming of TAMs to enhance T cell activity.
Virtakoivu, R. et al. Systemic blockade of clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: results from a phase I/II clinical trial. Clin. Cancer Res. 27, 4205–4220 (2021).
Bottazzi, B. et al. Regulation of the macrophage content of neoplasms by chemoattractants. Science 220, 210–212 (1983). This study is the first demonstration that macrophages are recruited in tumour tissues by tumour-derived chemotactic factors, later identified as CCL2.
Bain, C. C. et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat. Commun. 7, ncomms11852 (2016).
Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016).
Zhu, Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338 (2017).
Etzerodt, A. et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 217, e20191869 (2020).
Blériot, C., Chakarov, S. & Ginhoux, F. Determinants of resident tissue macrophage identity and function. Immunity 52, 957–970 (2020).
Gutmann, D. H. & Kettenmann, H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 104, 442–449 (2019).
Müller, A., Brandenburg, S., Turkowski, K., Müller, S. & Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 137, 278–288 (2015).
Dumas, A. A. et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 39, e103790 (2020).
Gangoso, E. et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184, 2454–2470 (2021).
Casanova-Acebes, M. et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584 (2021). This study dissected the role of tissue-resident macrophages and monocyte-derived macrophages in lung cancer, showing that tissue-resident macrophages promote early epithelial–mesenchymal transition and tumour invasiveness, and a potent regulatory T cell response.
Pombo Antunes, A. R. et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610 (2021).
Martinez-Usatorre, A. et al. Overcoming microenvironmental resistance to PD-1 blockade in genetically engineered lung cancer models. Sci. Transl Med. 13, eabd1616 (2021).
Singhal, S. et al. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci. Transl Med. 11, eaat1500 (2019).
Biswas, S. K. & Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11, 889–896 (2010).
Martinez, F. O. et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–e69 (2013).
Leader, A. M. et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 39, 1594–1609 (2021).
Li, H. et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell 40, 36–52 (2022).
Pushalkar, S. et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8, 403–416 (2018).
Dzutsev, A. et al. Microbes and cancer. Annu. Rev. Immunol. 35, 199–228 (2017).
Zhang, Q. et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov. 11, 1248–1267 (2021).
Roy, S. & Trinchieri, G. Microbiota: a key orchestrator of cancer therapy. Nat. Rev. Cancer 17, 271–285 (2017).
Movahedi, K. et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 70, 5728–5739 (2010).
Lavin, Y. et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765 (2017).
Lambrechts, D. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Zilionis, R. et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334 (2019).
Klemm, F. et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–1660 (2020).
Friebel, E. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181, 1626–1642 (2020).
Azizi, E. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308 (2018).
Puram, S. V. et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624 (2017).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Chevrier, S. et al. An immune atlas of clear cell renal cell carcinoma. Cell 169, 736–749 (2017).
Donadon, M. et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J. Exp. Med. 217, e20191847 (2020).
Braun, D. A. et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 39, 632–648 (2021).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459 (2020).
Pelka, K. et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752 (2021).
Cortese, N., Carriero, R., Laghi, L., Mantovani, A. & Marchesi, F. Prognostic significance of tumor-associated macrophages: past, present and future. Semin. Immunol. 48, 101408 (2020).
Forssell, J. et al. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 13, 1472–1479 (2007).
Malesci, A. et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 6, e1342918 (2017).
Quail, D. F. & Dannenberg, A. J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 15, 139–154 (2019).
Ruffell, B. & Coussens, L. M. Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015).
Galluzzi, L., Humeau, J., Buqué, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Germano, G. et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23, 249–262 (2013). This study demonstrates that the registered anti-tumour agent trabectedin has selective cytotoxicity against the monocyte–macrophage lineage, which contributes to its anti-tumour efficacy.
Di Caro, G. et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 65, 1710–1720 (2016).
Heath, O. et al. Chemotherapy induces tumor-associated macrophages that aid adaptive immune responses in ovarian cancer. Cancer Immunol. Res. 9, 665–681 (2021).
Iida, N. et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970 (2013).
Shiao, S. L. et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell 39, 1202–1213.e6 (2021).
Singh, S. et al. FDA approval summary: lurbinectedin for the treatment of metastatic small cell lung cancer. Clin. Cancer Res. 27, 2378–2382 (2021).
Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 19, 667–685 (2019).
Li, J. et al. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc. Natl Acad. Sci. USA 118, e2009290118 (2021).
Paulus, P., Stanley, E. R., Schafer, R., Abraham, D. & Aharinejad, S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 66, 4349–4356 (2006).
DeNardo, D. G. et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67 (2011).
Mitchem, J. B. et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141 (2013).
Salvagno, C. et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat. Cell Biol. 21, 511–521 (2019).
Gomez-Roca, C. A. et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 30, 1381–1392 (2019).
Gül, N. & van Egmond, M. Antibody-dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 75, 5008–5013 (2015).
DiLillo, D. J. & Ravetch, J. V. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol. Res. 3, 704–713 (2015).
Uchida, J. et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 199, 1659–1669 (2004).
Weng, W. K. & Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947 (2003).
Musolino, A. et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796 (2008).
Bibeau, F. et al. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 27, 1122–1129 (2009).
Lapenna, A., De Palma, M. & Lewis, C. E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 18, 689–702 (2018).
Greenberg, J. I. et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456, 809–813 (2008).
Peterson, T. E. et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl Acad. Sci. USA 113, 4470–4475 (2016).
Kloepper, J. et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl Acad. Sci. USA 113, 4476–4481 (2016).
Stender, J. D. et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol. Cell 65, 1122–1135 (2017).
Siersbæk, R. et al. IL6/STAT3 signaling hijacks estrogen receptor α enhancers to drive breast cancer metastasis. Cancer Cell 38, 412–423 (2020).
Cioni, B. et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat. Commun. 11, 4498 (2020).
Calcinotto, A. et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 559, 363–369 (2018). This study shows that IL-23 secreted by myeloid cells acts on prostate cancer cells and promotes the development of castration-resistant prostate cancer, linking inflammatory pathways to androgen therapy.
Formenti, S. C. & Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 10, 718–726 (2009).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). A seminal review improving our understanding of the mechanisms limiting cancer immunotherapy.
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009).
Bloch, O. et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 19, 3165–3175 (2013).
Topalian, S. L., Taube, J. M., Anders, R. A. & Pardoll, D. M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016).
Havel, J. J., Chowell, D. & Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 (2019).
Tang, H. et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest. 128, 580–588 (2018). In this paper, the relevant therapeutic target of ICB in preclinical models was PDL1 expressed by myeloid cells and not by tumour cells, suggesting that host expression of checkpoint molecules has an essential role in response and resistance.
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Strauss, L. et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 5, eaay1863 (2020).
Laba, S., Mallett, G. & Amarnath, S. The depths of PD-1 function within the tumor microenvironment beyond CD8+ T cells. Semin. Cancer Biol. https://doi.org/10.1016/j.semcancer.2021.05.022 (2021).
Wang, L. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 208, 577–592 (2011). This study identifies a novel and structurally distinct member of the immunoglobulin superfamily inhibitory ligands, homologous to PDL1, produced by myeloid cells and whose blockade interferes with suppression of T cell responses.
Johnston, R. J. et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570 (2019).
Rogers, B. M. et al. VISTA is an activating receptor in human monocytes. J. Exp. Med. 218, e20201601 (2021).
Yu, J. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 27, 152–164 (2021). This work shows that tissue contexture dictates the role of macrophages in cancer given that liver tumours but not lung metastasis induced macrophage-mediated resistance to ICB.
Chow, A. et al. Tim-4+ cavity-resident macrophages impair anti-tumor CD8+ T cell immunity. Cancer Cell 39, 973–988 (2021).
Krishna, C. et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662–677 (2021).
Koh, M. Y., Sayegh, N. & Agarwal, N. Seeing the forest for the trees-single-cell atlases link CD8. Cancer Cell 39, 594–596 (2021).
Vétizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Fridlender, Z. G. et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 70, 109–118 (2010).
Zhu, Y. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069 (2014).
Peranzoni, E. et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl Acad. Sci. USA 115, E4041–E4050 (2018).
Neubert, N. J. et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl Med. 10, eaan3311 (2018).
Beltraminelli, T. & De Palma, M. Biology and therapeutic targeting of tumour-associated macrophages. J. Pathol. 250, 573–592 (2020).
Argyle, D. & Kitamura, T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol. 9, 2629 (2018).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Ries, C. H. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859 (2014). Together with Pyonteck et al., these authors demonstrate that monocytes/macrophages can be targeted using small molecule inhibitors or antibodies to the CSF1 receptor, inhibiting tumour progression in preclinical models.
Pfirschke, C. et al. Macrophage-targeted therapy unlocks antitumoral cross-talk between IFNγ-secreting lymphocytes and IL12-producing dendritic cells. Cancer Immunol. Res. 10, 40–55 (2022).
Cassier, P. A. et al. Long-term clinical activity, safety and patient-reported quality of life for emactuzumab-treated patients with diffuse-type tenosynovial giant-cell tumour. Eur. J. Cancer 141, 162–170 (2020).
Bissinger, S. et al. Macrophage depletion induces edema through release of matrix-degrading proteases and proteoglycan deposition. Sci. Transl Med. 13, eabd4550 (2021).
Rodriguez-Garcia, A. et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 12, 877 (2021).
Etzerodt, A. et al. Specific targeting of CD163 mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 216, 2394–2411 (2019).
Reis, E. S., Mastellos, D. C., Ricklin, D., Mantovani, A. & Lambris, J. D. Complement in cancer: untangling an intricate relationship. Nat. Rev. Immunol. 18, 5–18 (2018).
Pio, R., Ajona, D., Ortiz-Espinosa, S., Mantovani, A. & Lambris, J. D. Complementing the cancer-immunity cycle. Front. Immunol. 10, 774 (2019).
Markiewski, M. M. et al. Modulation of the antitumor immune response by complement. Nat. Immunol. 9, 1225–1235 (2008). This is a pioneering study showing that the generation of complement C5a in a tumour microenvironment enhances tumour growth by promoting the recruitment of MDSCs into tumours and their T cell-directed suppressive abilities.
Magrini, E. et al. Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression. Nat. Cancer 2, 218–232 (2021). This study systematically assessed the role of complement activation and effector pathways in preclinical sarcoma models, showing that the lectin pathway and C3a–C3aR axis are key components of complement and macrophage-mediated sarcoma promotion and immunosuppression.
Medler, T. R. et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell 34, 561–578 (2018).
Laskowski, J. et al. Complement factor H-deficient mice develop spontaneous hepatic tumors. J. Clin. Invest. 130, 4039–4054 (2020).
Daugan, M. V. et al. Intracellular factor H drives tumor progression independently of the complement cascade. Cancer Immunol. Res 9, 909–925 (2021).
Roumenina, L. T., Daugan, M. V., Petitprez, F., Sautès-Fridman, C. & Fridman, W. H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 19, 698–715 (2019).
Roumenina, L. T. et al. Tumor cells hijack macrophage-produced complement C1q to promote tumor growth. Cancer Immunol. Res. 7, 1091–1105 (2019).
Bonavita, E. et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 160, 700–714 (2015). This is the first study showing that a humoral innate immunity effector molecule, PTX3, acts as an extrinsic oncosuppressor gene in mouse and human by regulating complement-dependent, macrophage-sustained, tumour-promoting inflammation.
Rubino, M. et al. Epigenetic regulation of the extrinsic oncosuppressor PTX3 gene in inflammation and cancer. Oncoimmunology 6, e1333215 (2017).
Vadrevu, S. K. et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 74, 3454–3465 (2014).
Ajona, D. et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. 7, 694–703 (2017).
Mantovani, A., Dinarello, C. A., Molgora, M. & Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 50, 778–795 (2019).
Ridker, P. M. et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017). This study proved that IL-1 represents a driver of tumour progression in human lung cancer by showing that an anti-inflammatory therapy with canakinumab targeting the IL-1β innate immunity pathway significantly reduced incident lung cancer and lung cancer mortality in a high-risk atherosclerosis population.
Garlanda, C. & Mantovani, A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 39, 1023–1027 (2021).
Kaplanov, I. et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl Acad. Sci. USA 116, 1361–1369 (2019).
Aggen, D. H. et al. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multidimensional analyses. Clin. Cancer Res. 27, 608–621 (2021).
Wong, C. C. et al. Inhibition of IL1β by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. 80, 5597–5605 (2020).
Tengesdal, I. W. et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl Acad. Sci. USA 118, e2000915118 (2021). This study showed that NLRP3 activation occurs in human metastatic melanoma and that it drives melanoma progression in mice by inducing IL-1β-dependent inflammation and immunosuppression, demonstrating that NLRP3 is a promising target in patients with melanoma treated with immunotherapy.
Zhang, F. et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 10, 3974 (2019).
Curtale, G., Rubino, M. & Locati, M. MicroRNAs as molecular switches in macrophage activation. Front. Immunol. 10, 799 (2019).
Zang, X. et al. Targeted delivery of miRNA 155 to tumor associated macrophages for tumor immunotherapy. Mol. Pharm. 16, 1714–1722 (2019).
Baer, C. et al. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 18, 790–802 (2016).
Lopez-Yrigoyen, M., Cassetta, L. & Pollard, J. W. Macrophage targeting in cancer. Ann. NY Acad. Sci. 1499, 18–41 (2021).
Allavena, P., Anfray, C., Ummarino, A. & Andón, F. T. Therapeutic manipulation of tumor-associated macrophages: facts and hopes from a clinical and translational perspective. Clin. Cancer Res. 27, 3291–3297 (2021).
Beatty, G. L. et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 19, 6286–6295 (2013).
Huffman, A. P., Lin, J. H., Kim, S. I., Byrne, K. T. & Vonderheide, R. H. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 5, e137263 (2020).
Hoves, S. et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 215, 859–876 (2018).
O’Hara, M. H. et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 22, 118–131 (2021).
Ji, N. et al. Percutaneous BCG enhances innate effector antitumor cytotoxicity during treatment of bladder cancer: a translational clinical trial. Oncoimmunology 8, 1614857 (2019).
McWhirter, S. M. & Jefferies, C. A. Nucleic acid sensors as therapeutic targets for human disease. Immunity 53, 78–97 (2020).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Pettenati, C. & Ingersoll, M. A. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 15, 615–625 (2018).
Sun, L. et al. Activating a collaborative innate-adaptive immune response to control metastasis. Cancer Cell 39, 1361–1374 (2021).
Anfray, C. et al. Intratumoral combination therapy with poly(I:C) and resiquimod synergistically triggers tumor-associated macrophages for effective systemic antitumoral immunity. J. Immunother. Cancer 9, e002408 (2021).
Frega, G. et al. Trial Watch: experimental TLR7/TLR8 agonists for oncological indications. Oncoimmunology 9, 1796002 (2020).
Márquez-Rodas, I. et al. Intratumoral nanoplexed poly I:C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci. Transl Med. 12, eabb0391 (2020).
Kyi, C. et al. Therapeutic immune modulation against solid cancers with intratumoral poly-ICLC: a pilot trial. Clin. Cancer Res. 24, 4937–4948 (2018).
Goldberg, M. S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 19, 587–602 (2019).
Irvine, D. J. & Dane, E. L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 20, 321–334 (2020).
Lu, R. et al. Formulation and preclinical evaluation of a toll-like receptor 7/8 agonist as an anti-tumoral immunomodulator. J. Control. Rel. 306, 165–176 (2019).
Ribas, A. et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. 11, 2998–3007 (2021).
Ackerman, S. E. et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat. Cancer 2, 18–33 (2021).
Chow, L. Q. M. et al. Phase Ib trial of the toll-like receptor 8 agonist, motolimod (VTX-2337), combined with cetuximab in patients with recurrent or metastatic SCCHN. Clin. Cancer Res. 23, 2442–2450 (2017).
Ferris, R. L. et al. Effect of adding motolimod to standard combination chemotherapy and cetuximab treatment of patients with squamous cell carcinoma of the head and neck: the active8 randomized clinical trial. JAMA Oncol. 4, 1583–1588 (2018).
Frank, M. J. et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 8, 1258–1269 (2018).
Karapetyan, L., Luke, J. J. & Davar, D. Toll-like receptor 9 agonists in cancer. OncoTargets Ther. 13, 10039–10060 (2020).
Li, J. K., Balic, J. J., Yu, L. & Jenkins, B. TLR agonists as adjuvants for cancer vaccines. Adv. Exp. Med. Biol. 1024, 195–212 (2017).
Vermaelen, K. Vaccine strategies to improve anti-cancer cellular immune responses. Front. Immunol. 10, 8 (2019).
Kalbasi, A. et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl Med. 12, eabb0152 (2020).
Smith, M. et al. Trial watch: toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 7, e1526250 (2018).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Vanpouille-Box, C., Hoffmann, J. A. & Galluzzi, L. Pharmacological modulation of nucleic acid sensors- therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 18, 845–867 (2019).
Le Naour, J., Zitvogel, L., Galluzzi, L., Vacchelli, E. & Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 9, 1777624 (2020).
Zhou, S., Kestell, P., Baguley, B. C. & Paxton, J. W. 5,6-dimethylxanthenone-4-acetic acid (DMXAA): a new biological response modifier for cancer therapy. Invest. New Drugs 20, 281–295 (2002).
Motedayen Aval, L., Pease, J. E., Sharma, R. & Pinato, D. J. Challenges and opportunities in the clinical development of STING agonists for cancer immunotherapy. J. Clin. Med. 9, 3323 (2020).
Zou, S. S. et al. Intrinsic strategies for the evasion of cGAS-STING signaling-mediated immune surveillance in human cancer: how therapy can overcome them. Pharmacol. Res. 166, 105514 (2021).
Pan, B. S. et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 369, eaba6098 (2020).
Lam, K. C. et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 184, 5338–5356 (2021).
Barkal, A. A. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396 (2019).
Mantovani, A. & Longo, D. L. Macrophage checkpoint blockade in cancer - back to the future. N. Engl. J. Med. 379, 1777–1779 (2018).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009). Together with Jaiswal, S. et al., this paper demonstrates that cancer cells with high levels of the protein CD47 have a survival advantage and avoid phagocytosis by macrophages, inspiring the generation of therapeutic inhibitors to block CD47 and its receptor SIRPα.
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009). Together with Majeti, R. et al., this paper demonstrates that cancer cells with high levels of the protein CD47 have a survival advantage and avoid phagocytosis by macrophages, inspiring the generation of therapeutic inhibitors to block CD47 and its receptor SIRPα.
Gholamin, S. et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl Med. 9, eaaf2968 (2017).
Tseng, D. et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl Acad. Sci. USA 110, 11103–11108 (2013).
Ring, N. G. et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl Acad. Sci. USA 114, E10578–E10585 (2017).
Zhang, M. et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS ONE 11, e0153550 (2016).
Weiskopf, K. et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest. 126, 2610–2620 (2016).
Upton, R. et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl Acad. Sci. USA 118, e2026849118 (2021).
van Bommel, P. E. et al. CD20-selective inhibition of CD47-SIRPα “don’t eat me” signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. Oncoimmunology 7, e1386361 (2018).
Wang, Y. et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 70, 365–376 (2021).
Hendriks, M. A. J. M. et al. Bispecific antibody approach for EGFR-directed blockade of the CD47-SIRPα “don’t eat me” immune checkpoint promotes neutrophil-mediated trogoptosis and enhances antigen cross-presentation. Oncoimmunology 9, 1824323 (2020).
Tian, L. et al. Targeting Fc receptor-mediated effects and the “don’t eat me” signal with an oncolytic virus expressing an anti-CD47 antibody to treat metastatic ovarian cancer. Clin. Cancer Res. 28, 201–214 (2022).
Jing, W. et al. Breast cancer cells promote CD169+ macrophage-associated immunosuppression through JAK2-mediated PD-L1 upregulation on macrophages. Int. Immunopharmacol. 78, 106012 (2020).
Ibarlucea-Benitez, I., Weitzenfeld, P., Smith, P. & Ravetch, J. V. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor immunity. Proc. Natl Acad. Sci. USA 118, e2107424118 (2021).
Festenstein, H. & Garrido, F. MHC antigens and malignancy. Nature 322, 502–503 (1986).
Barkal, A. A. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 19, 76–84 (2018).
Chen, H. M. et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J. Clin. Invest. 128, 5647–5662 (2018).
Sharma, N., Atolagbe, O. T., Ge, Z. & Allison, J. P. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J. Exp. Med. 218, e20201811 (2021).
Jensen, T. O. et al. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J. Clin. Oncol. 27, 3330–3337 (2009).
Lau, S. K., Chu, P. G. & Weiss, L. M. CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am. J. Clin. Pathol. 122, 794–801 (2004).
Allavena, P. et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin. Dev. Immunol. 2010, 547179 (2010).
Chieppa, M. et al. Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J. Immunol. 171, 4552–4560 (2003).
Heinsbroek, S. E. et al. miR-511-3p, embedded in the macrophage mannose receptor gene, contributes to intestinal inflammation. Mucosal Immunol. 9, 960–973 (2016).
Rahabi, M. et al. Divergent roles for macrophage C-type lectin receptors, dectin-1 and mannose receptors, in the intestinal inflammatory response. Cell Rep. 30, 4386–4398 (2020).
Jaynes, J. M. et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl Med. 12, eaax6337 (2020).
Georgoudaki, A. M. et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15, 2000–2011 (2016).
La Fleur, L. et al. Targeting MARCO and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res. 81, 956–967 (2021).
Molgora, M. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551, 110–114 (2017).
Eisinger, S. et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc. Natl Acad. Sci. USA 117, 32005–32016 (2020).
Sa, J. K. et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 21, 216 (2020).
Palani, S. et al. Stabilin-1/CLEVER-1, a type 2 macrophage marker, is an adhesion and scavenging molecule on human placental macrophages. Eur. J. Immunol. 41, 2052–2063 (2011).
Viitala, M. et al. Immunotherapeutic blockade of macrophage clever-1 reactivates the CD8. Clin. Cancer Res. 25, 3289–3303 (2019). In this study, blocking Clever 1 expressed by M2-like macrophages unleashed macrophage and T cell-mediated antitumour immunity, pointing to Clever 1 as a novel ‘don’t eat me’ signal.
Molgora, M. et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell 182, 886–900 (2020).
Katzenelenbogen, Y. et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 182, 872–885 (2020).
Binnewies, M. et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 37, 109844 (2021).
Turnbull, I. R. et al. Cutting edge: TREM2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 (2006).
Nguyen, P. et al. 862 Targeting PSGL-1, a novel macrophage checkpoint, repolarizes suppressive macrophages, induces an inflammatory tumor microenvironment, and suppresses tumor growth. J. Immunother. Cancer 8, https://doi.org/10.1136/jitc-2020-SITC2020.0862 (2020).
Biswas, S. K. Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449 (2015).
Mehla, K. & Singh, P. K. Metabolic regulation of macrophage polarization in. Cancer Trends Cancer 5, 822–834 (2019).
Murray, P. J. Macrophage polarization. Annu. Rev. Physiol. 79, 541–566 (2017).
Guerriero, J. L. et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432 (2017).
Ossenkoppele, G. J. et al. A phase I first-in-human study with tefinostat - a monocyte/macrophage targeted histone deacetylase inhibitor-in patients with advanced haematological malignancies. Br. J. Haematol. 162, 191–201 (2013).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Zhao, Q. et al. 2-Deoxy-d-glucose treatment decreases anti-inflammatory M2 macrophage polarization in mice with tumor and allergic airway inflammation. Front. Immunol. 8, 637 (2017).
Wang, S. et al. Low-dose metformin reprograms the tumor immune microenvironment in human esophageal cancer: results of a phase II clinical trial. Clin. Cancer Res. 26, 4921–4932 (2020).
Di Biase, S. et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 30, 136–146 (2016).
Pietrocola, F. et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell 30, 147–160 (2016).
Devalaraja, S. et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell 180, 1098–1114 (2020).
Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015).
Oh, M. H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 130, 3865–3884 (2020).
Menga, A. et al. Glufosinate constrains synchronous and metachronous metastasis by promoting anti-tumor macrophages. EMBO Mol. Med. 12, e11210 (2020).
Labadie, B. W., Bao, R. & Luke, J. J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin. Cancer Res. 25, 1462–1471 (2019).
Van den Eynde, B. J., van Baren, N. & Baurain, J. F. Is there a clinical future for IDO1 inhibitors after the failure of epacadostat in melanoma? Annu. Rev. Cancer Biol. 4, 241–256 (2020).
Hong, C. & Tontonoz, P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov. 13, 433–444 (2014).
Bonavita, E. et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity 53, 1215–1229 (2020). This study showed that tumour-derived PGE2 plays a key role in inhibiting NK cell-driven myeloid cell reprogramming and enabling immune evasion in mice; the signature associated with the COX2–PGE2 axis and NK cell activity predicted patient survival and response to ICB.
Porta, C. et al. Tumor-derived prostaglandin E2 promotes p50 NF-κB-dependent differentiation of monocytic MDSCs. Cancer Res. 80, 2874–2888 (2020).
Pelly, V. S. et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 11, 2602–2619 (2021).
Allavena, P. et al. PLGA based nanoparticles for the monocyte-mediated anti-tumor drug delivery system. J. Biomed. Nanotechnol. 16, 212–223 (2020).
De Palma, M. et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 14, 299–311 (2008). In this pioneering study, monocytes were transduced with the IFNA gene and used as cellular vehicles to deliver the anti-tumour cytokine IFNα to the immune microenvironment of tumours.
Shields, C. W. et al. Cellular backpacks for macrophage immunotherapy. Sci. Adv. 6, eaaz6579 (2020).
Kaczanowska, S. et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 184, 2033–2052 (2021).
Sunseri, N., O’Brien, M., Bhardwaj, N. & Landau, N. R. Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J. Virol. 85, 6263–6274 (2011).
Bobadilla, S., Sunseri, N. & Landau, N. R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 20, 514–520 (2013).
Zhang, W. et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 121, 837–845 (2019).
Biglari, A., Southgate, T. D., Fairbairn, L. J. & Gilham, D. E. Human monocytes expressing a CEA-specific chimeric CD64 receptor specifically target CEA-expressing tumour cells in vitro and in vivo. Gene Ther. 13, 602–610 (2006).
Morrissey, M. A. et al. Chimeric antigen receptors that trigger phagocytosis. Elife 7, e36688 (2018).
Sloas, C., Gill, S. & Klichinsky, M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front. Immunol. 12, 783305 (2021).
Zhang, Q. W. et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE 7, e50946 (2012).
Pienta, K. J. et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 31, 760–768 (2013).
Nywening, T. M. et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 17, 651–662 (2016).
Haag, G. M. et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer - The PICCASSO phase I trial. Eur. J. Cancer 167, 112–122 (2022).
Manji, G. A. et al. A phase I study of the combination of pexidartinib and sirolimus to target tumor-associated macrophages in unresectable sarcoma and malignant peripheral nerve sheath tumors. Clin. Cancer Res. 27, 5519–5527 (2021).
Lin, C. Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 80 (Suppl. 16), CT171 (2020).
Razak, A. R. et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 8, e001006 (2020).
Fisher, G. A. et al. A phase Ib/II study of the anti-CD47 antibody magrolimab with cetuximab in solid tumor and colorectal cancer patients. JCO Gastointestinal Cancers Symp. 38 (Suppl. 4), 114 (2020).
Marquez-Rodas, I. et al. Combination of radiomic and biomarker signatures as exploratory objective in a phase II trial with intratumoral BO-112 plus pembrolizumab for advanced melanoma. J. Clin. Oncol. https://doi.org/10.1200/JCO.2021.39.15_suppl.TPS9586 (2021).
Sharma, M. et al. Preliminary results from a phase 1/2 study of BDC-1001, a novel HER2 targeting TLR7/8 immune-stimulating antibody conjugate (ISAC), in patients (pts) with advanced HER2-expressing solid tumors. J. Clin. Oncol. 39 (Suppl. 15), 2549 (2021).
Finocchiaro, G. et al. A phase I-IIa study of genetically modified Tie-2 expressing monocytes in patients with glioblastoma multiforme (TEM-GBM Study). J. Clin. Oncol. 39 (Suppl. 15), 2532 (2021).
Guilliams, M., Mildner, A. & Yona, S. Developmental and functional heterogeneity of monocytes. Immunity 49, 595–613 (2018).
Gosselin, D. et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014).
Okabe, Y. & Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157, 832–844 (2014).
Amit, I., Winter, D. R. & Jung, S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 17, 18–25 (2015).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 (2012).
Zhang, B. et al. B cell-derived GABA elicits IL-10+ macrophages to limit anti-tumor immunity. Nature 599, 471–476 (2021).
Scala, S. & Aiuti, A. In vivo dynamics of human hematopoietic stem cells: novel concepts and future directions. Blood Adv. 3, 1916–1924 (2019).
Jaillon, S. et al. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 20, 485–503 (2020).
Güç, E. & Pollard, J. W. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity 54, 885–902 (2021).
Kitamura, T. et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 212, 1043–1059 (2015).
Ma, R. Y. et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med. 217, e20191820 (2020).
Bieniasz-Krzywiec, P. et al. Podoplanin-expressing macrophages promote lymphangiogenesis and lymphoinvasion in breast cancer. Cell Metab. 30, 917–936 (2019).
Singh, R. & Choi, B. K. Siglec1-expressing subcapsular sinus macrophages provide soil for melanoma lymph node metastasis. Elife 8, e48916 (2019).
Colombo, N. et al. Anti-tumor and immunomodulatory activity of intraperitoneal IFN-gamma in ovarian carcinoma patients with minimal residual tumor after chemotherapy. Int. J. Cancer 51, 42–46 (1992).
Marchesi, F., Piemonti, L., Mantovani, A. & Allavena, P. Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor. Rev. 21, 77–82 (2010).
Obradovic, A. et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988–3005 (2021).
Combes, A. J. et al. Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 185, 184–203 (2022).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809 (2021).
Raghavan, S. et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137 (2021).
Acknowledgements
The research leading to the results here reviewed has received funding from Associazione Italiana Ricerca Cancro (AIRC): AIRC 5×1000 21147 to A.M.; AIRC-IG 23465 to A.M.; AIRC-IG 21714 to C.G.; the Italian Ministry of Health (Ricerca Finalizzata: RF-2019-12369142 to F.M.; RF-2013-02355470 to C.G.) and EuroNanoMed III-2-INTRATARGET project (PCIN-2017-129/AEI) to P.A. The funding agency had no role in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
The authors have equally contributed to this review.
Corresponding author
Ethics declarations
Competing interests
A.M. has been a recipient of commercial research grants from Sigma Tau, Roche, Novartis, Compugen and Efranat, and has been a consultant/advisory board member/lecturer for Novartis, Roche, Ventana, Pierre Fabre, Verily, Abbvie, BMS, J&J, Compugen, Imcheck, Macrophage Therapeutics, AstraZeneca, Biovelocita, BG Fund, Third Rock, Verseau Therapeutics and Olatec Therapeutics. C.G. and P.A. are recipients of research grants from Imcheck and Macrophage Therapeutics. A.M. and C.G. are inventors of patents related to PTX3 and other innate immunity molecules. A.M., C.G. and P.A. receive royalties for reagents related to innate immunity.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks Jeffrey Pollard, Michele De Palma, and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Myeloid Therapeutics technology platforms: https://www.myeloidtx.com/our-science
Glossary
- Myelomonocytic cells
-
Haematopoietic cells, including monocytes, macrophages and monocyte-derived dendritic cells.
- Desmoplastic reaction
-
A dense, fibrous tissue that is formed in some tumours secondary to the tissue repair response orchestrated by inflammatory cells.
- Regulatory T cells
-
A specialized population of T cells that suppress the activation of other T cells and maintain peripheral tolerance to self-antigens.
- Polarized type 2 adaptive immune responses
-
An adaptive immune response induced by cytokines from T helper 2 cells, such as IL-4, IL-5 and IL-13, and characterized by alternative macrophage activation that is important for tissue repair and fibrosis.
- Coelomic cavities
-
They include pleural, peritoneal and pericardial cavities that originate during embryogenesis in the gastrulation phase. These cavities are lined by specialized mesodermal cells called mesothelium.
- Inflammasome
-
A critical inflammatory component, composed of an intracellular oligomeric protein complex. Its activation by both pathogen and danger signals results in the assembly of the complex and leads to caspase-dependent cleavage of IL-1 and IL-18 precursors.
- Scavenger receptors
-
A heterogeneous family of cell-surface receptors expressed on phagocytes that allow recognition of pathogen-related and danger-associated molecular moieties of various nature (such as sugars and modified lipoproteins), instrumental for the defence against infections and tissue damage.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mantovani, A., Allavena, P., Marchesi, F. et al. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov 21, 799–820 (2022). https://doi.org/10.1038/s41573-022-00520-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00520-5
This article is cited by
-
Prognostic role of macrophages and mast cells in the microenvironment of hepatocellular carcinoma after resection
BMC Cancer (2024)
-
First effectiveness data of lenvatinib and pembrolizumab as first-line therapy in advanced anaplastic thyroid cancer: a retrospective cohort study
BMC Endocrine Disorders (2024)
-
Exosome-based delivery strategies for tumor therapy: an update on modification, loading, and clinical application
Journal of Nanobiotechnology (2024)
-
Single-cell RNA sequencing reveals recruitment of the M2-like CCL8high macrophages in Lewis lung carcinoma-bearing mice following hypofractionated radiotherapy
Journal of Translational Medicine (2024)
-
Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies
Molecular Cancer (2024)
