
Abstract
CYLD is a gene mutated in familial cylindromatosis and related diseases, leading to the development of skin appendages tumors. Although the deubiquitinase CYLD is a skin tumor suppressor, its role in skin physiology is unknown. Using skin organotypic cultures as experimental model to mimic human skin, we have found that CYLD acts as a regulator of epidermal differentiation in humans through the JNK signaling pathway. We have determined the requirement of CYLD for the maintenance of epidermal polarity, keratinocyte differentiation and apoptosis. We show that CYLD overexpression increases keratinocyte differentiation while CYLD loss of function impairs epidermal differentiation. In addition, we describe the important role of CYLD in the control of human non-melanoma skin cancer progression. Our results show the reversion of the malignancy of human squamous cell carcinomas that express increased levels of CYLD, while its functional inhibition enhances the aggressiveness of these tumors which progress toward spindle cell carcinomas. We have found that the mechanisms through which CYLD regulates skin cancer progression include the control of tumor differentiation, angiogenesis and cell survival. These findings of the role of CYLD in human skin cancer prognosis make our results relevant from a therapeutic point of view, and open new avenues for exploring novel cancer therapies.
Similar content being viewed by others


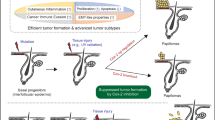
Main
The epidermis is a stratified squamous epithelium composed mainly of keratinocytes. Basal keratinocytes proliferate and give rise to differentiated cells, which, upon full maturation, undergo enucleation and generate the squamous cornified layer.1 Several molecules have been found to develop important roles in this differentiation process. Among them, the c-Jun-NH2-terminal kinase (JNK) signaling pathway has an emergent role,2, 3 as the inhibition of JNK activity in keratinocyte induces epidermal differentiation (stratification and production of cornified envelopes), while augmented JNK activity delays or inhibits keratinocyte differentiation.2 To be functional, keratinocytes must also maintain an apico-basal polarization, β-catenin being an important molecule to the generation of a polarized epithelial phenotype;4, 5 that is, perturbed β-catenin location (cytoplasmic localization and reduced membranous expression) is associated to lack of differentiation, invasive potential and aggressive behaviour6 in tumors. Disturbances of the balance proliferation/differentiation/apoptosis of the epidermis result in epidermal disorders, such as hyperplasia, psoriasis and skin cancer. Non-melanoma skin cancers (NMSC): basal cell carcinomas (BCC) and squamous cell carcinomas (SCC) are the most common human malignancies.
CYLD is a tumor suppressor gene that was originally identified as a gene mutated in familial cylindromatosis, a genetic condition that predisposes patients for the development of skin appendages tumors.7 CYLD deubiquitinating activity removes lysine-63 polyubiquitin chains.7 Most of the mutations within the CYLD locus are located toward the carboxyl terminus of the protein, the position of the catalytic residues of ubiquitin hydrolase.8, 9 Depending on the cellular context CYLD negatively regulates NF-κB and/or JNK-signaling pathways, resulting in suppression of cell proliferation.10, 11, 12, 13 In keratinocytes, JNK activation promotes cell proliferation and inhibits differentiation,2 whereas NF-κB appears uninvolved in epidermal differentiation.14, 15
Besides its role in the familial cylindromatosis, additional studies have associated CYLD downregulation with the development of other types of human cancer including tumors of colon, lung and kidney, as well as melanomas and cervical and hepatocellular carcinomas.16, 17, 18, 19, 20 Recently, we have demonstrated a relationship between the inhibition of CYLD function and an increase in aggressiveness of mouse NMSC.21 However, although CYLD is ubiquitously expressed and its function as tumor suppressor is being actively investigated, little is known about its role in the homeostasis of the different tissues; particularly in the skin.
In this study we have analyzed the role of CYLD in the homeostasis and carcinogenesis of human skin. We have used skin organotypic cultures (also named skin equivalents) as experimental model to mimic human skin in vitro. Similar to the epidermis, the epithelium of the skin organotypic culture stratifies and differentiates. This model has allowed us to increase the expression or to inhibit the function of CYLD in a human-like context. As a result, we have found that forced expression of wild-type CYLD (CYLDWT) both in HaCaT keratinocytes and in the skin equivalents enhances keratinocyte differentiation, while the inhibition of CYLD function by expression of a catalytically inactive form of CYLD impairs epidermal differentiation. CYLDWT overexpression is also able to induce the differentiation of human A431 squamous carcinoma (SCC) cell line. Our results show that CYLD controls skin differentiation through the JNK signaling pathway. In addition, using an in vivo xenograft model of skin carcinogenesis, we describe the important role of CYLD in the control of human NMSC progression.
Results
Forced expression of CYLDWT in human keratinocytes
The HaCaT cell line, a non-tumoral immortalized human epidermal cell line commonly used in epidermal biology studies,22 was employed. HaCaT cells maintain full epidermal differentiation capacity: when cultured in the absence of serum, they form stratification domes and express the early differentiation marker keratin K10.22 Moreover, they are able to grow in organotypic cultures.23 HaCaT cells were stably transfected with the CYLDWT cDNA under the control of the β-actin promoter (β-actin-CYLDWT) or with the empty vector. Pooled stable transfectants carrying either construct were used to minimize any potential effect of clonal selection. They were designated as H-CYLDWT and H-control cells (expressing the CYLDWT transgene and the empty vector, respectively). Western blotting against CYLD showed that H-CYLDWT cells had increased levels of CYLD (Figure 1a) compared with the endogenous levels of CYLD in the H-control cells.

Properties of transfected HaCaT CYLD cells. (a) Western blot showing CYLD overexpression in one of the H-CYLDWT pools (H-Co, H-Control cells). Tubulin was used as loading control. (b) H-control and H-CYLDWT cells differentiation cultures. Red circles: stratification domes (c) (top) keratin K10 RNA levels in H-control and H-CYLDWT cells detected by northern blotting at the stated times (days); 7S, loading control. (bottom) Graphical representation of the northern blotting signals relative to those of loading controls (arbitrary units). The difference in K10 content between cells of both genotypes is statistically significant at 6, 10 and 13 days of differentiation. (d) Western blot of proteins from H-control, H-CYLDWT and H-CYLDC/S cells in the basal state or after UV stimulation showing an increase in JNK activation in the H-CYLDC/S cells and a reduction in the H-CYLDWT. Analysis of Bax expression. Actin was used as loading control. Western blots were performed 3–4 times
Increased CYLDWT expression enhances the differentiation of human keratinocytes in culture
We analyzed the differentiation properties of H-control and H-CYLDWT cells. By day 7 of growth in absence of serum all the H-CYLDWT pools of cells showed well-formed stratification domes, while in H-control cells the first (still rudimentary) domes were detected at day 14 (Figure 1b). The analysis of the early differentiation marker keratin K10, not expressed in undifferentiated cells, shows that at day 6 of differentiation the H-CYLDWT cells showed very high levels of K10, while the H-control cells did not show these levels of K10 expression until day 13 of differentiation (Figure 1c). Abundant cell death was observed in the supernatants of both, differentiated and conventional H-CYLDWT monolayer cultures; the analysis of the pro-apoptoctic protein Bax showed that it was highly expressed in the H-CYLDWT cells compared with the low levels of expression found in the H-control cells (Figure 1d). Moreover, after UV light stimulation (an inductor of apoptosis in keratinocytes),24 Bax was highly induced in the H-CYLDWT cells (Figure 1d). Therefore, our results indicate that H-CYLDWT cells are prone to differentiation in addition to presenting a high rate of apoptosis.
Increased CYLD expression enhances the differentiation of keratinocytes in human skin equivalents
Organotypic skin cultures were established with both H-control and H-CYLDWT cells. H-control skin (6-day) equivalents were composed by 3–4 layers of keratinocytes distributed in well distinguishable strata: a basal layer with cylindrical cells of large nuclei; 1–2 layers of suprabasal cells of small nuclei and a superficial stratum containing flattened cells with the nuclei parallel to the basal membrane (Figure 2a). More engrossed epithelium was found in the H-CYLDWT skin equivalents that contain 6–7 layers of keratinocytes, including higher number of flattened suprabasal cells (3–4 layers), which suggested an increased differentiation (Figure 2c). According to these histological characteristics, the differentiation marker keratin K1 was widely expressed in the 6-day H-CYLDWT skin equivalents, (Figure 2d) whereas there was scarce K1 expression in 6-day-H-control organotypic cultures (Figure 2b). By 12 days of growth in air–liquid interface, H-control skin equivalents showed increased stratification (with respect to that observed at 6 days of culture), showing a higher number of suprabasal cells containing abundant cytoplasm and some apoptotic cells (Figure 2e). By this time point, CYLDWT organotypic cultures also have increased their number of suprabasal cells, and strikingly, there were very high number of apoptotic cells (Figure 2g). Cleaved caspase 3 staining confirmed the abnormally high number of apoptotic cells present in the suprabasal and the intermediate cell layers of H-CYLDWT skin equivalents (Figures 2f and h).

Increased differentiation and apoptosis in H-CYLDWT skin equivalents. (a–d) Skin (6-day) equivalents showing increased differentiation of H-CYLDWT organotypic cultures by histological examination (a and c) and by K1 staining (b and d). Arrow in (a): example of stratified cell; sl, suprabasal layers. (e–h) Skin (12-day) equivalents showing increased apoptosis in the H-CYLDWT skin equivalents by histological observation (e and g) and by anticleaved caspase 3 staining (C3C) (f and h). Arrows in e and g: apoptotic nuclei
The decreased JNK pathway activation in the H-CYLDWT cells appears as a plausible mechanism for CYLD induction of keratinocytes differentiation
JNK signaling is a relevant pathway for keratinocyte differentiation and it is negatively regulated by CYLD.13 The analysis of JNK phosphorylation (activation) in H-CYLDWT cells showed decreased activation of JNK with respect to that of H-control cells in the non-stimulated state (Figure 1d, middle row). No differences were found between H-control and H-CYLDWT cells in response to UV stimulation (Figure 1d). Our results showing increased differentiation of the H-CYLDWT cells that present a diminished JNK activation, are in agreement with the previous studies reporting increased keratinocyte differentiation as a consequence of JNK activity inhibition.2
The inhibition of CYLD function in keratinocytes enhances the activation of the JNK pathway
To further confirm the CYLD function as regulator of keratinocyte differentiation and the relevance of the JNK pathway activation as a mechanism through which it may exerts its function, we generated HaCaT cells expressing a catalytically deficient CYLD mutant (CYLDC/S) that carries the 601C/S point mutation in the cysteine box of the deubiquitinase.10 The expression of this transgene results in a catalytically inactive protein that competes with the endogenous CYLD.10, 12, 20, 21, 25 HaCaT cells were stably transfected with CYLDC/S cDNA under the control of the β-actin promoter or with the empty vector, and different pooled stable transfectants were collected (designated as H-CYLDC/S and H-control cells). Western blot analysis showed increased levels of CYLD in the H-CYLDC/S cells (Figure 3a).

Functional analysis of H-CYLDC/S cells. (a) Western blot showing CYLD overexpression in two H-CYLDC/S pools (C/S 1 and C/S 2). (b) Deubiquitination of IKKγ/NEMO (by CYLD) in H-control and H-CYLDC/S cells before (0) or after (30′ and 60′) TNF-α stimulation. IKKγ-immunoprecipitated samples were probed with an antiubiquitin antibody (upper panel) then reprobed with an antibody against IKKγ (lower panel). Observe that TNF-α stimulation failed to increase the polyubiquitination of IKKγ in the H-control cells. By contrast, TNF-α stimulation resulted in significant polyubiquitination of IKKγ in the H-CYLDC/S cells. (c–e) Hematoxylin/eosin staining of 10-day skin equivalents showing the altered morphology of H-CYLDC/S skin equivalents (d and e) versus H-control organotypic cultures (c); arrows in (e and j): foci of invasion. (g) Impaired differentiation of H-CYLDC/S organotypic cultures as studied by involucrin (invol) staining compared with the differentiated H-control skin equivalents (f). (i–j) Cleaved caspase 3 (C3C) immunostaining showing the resistance to apoptosis of the H-CYLDC/S skin equivalents compared with H-control organotypic cultures (h). (k and l) β-catenin expression in H-control, H-CYLDWT and H-CYLDC/S keratinocytes growing as monolayers (l) or in the skin equivalents (k)
We proved the lack of deubiquitination function of CYLD in the H-CYLDC/S cells (Figure 3b). We then studied the JNK activation pathway in the H-CYLDC/S cells. Both H-control and H-CYLDC/S cells were UV-irradiated and JNK activation in response to irradiation was analyzed. We found that JNK was highly activated after UV stimulation in the H-CYLDC/S cells (Figure 1d). Therefore, our results show that in human keratinocytes the activation of JNK pathway was increased by the functional inhibition of CYLD while, consistently, it was diminished by the overexpression of CYLDWT (Figure 1d).
The diminished function of CYLD impairs the differentiation of keratinocytes in human HaCaT skin equivalents
H-CYLDC/S skin equivalents were established. By 10 days of air-phase growth, H-control organotypic cultures formed a well-arranged tissue structure (Figure 3c) with a basal layer formed by cylindrical cells of large nuclei and 4–6 layers of suprabasal cells, being the most superficial flattened cells with small nuclei in parallel to the basal membrane (Figure 3c). In contrast, 10-days H-CYLDC/S skin-equivalents showed a higher number of cell layers than the H-control skin equivalents, presenting heterogeneity in size, shape and orientation of the nuclei, resembling a disorganized epidermis with loss of cell polarity (Figure 3d). Cells invading the subjacent matrix were also frequently seen in the H-CYLDC/S skin equivalents (Figures 3e and j). To ascertain the possible cause of the loss of cell polarity observed in the CYLDC/S keratinocytes, we analyzed the expression of β-catenin, as this is a relevant protein for the generation of a polarized epithelial phenotype.4, 5 We found a normal, membranous localization of β-catenin at the cell–cell contacts of almost all the cell layers in both H-control and H-CYLDWT skin equivalents (Figure 3k); by contrast, H-CYLDC/S skin equivalents showed a disorganized β-catenin location, that even was absent in most of the suprabasal cells (Figure 3k). Even more, we also found that in H-CYLDC/S monolayer cultures, β-catenin appeared forming dots or diffusely distributed in the cytoplasm of most of the cells (Figure 3l), in sharp contrast with the membranous pattern of β-catenin staining at the cell–cell contacts found in both H-control and H-CYLDWT cells.
To analyze the differentiation of both the H-CYLDC/S and H-control skin-equivalents, we studied the expression of involucrin, a marker of early differentiation. We found an extensive staining in the suprabasal layers of H-control organotypic cultures (Figure 3f). However, H-CYLDC/S skin equivalents only showed a very scarce involucrin staining, indicating their lack of differentiation (Figure 3g).
Cleaved caspase 3 immunostaining showed an important reduction in the number of apoptotic cells in H-CYLDC/S organotypic cultures (Figures 3i and j), compared with H-control skin equivalents (Figure 3h), suggesting that CYLDC/S confers protection against cell death by apoptosis in human keratinocytes. Remarkably, these features found in H-CYLDC/S skin equivalents, that is, loss of cell polarity, altered β-catenin distribution, lack of differentiation, presence of invasion foci and cell resistance to apoptosis, are alterations that could evolve to epithelial dysplasia and pre-malignant changes.
Generation of human A431 squamous carcinoma cells expressing increased levels of CYLDWT or the catalytically inactive CYLDC/Smutant
To investigate the role of CYLD in the progression of human skin cancer, we generated A431 cells expressing the β-actin-CYLDWT and β-actin-CYLDC/S constructs (Figure 4a; Supplementary Information SI-1a). They were designated as A-CYLDWT, A-CYLDC/S and A-control cells. We checked that the A431-CYLDC/S cells have impaired the CYLD deubiquitination activity (Supplementary Figure SI-1b).

Characterization of A-CYLDWT and A-CYLDC/S cells. (a) Western blot analysis of CYLD and Bax expression in total protein extracts from A431 cells of the three genotypes. (b) Monolayer culture of A-control, A-CYLDWT and A-CYLDC/S cells. Red circles: foci of spontaneous differentiation. Note the scattered phenotype of A-CYLDC/S cells and its loose growth in culture. (c) Immunofluorescence showing expression of K10 in A-CYLDWT cells. (d) Western blot of protein extracts from A-control, A-CYLDWT and A-CYLDC/S cells in unstimulated or after UV stimulation. Note the increase in JNK activation in A-CYLDC/S cells and the reduction in the A-CYLDWT cells
Increased CYLDWT expression in the A431 human squamous carcinoma cells induces a differentiated phenotype of the cells in vitro and an increase in cell death by apoptosis
Stratification domes were formed in conventional cultures of A-CYLDWT cells (Figure 4b), suggesting the spontaneous differentiation of the A-CYLDWT cells. The presence of K10 staining in the A-CYLDWT cells and its absence in both, A-control and A-CYLDC/S cultures, confirmed their more differentiated phenotype (Figure 4c). Additionally, abundant dead cells were found in the supernatants of A-CYLDWT cells coincident with elevated levels of expression of the pro-apoptotic protein Bax, while low expression or absence of Bax were detected in A-control and A-CYLDC/S cells, respectively (Figure 4a).
JNK activity is enhanced in A-CYLDC/S cells and inhibited in A-CYLDWTcells
We analyzed the JNK pathway and found that in the non-stimulated state, the A-CYLDC/S cells presented an enhanced JNK activity (Figure 4d). In addition, A-CYLDC/S cells also showed increased levels of P-JNK upon TNF-α stimulation. On the contrary, CYLDWT overexpression leads to a diminished JNK activation both in non-stimulated and in TNF-α-stimulated A-CYLDWT cells (Figure 4d). The fact that the overexpression of CYLD in A431 cells and its functional inactivation lead to opposite effects in JNK activation, reinforces the relevance of this signaling pathway as mediator of CYLD functions in the A431 cells and is in agreement with the results found in the HaCaT keratinocytes.
Increased CYLDWT expression in human squamous carcinoma cells improves the malignant phenotype of the SCCs while CYLDC/S enhances their aggressiveness
We performed xenograft carcinogenesis assays of the A431 cells. A-control, A-CYLDWT and A-CYLDC/S cells were injected into nude mice. The latency of macroscopic tumor appearance was similar, approximately 1 week, in the three cases (data not shown). Histologically, A-control tumors were solid SCCs and poorly differentiated (Figures 5a–c). By contrast, tumors originated from A-CYLDWT were of better prognosis as in addition to poorly differentiated areas, they exhibit extensive regions of necrosis in the core of the tumor (Figures 5d–f); moreover, we also found A-CYLDWT tumors consisting of benign cysts (Figures 5g–i). These features are compatible with the regression of the tumor. On the other hand, A-CYLDC/S tumors showed worse prognosis than A-control tumors, as they were anaplastic SCCs with high grade of cellular atypia and fusocellular morphology, resembling sarcomas or spindle-cell carcinomas (Figures 5j–k).

Hematoxilin/eosin staining of A-control, A-CYLDWT and CYLDC/S tumors. (a–c) A-control tumors are solid undifferentiated SCCs. (d–f) A-CYLDWT tumors contain large cavities as a result of tumor degeneration (arrow in d) (g–i); often they also form cystic, differentiated structures. (j–k) A-CYLDC/S SCCs are spindle-shape tumors. Compare the cords of fibroblasts-like cells forming the mutant tumors with the keratinocytes of the A-control SCCs (c). Tumors (10–12) of each type were analyzed
The immunohistochemical analysis showed increased expression of CYLD in both A-CYLDWT and A-CYLDC/S tumors compared with the expression in A-control SCCs (Figure 6). Low levels of expression of keratins K10 and K1 –reliable markers of well-differentiated tumors– were noted in A-control and A-CYLDWT SCCs, and they were not detected in A-CYLDC/S carcinomas (Figure 6 and data not shown). Keratin K8, a marker of malignancy in skin tumors26 showed extensive staining in both A-control and A-CYLDC/S carcinomas, while it was scarcely expressed in A-CYLDWT tumors (Figure 6). K5 is a keratin typical of stratified epithelial tissues, expressed by normal skin and skin tumors that are lost in the later stages of tumor progression, that is, in spindle cell carcinomas. In agreement to their spindle-like phenotype, A-CYLDC/S tumors exhibited areas lacking K5 expression (Figure 6). CD31 staining analysis of tumor blood vessels showed a network of large and lacunar vessels in A-CYLDC/S tumors while those found in A-CYLDWT were predominantly small and narrow; blood vessels developed in A-control carcinomas had an intermediate lumen diameter (Figures 7a–c). The analysis of α-smooth muscle actin (SMA), a marker of smooth muscle cells and pericytes, showed a discontinuous expression in the A-CYLDC/S tumors indicating the immature and leaky nature of the blood vessels (Figure 7f). By contrast, the vessels of the A-CYLDWT tumors were strongly stained demonstrating a mature and impermeable vascular phenotype (Figure 7d). A-control tumors presented weak SMA staining although predominantly in a continuous way (Figure 7e). Therefore, the vasculature analysis indicates an increased angionesis in A-CYLDC/S SCCs and consequently, poor prognosis. Finally we also check that A-CYLDWT tumors were prone to undergo apoptosis (SI-2).

Immunocharacterization of A-control, A-CYLDWT and A-CYLDC/S tumors. Staining with specific antibodies against CYLD, K10, K8 and K5. The increased K8 expression in the A-CYLDC/S tumors, together with lack of K5 and K10 staining are indicative of an aggressive phenotype. Tumors (10–12) of each type were analyzed

CYLDWT expression decreases tumor angiogenesis, while loss of CYLD function enhances the vascularization of skin tumors. (a–c) CD31 staining of blood vessels. Note the smaller size of the vessels in the A-CYLDWT tumors compared with that of A-CYLDC/S SCCs. (d–f) SMA staining. Representative tumors are shown in each panel. Similar data were obtained in five tumors of each genotype. Arrows in (e) indicate the blood vessels. Arrowheads in (f) mark the extremes of the blood vessels. Six tumors of each type were analyzed
Discussion
Here we describe that CYLD acts as regulator of epidermal differentiation in humans. Our data demonstrate that alterations in CYLD expression in keratinocytes disrupt normal epidermal homeostasis: the forced expression of CYLDWT in human HaCaT keratinocytes and the skin equivalents enhance keratinocyte differentiation. By contrast, the expression of a mutant CYLDC/S that inhibits the endogenous CYLD-deubiquitination function impairs keratinocyte differentiation. We have found that JNK signaling pathway could be mediating the effects of CYLD in epidermal differentiation. In fact, it was previously reported that the inhibition of JNK activation in keratinocyte cultures in vitro induces almost all aspects of epidermal differentiation in vivo, including stratification and even production of cornified envelopes.2 By contrast, augmented JNK activity delays or inhibits keratinocyte differentiation.2 In line with these results, we have found diminished JNK activation in keratinocytes expressing CYLDWT, concomitant with an enhanced epidermal differentiation of the CYLDWT skin equivalents; additionally, we have found increased JNK activation in the CYLDC/S keratinocytes associated to an impaired epidermal differentiation of the CYLDC/S skin equivalents. This finding is relevant for our understanding of both the normal epidermal differentiation and its perturbations during pathological conditions.
Importantly, we have discovered that CYLDWT also promotes the differentiation of A431 tumoral keratinocytes through inhibition of JNK activation. It may be significant clinically, because attenuating JNK activity could become a differentiation therapy-based approach for skin cancer in general and for tumors developed by familial cylindromatosis patients in particular.
Another finding of this study is the increased rate of apoptosis induced by CYLDWT both in tumoral and non-tumoral human keratinocytes. This effect could be the consequence of the lack of JNK-mediated cell survival signaling due to the diminished JNK activation in these cells.27 Our present results showing increased apoptosis in cells, and importantly, in skin tumors overexpressing CYLDWT, constitute an important finding for the identification of the mechanisms through which CYLD exerts its role as a tumor suppressor.
In addition to the impairment of keratinocyte differentiation, we have found that CYLDC/S expression provokes other alterations in skin equivalents such as the loss of cell polarity, likely due to the loss of β-catenin membranous distribution and its cytoplasmic accumulation. Maintenance of cell polarity is an essential requirement for the functionality and homeostasis of epithelial tissues, being β-catenin a key molecule in the maintenance of epithelial architecture as it regulates the polarity of cells and tissues.28 Other features of the H-CYLDC/S skin equivalents are the formation of foci of basal invasion and resistance to apoptosis. Altogether, these changes could lead to epithelial dysplasia and reflect pre-malignant modifications, such as loss of epithelial differentiation and cell polarity, and the acquisition of invasive properties are frequently observed in malignant transformation. According to these pathological alterations found in H-CYLDC/S skin equivalents, CYLDC/S expression in A431 squamous carcinoma cells induces an enhancement of their malignancy in carcinogenesis assays.
Several mechanisms have been proposed through which loss of CYLD may exert a role in tumor promotion. One of them is the hyperactivation of antiapoptotic signals in absence of functional CYLD in tumoral cells;29 previously we described that CYLDC/S induced resistance to apoptosis in mouse skin tumors;21 here we also extend this property to human skin cancer. We have also showed that CYLDC/S expression enhances angiogenesis in mouse skin carcinomas,21 what constitutes a prominent feature of skin tumor progression.30 Here we have also confirmed that in human SCCs CYLDC/S expression leads to a strong angiogenesis, increasing the size and permeability of blood vessels. Importantly, we have found as well that CYLDWT overexpression in SCCs reduces the diameter of the blood vessels and augments its maturity, which possibly make them unable to supply the nutrients needed for tumor progression.
Therefore our study is relevant because it established the role that CYLD has in normal skin physiology. The use of our skin equivalent model has allowed us to determine the importance of CYLD for the maintenance of epidermal polarity, keratinocyte differentiation and apoptosis. In addition, our results show the reversion of the malignancy of the SCCs that express increased levels of CYLDWT. The increase in apoptotic cell death of the A-CYLDWT tumors, together with the reduced angiogenesis could be responsible of the A-CYLDWT tumor regression observed in the carcinogenesis assays. Accordingly, SCCs defective in CYLD function are very aggressive as revealed by their progression toward spindle cell carcinomas, resistance to apoptosis and increased angiogenesis. These findings of the in vivo influence of CYLD in cancer prognosis make our results relevant from a therapeutic point of view, and open new avenues for exploring novel cancer therapies.
Materials and Methods
DNA constructs
CYLDC/S cDNA was kindly provided by Dr. R Bernards and subcloned under the control of β-actin promoter.21 β-actin-control construct has been described.21
Cells and culture conditions
HaCaT and A431 cells were cultured in DMEM and 10% FCS. Cells were permanently transfected using the calcium phosphate method. In all transfections, the corresponding empty vectors were used as controls. Resistant colonies were selected using G418 and pooled (each pool was formed from approximately 20 different colonies). To allow HaCaT in vitro differentiation, cells were seeded and after reaching 60–80% confluence were grown in culture medium without FCS for different days.22 Experiments were performed with 2–3 different pools of transfected cells of each genotype.
TNF-α and UV stimulation
When required, cells were treated with human TNFα (50 ng/ml; Sigma, Saint-Louis, Missouri, USA) or irradiated with UVB for the indicated times. For UVB irradiation, a Waldmann lamp (UV236B, TECNOSA, Nuevas Tecnologías SA, Barcelona, Spain)) was used. The light emitted was within the UVB range (280–320 nm), with a peak emission at 312 nm. HaCaT cells in PBS were irradiated with 800 mJ/cm2 dose of UVB for 2 min. After irradiation, the cells were put in pre-warmed medium (37°C) and collected 3 hs after irradiation.
Immunoblots
Antibodies used were: Actin, Bax, P-JNK, CYLD, tubulin (Sigma); ubiquitin (Cell Signaling Technology, Danvers, MA, USA); IKKγ/NEMO (IMGENEX, San Diego, CA, USA). For immunoprecipitation, total lysates were obtained from untreated or TNFα-treated (50 ng/ml; Sigma) HaCaT and A431 cells. A total of 300 μg of lysate were immunoprecipated with and the anti-IKKγ antibody and resolved on an SDS gel; probed with an antiubiquitin antibody, then stripped and reprobed with an antibody against IKKγ (to show the amounts of IKKγ immunoprecipitated).
Skin equivalents preparation
Human dermal fibroblasts were obtained according to Meana et al.31 To generate skin equivalents, 1.5 ml of fibrinogen solution (from cryoprecipitated pig blood) was added to 5 ml of keratinocyte growth medium containing 2.5 × 105 dermal fibroblasts. Immediately after that, 0.5 ml of 0.025 mM Cl2Ca, with 5.5 IU of bovine thrombin (Sigma-Aldrich Co., St. Louis, MO, USA) was added. The mixture was then placed on polycarbonate inserts (4 μM porous) in a six-well culture plate (Corning Costar Corp., Cambridge, MA, USA) and allowed to solidify at 37 °C for 2 h; 106 HaCaT cells were seeded on the fibrin matrix and grown to confluence. After 48 h of seeding, organotypic cultures were raised to the air–liquid interface for 6–12 days to achieve epidermal stratification.
Northern blotting
Total RNA was isolated by Trizol extraction (Gibco-BRL, San Francisco, CA, USA) following the manufacturer's instructions; 10 μg/lane RNA were probed for the expression of keratin K10.22 A ribosomal 7S RNA probe was used as a loading control.
Xenograft model of carcinogenesis
Tumors were induced in nude mice (Hsd-Athymic Nude, Harlan Europe, Barcelona, Spain) by subcutaneous flank inoculation of 106 A431 control, CYLDWT or CYLDC/S cells. In all, 6–8 mice were inoculated in both flanks with each type of cells. Tumors were harvested 18 days post-cell injection. Experimental procedures were performed according to European and Spanish laws.
Histology and immunohistochemistry
Skin equivalents were fixed in formalin and embedded in paraffin. A431 tumors were fixed in methanol/acetone (1/1); in 70% ethanol or in formalin. Sections were stained with H&E for histopathological evaluation. Cells and tissue sections were stained with antibodies against keratins K1 (Covance, San Diego, CA, USA); K8, K10, involucrin and SMA (Sigma-Aldrich); β-catenin, CD31 (BD Biosciences, San Diego, CA, USA) and cleaved caspase 3 (Cell Signaling Technology).
Immunofluorescence
Cells were fixed in methanol/acetone (1/1). Alexa Fluor-594 goat antimouse IgG(H+L) (Life Technologies SA, Paisley, UK) was used as fluorochrome.
Statistics
Statistical significance of data was assessed using the t-test and the Mann–Whitney (Wilcoxon) W test.
Abbreviations
- NMSC:
-
non-melanoma skin cancer
- BCC:
-
basal cell carcinoma
- SCC:
-
squamous cell carcinoma
References
Fuchs E . Skin stem cells: rising to the surface. J Cell Biol 2008; 180: 273–284.
Gazel A, Banno T, Walsh R, Blumenberg M . Inhibition of JNK promotes differentiation of epidermal keratinocytes. J Biol Chem 2006; 281: 20530–20541.
Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ . The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proc Natl Acad Sci USA 2004; 101: 14114–14119.
Wheelock MJ, Jensen PJ . Regulation of keratinocyte intercellular junction organization and epidermal morphogenesis by E-cadherin. J Cell Biol 1992; 117: 415–425.
Gumbiner B, Stevenson B, Grimaldi A . The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol 1988; 107: 1575–1587.
Chandrashekar C, Angadi PV, Krishnapillai R . B-catenin expression in benign and malignant salivary gland tumors. Int J Surg Pathol 2009; 24: 24.
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000; 25: 160–165.
Massoumi R, Paus R . Cylindromatosis and the CYLD gene: new lessons on the molecular principles of epithelial growth control. Bioessays 2007; 29: 1203–1214.
Almeida S, Maillard C, Itin P, Hohl D, Huber M . Five new CYLD mutations in skin appendage tumors and evidence that aspartic acid 681 in CYLD is essential for deubiquitinase activity. J Invest Dermatol 2008; 128: 587–593.
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R . Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003; 424: 797–801.
Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G . The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003; 424: 801–805.
Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G . CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003; 424: 793–796.
Reiley W, Zhang M, Sun SC . Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem 2004; 279: 55161–55167.
Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M . IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature 2001; 410: 710–714.
Seitz CS, Lin Q, Deng H, Khavari PA . Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci USA 1998; 95: 2307–2312.
Hirai Y, Kawamata Y, Takeshima N, Furuta R, Kitagawa T, Kawaguchi T et al. Conventional and array-based comparative genomic hybridization analyses of novel cell lines harboring HPV18 from glassy cell carcinoma of the uterine cervix. Int J Oncol 2004; 24: 977–986.
Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK . Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 2007; 28: 21–27.
Zhong S, Fields CR, Su N, Pan YX, Robertson KD . Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene 2007; 26: 2621–2634.
Hashimoto K, Mori N, Tamesa T, Okada T, Kawauchi S, Oga A et al. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod Pathol 2004; 17: 617–622.
Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med 2009; 206: 221–232.
Alameda JP, Moreno-Maldonado R, Navarro M, Bravo A, Ramírez A, Page A et al. An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene 2010; 29: 6522–6532.
Moreno-Maldonado R, Ramirez A, Navarro M, Fernández-Aceñero MJ, Villanueva C, Page A et al. IKKalpha enhances human keratinocyte differentiation and determines the histological variant of epidermal squamous cell carcinomas. Cell Cycle 2008; 7: 2021–2029.
Carretero M, Del Rio M, Garcia M, Escámez MJ, Mirones I, Rivas L et al. A cutaneous gene therapy approach to treat infection through keratinocyte-targeted overexpression of antimicrobial peptides. Faseb J 2004; 18: 1931–1933.
Young AR . The sunburn cell. Photodermatol 1987; 4: 127–134.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R . Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 2006; 125: 665–677.
Casanova ML, Bravo A, Martinez-Palacio J, Fernández-Aceñero MJ, Villanueva C, Larcher F et al. Epidermal abnormalities and increased malignancy of skin tumors in human epidermal keratin 8-expressing transgenic mice. Faseb J 2004; 18: 1556–1558.
Singh N, Khanna N, Sharma H, Kundu S, Azmi S . Insights into the molecular mechanism of apoptosis induced by TNF-alpha in mouse epidermal JB6-derived RT-101 cells. Biochem Biophys Res Commun 2002; 295: 24–30.
Kajino Y, Yamaguchi A, Hashimoto N, Matsuura A, Sato N, Kikuchi K . beta-Catenin gene mutation in human hair follicle-related tumors. Pathol Int 2001; 51: 543–548.
Urbanik T, Kohler BC, Boger RJ, Wörns MA, Heeger S, Otto G et al. Down-regulation of CYLD as a trigger for NF-kappaB activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int J Oncol 2010; 38: 121–131.
Bolontrade MF, Stern MC, Binder RL, Zenklusen JC, Gimenez-Conti IB, Conti CJ . Angiogenesis is an early event in the development of chemically induced skin tumors. Carcinogenesis 1998; 19: 2107–2113.
Meana A, Iglesias J, Del Rio M, Larcher F, Madrigal B, Fresno MF et al. Large surface of cultured human epithelium obtained on a dermal matrix based on live fibroblast-containing fibrin gels. Burns 1998; 24: 621–630.
Acknowledgements
We thank Dr. R Bernards for providing the CYLDC/S cDNA; I Santos and F Sánchez-Sierra for histological and immunohistochemical analyses; F Larcher for helpful discussion of the skin equivalents results; A Holguín and M García for advice in skin equivalents preparation; and CIEMAT Animal Unit for care of mice. This work was funded by grants from the Ministerio de Ciencia e Innovación PI06/1233 and PI10/01480 to MLC, and SAF2010-22156 to AR.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by V De Laurenzi
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Alameda, J., Fernández-Aceñero, M., Moreno-Maldonado, R. et al. CYLD regulates keratinocyte differentiation and skin cancer progression in humans. Cell Death Dis 2, e208 (2011). https://doi.org/10.1038/cddis.2011.82
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.82
Keywords
This article is cited by
-
Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model
Oncogenesis (2019)
-
Post-translational modification of the death receptor complex as a potential therapeutic target in cancer
Archives of Pharmacal Research (2019)
-
Dishevelled proteins and CYLD reciprocally regulate each other in CML cell lines
Molecular Biology Reports (2017)
-
Functional Inactivation of CYLD Promotes the Metastatic Potential of Tumor Epidermal Cells
Journal of Investigative Dermatology (2013)
-
Novel transglutaminase 1 mutations in patients affected by lamellar ichthyosis
Cell Death & Disease (2012)
