
Abstract
DNA interstrand cross-links (ICLs) are critical cytotoxic lesions produced by cancer chemotherapeutic agents such as the nitrogen mustards and platinum drugs; however, the exact mechanism of ICL-induced cell death is unclear. Here, we show a novel mechanism of p53-independent apoptotic cell death involving prolonged cell-cycle (G2) arrest, ICL repair involving HR, transient mitosis, incomplete cytokinesis, and gross chromosomal abnormalities resulting from ICLs in mammalian cells. This characteristic ‘giant’ cell death, observed by using time-lapse video microscopy, was reduced in ICL repair ERCC1- and XRCC3-deficient cells. Collectively, the results illustrate the coordination of ICL-induced cellular responses, including cell-cycle arrest, DNA damage repair, and cell death.
Similar content being viewed by others


Main
DNA interstrand cross-linking agents such as the nitrogen mustards, platinum drugs, chloroethylnitrosoureas, alkylalkanesuphonates, and the natural product mitomycin-C are still widely used in cancer chemotherapy.1 Bifunctional covalent modification of DNA (cross-linking) is essential for antitumour activity as monofunctional analogues are generally inactive.2 Cross-links can be formed on the same strand of DNA (intrastrand), between the opposite strands of DNA (interstrand), or between DNA and a reactive protein (DNA–protein).3 Among these, interstrand cross-links (ICLs), which normally account for a small proportion (1–10%) of total adducts, are critical for cytotoxicity4, 5 and also, if unrepaired, the efficacy of treatment6, 7, 8 by preventing any process requiring the separation of the DNA strands.9
Clinical inherent sensitivity of tumours to ICL agents may result from a defect in recognition or repair, and enhanced repair can result in clinical acquired resistance,6, 7 although the precise mechanism of ICL repair in mammalian cells has still to be fully elucidated.3, 10 Extensive genetic and biochemical studies suggest that ICL repair involves the initial incision and ‘unhooking’ of ICLs by Mus81-Eme1 and XPF-ERCC1 endonucleases,11 and subsequent processing by homologous recombination (HR)2 and translesion synthesis12 involving XRCC2, XRCC3, WRN, BRCA2, Fanconi anaemia (FA),13, 14, 15 and DNA pol-ζ16 depending on the phase of the cell cycle.17
As an ICL can lead to a stalled replication fork in S-phase,9 ICL damaged cells likely activate cell-cycle checkpoint and arrest at late S to G218 to attempt to repair the DNA damage.19 The cellular fate after the ICL-induced G2 arrest is, however, poorly understood. Cells can continuously arrest or die in G2.20 Alternatively, cells may progress through G2 with the induction of cell death in mitosis,21 or the subsequent G1 or S phase.12 The detailed mechanism and timing of ICL-induced cell death are still to be elucidated.
Cell death has an important role in the elimination of damaged cells. Necrosis and apoptosis are often observed following extensive DNA damage. While necrosis characteristically results in cell swelling and plasma membrane rupture, apoptosis produces nuclear condensation, cell shrinkage, loss of cell–cell or cell–substrate contact,22 mitochondrial release of cytochrome-c, and activation of caspase cascades leading to DNA fragmentation.23 Apoptotic-like G220 and mitotic cell death, which is linked to abnormal chromosomal segregation and failure of mitosis,21 are observed following mitotic-disrupting drugs. Escaping cell death at G2/M may result in tetraploidy24 or polyploidy.25
The aim of this study was to elucidate the mechanism of cell death resulting from ICL by the clinically used nitrogen mustard mechlorethamine (HN2). This is evaluated in relation to cell-cycle response and repair of ICL damage using DNA-repair-deficient Chinese hamster ovary (CHO) cell lines and human cervix carcinoma (HeLa) cells. The fate of cells following ICL damage is investigated by using time-lapse video microscopy. Collectively, these studies show the coordination of ICL-induced cell-cycle arrest, DNA damage repair, and cell death. Our data more fully define the mechanism of ICL-induced cell death following cell-cycle arrest and failure of ICL repair.
Results
DNA ICLs induce cell death associated with prolonged G2 cell-cycle arrest
DNA ICLs are critical cytotoxic lesions, and the number of ICLs formed increases with the concentration of bifunctional ICL agents (Supplementary Figure S1).2, 26 To investigate the relationship between cytotoxicity and cell-cycle response resulting from ICL formation, we performed clonogenic cell survival and cell-cycle analyses in wild-type CHO (AA8) cells after exposure to HN2 for 1 h. A dose-dependent decrease in cell survival was observed, with an LD50 (lethal dose, 50%) of 5 μM (Figure 1a). Moreover, a dose-dependent induction of G2 arrest at 12 h was observed in AA8 cells following exposure to HN2 (Figure 1b).

HN2-induced cell death associated with prolonged G2 cell-cycle arrest. (a) HN2 concentration-dependent induction of cell death. Percentage cell survival of wild-type CHO (AA8) cells treated with HN2 as determined by clonogenic assay. The data are represented as mean±S.E.M. (b) HN2 concentration-dependent induction of G2 cell-cycle arrest. Cell-cycle histograms (DNA content versus cell count) of AA8 cells treated with HN2 (10 μM) were acquired by flow-cytometric analysis. Cells that had undergone apoptosis and produced a sub-G1 population (floating cells) were excluded from this analysis. (c) Maintenance of G2 arrest in AA8 cells treated with HN2. BrdU cell-cycle analysis of AA8 cells treated with HN2 (10 μM). The dot plots represent incorporation of BrdU (y-axis) against DNA content (x-axis) of untreated control and HN2-treated cells. BrdU-positive cells were gated and their DNA profiles are shown. Floating cells were excluded from this analysis. (d) Difference in sensitivity to HN2 between cells in the G1 and G2 phases of the cell cycle. Hoechst-33342-stained live G1 and G2 cells were sorted according to DNA content by using a FACS Vantage cell sorter (BD BioSciences). After HN2 treatment, subsequent clonogenic cell survival of G1 and G2 cells was determined. The data are presented as mean±S.E.M.
To examine induction and prolongation of G2 arrest, we performed phase-dependent cell-cycle analysis in bromodeoxyuridine (BrdU) pulse-labelled cells by using flow cytometry following 10 μM (LD80) HN2 treatment (Figure 1c). It was observed that cells treated with HN2 during any phase of the cell cycle were subsequently arrested in G2 and this persisted for up to 48 h. HN2 treatment of cells in G1, S, and G2 induced G2 arrest but at different time points (Figure 1c). HN2-treated cells in G1 progressed through S-phase and were subsequently arrested in G2 by 24 h, which persisted up to 48 h. Cells treated in S-phase (Figure 1c, bottom) were also arrested at the subsequent G2 phase as early as 12 h after treatment and persisted up to 48 h. Interestingly, the percentage of G2 cells relative to G1 cells in BrdU-negative cell fractions at 0 h (19.4%) was slightly decreased at 6 h (16.8%) as compared with a significant increase at later time points (66.59% at 12 h, 90.58% at 24 h, and 81.9% at 48 h). This may indicate that cells in G2 at the time of HN2 treatment were not arrested in this phase and either completed the first cytokinesis or underwent cell death (Figure 1c and Supplementary Figure S2).
To determine any difference in sensitivity to ICLs depending on the cell-cycle phases, we performed Hoechst-33342 staining and live cells were sorted into G1 and late-S/G2 phases on the basis of their DNA profile at the time of HN2 treatment. Cells in late-S/G2 were approximately two-fold more resistant to HN2 than cells in G1 (Figure 1d), which suggests that the G1 cells died after the first S-phase because of the cytotoxicity induced as a result of attempted replication of cross-linked DNA. This may imply a failure to block cell-cycle progression even with unresolved DNA synthesis. By contrast, cells in G2 do not replicate their DNA until after division and therefore twice as many cells may contribute to the two-fold higher resistance of G2 cells to HN2.
Cellular fate following ICL damage
To investigate the fate of cells following ICL-induced G2 arrest, HN2-treated (10 μM) AA8 cells were observed for 72 h by time-lapse video microscopy (Figure 2). The number of HN2-treated cells was decreased by 72 h, indicating both suppression of cell division and induction of cell death (Supplementary Figure S3). Most of the HN2-treated cells (>90%) showed ‘giant’ cell formation. The HN2-treated cells did not undergo cell death in G2. Instead, they underwent pseudo-division (incomplete cytokinesis) after entry into a transient mitosis, which resulted in tetraploid (4n) G1 cell death associated with multinucleated DNA (Figure 2a). Remarkably, some giant cells progressed through mitosis and were capable of attempted cell division into two daughter cells. However, this cytokinesis was only temporary because both daughter cells were fused together and became a multinucleated, tetraploid G1 cell, which consequently resulted in tetraploid G1 cell death (Figure 2b).

Cell fate following ICL damage. (a) HN2-induced giant cell death following prolonged G2 arrest. Most of the 10-μM HN2-treated AA8 cells (red arrow) entered transient mitosis associated with incomplete cytokinesis (43 h to 45 h 55 min) and developed into giant cells containing polyploid nuclei (47 h 20 min to 56 h 20 min). This was followed by tetraploid G1 cell death (57 h 20 min to 62 h 30 min) as observed by time-lapse video microscopy. (b) HN2-induced giant cell death associated with fusion of two daughter cells. Some HN2-treated AA8 cells completed cytokinesis (43 h 25 min to 44 h 20 min) followed by cell fusion (49 h 30 min to 50 h 15 min) and consequently died in the tetraploid G1 phase (50 h 50 min to 54 h 10 min) as observed by time-lapse video microscopy. (c) HN2-treated H2B-GFP-expressing cells formed multinucleated tetraploid G1 cells following incomplete cytokinesis. Phase-contrast and/or epifluorescence images of HN2 (10 μM)-treated H2B-GFP AA8 cells observed by fluorescence video microscopy showing giant cell formation (0 h to 64 h 45 min), transient mitosis (65 h 35 min), exit from mitosis associated with incomplete cytokinesis (67 h 30 min to 68 h 20 min), and consequent formation of multinucleated tetraploid G1 cells (68 h 45 min to 71 h 55 min)
For a detailed observation of the chromosome dynamics during HN2-induced incomplete cytokinesis, we generated histone-H2B-GFP-expressing AA8 cells and continuously observed them by fluorescence video microscopy. HN2-treated cells demonstrated the formation of giant cells associated with an increase in the size of the interphase nucleus, chromosomal condensation, and transient mitosis without any evidence of proper metaphase chromosome alignment at the equator of the spindle (Figure 2c) compared with untreated control cells (Supplementary Figure S4). Thereafter, the HN2-treated cells exited from transient mitosis without showing proper anaphase characterised by synchronous chromatid separation; instead, small multi-nuclei and multinucleated G1 cells were formed after incomplete cytokinesis (Figure 2c). Therefore, after prolonged G2 arrest, the ICL-damaged cells underwent tetraploid G1 cell death associated with incomplete cytokinesis.
ICL induces apoptosis after prolonged G2 arrest
We next investigated the mechanism and the timing of giant cell death. Phosphatidylserine (PS) cell membrane externalisation, mitochondrial permeablisation, and activation of caspases were examined as typical features of apoptosis. In response to a 1-h HN2 treatment (10 μM), the number of apoptotic (Annexin-V-positive and PILow) and dead (Annexin-V-positive and PIHigh) cells were significantly increased between 24 and 72 h (Figures 3a–c). We then examined mitochondrial permeablisation by a decrease in LDS-751 intensity concomitant with a collapse in mitochondrial membrane potential. In agreement, the AA8 cells showed mitochondrial potential change (LDS-751Low and TO-PRO-3Low cells) and cell death associated with mitochondrial membrane potential collapse (LDS-751Low and TO-PRO-3High cells) between 24 and 72 h following HN2 treatment (Figures 3d–f). Moreover, activation of caspase-3 was detected at 48 h following HN2 treatment (Figures 4a and b).27 Together, these results suggest that cell death was associated with the typical morphological and biochemical features of apoptosis.

ICL-induced cell membrane externalisation and mitochondrial potential changes. PS cell membrane externalisation (Annexin-V versus PI staining) increases from 24 h in AA8 cells: Typical profiles of Annexin-V versus PI: Untreated control AA8 cells at 24 h (a) and HN2 (10 μM)-treated AA8 cells at 48 h (b) as measured by flow cytometry. The graph in panel c shows the percentages of committed cell death between 24 and 72 h after HN2 (10 μM) treatment. The fraction of Annexin-V-positive cells (%) was normalised by subtracting the number of dead cells from the number of untreated control cells. Floating cells were included in this analysis. Mitochondrial potential changes (TO-PRO-3 versus LDS-751) increase from 24 h in AA8 cells: Typical profiles of LDS-751 versus TO-PRO-3: Untreated control AA8 cells at 24 h (d) and HN2 (10 μM)-treated AA8 cells at 72 h (e) as measured by flow cytometry. The graph in panel f shows the percentages of committed cell death between 24 and 72 h after HN2 (10 μM) treatment in AA8 cells. The fraction of LDS-751Low cells (%) was normalised by subtracting the number of dead cells from the number of untreated control cells. Floating cells were included in this analysis

ICL-induced apoptosis after prolonged G2 arrest. HN2 activates caspase-3 in AA8 cells: Activation of caspase-3 was measured in untreated control AA8 cells (a) and HN2 (10 μM)-treated AA8 cells (b) at the indicated time points after treatment. Fragmentation of DNA is associated with prolonged G2 arrest in cells treated with bifunctional HN2 (LD80=10 μM) but not monofunctional HN1 (LD80=1000 μM) at equitoxic concentrations: Sub-G1 cell-cycle profiles of untreated control AA8 cells (c), HN1-treated AA8 cells (d), and HN2-treated AA8 cells (e). The fraction of cells in sub-G1 (%) was plotted against time after treatment (f). Floating cells were included in this analysis
To examine ICL-induced cell death and cell-cycle profiles, sub-G1 analysis was conducted following equitoxic monofunctional (HN1) and bifunctional (HN2) nitrogen mustard treatment. HN1 can extensively alkylate cellular DNA but can only produce monoadducts and only shows cytotoxicity at very high concentrations compared with bifunctional HN2 (the LD80 were 1000 and 10 μM for HN1 and HN2, respectively2). Although both HN1 and HN2 induced significant increases in the sub-G1 cells starting at 24 h following LD80 treatments, the cell-cycle responses between HN1- and HN2-treated cells were distinct (Figures 4c–f). The HN1-treated samples showed an accumulation of sub-G1 cells in non-phase-specific DNA fragmentation without induction of significant G2 arrest (Figure 4d). By contrast, the HN2-treated samples showed an accumulation of sub-G1 cells following G2 arrest with tetraploid (4n) DNA (Figure 4e). Taken together, these data suggest that apoptosis was induced following prolonged G2 arrest in an ICL-induced tetraploid G1 state.
Giant cells induce p53-independent apoptosis with chromosomal abnormalities
We examined interphase nuclei and metaphase chromosomes in HN2-treated AA8 cells during transient mitosis. We observed an enlargement of interphase nuclei and increased chromosomal aberrations such as chromosome crossing-over and ring structures at 48 h after HN2 treatment as compared with untreated control cells (Figure 5a). Further enlargement of the interphase nuclei and chromosomal amplification were observed at 72 h. Inhibition of cytokinesis has been reported following X-irradiation,28, 29 thus cells may override G2 the damage checkpoint following ICL damage, which may have resulted in chromosomal abnormalities and tetraploid G1 cell death associated with incomplete cytokinesis. This result also suggests that excess ICL-repair processing, such as by HR repair, participates in the formation of abnormal chromosomes and ICL-induced cell death.

p53-independent apoptosis of giant cells induced by chromosomal abnormalities. (a) ICL induces chromosomal aberrations in AA8 cells. Inverted fluorescence images of DAPI-stained giant interphase nucleus and abnormal metaphase chromosomes of untreated and HN2-treated (10 μM) AA8 cells at 48 and 72 h. Images were captured at the same magnification (objective, 100 × 10). (b) ICL induces p53-independent tetraploid G1 cell death in AA8 cells. Apoptotic Annexin-V-positive cells (left) and sub-G1 cells (right) in HN2 (10 μM)-treated vector control (vehicle) or dn-p53175 (dn-p53)-overexpressing AA8 cells. The data are presented as the means±S.E.M. (c) Hypersensitivity of HeLa cells to bifunctional HN2 as compared with monofunctional HN1. The data are presented as the mean±S.E.M. (d) HN2 induces giant cell death in HeLa cells. HN2 (1 μM)-treated H2B-GFP-expressing HeLa cells underwent giant cell death as observed by fluorescence video microscopy. (e) HN2-induced tetra/octaploid cell death in HeLa cells. The sub-G1 cell-cycle profiles of untreated cells, HN2 (1 μM)-treated cells (including both floating and adherent cells), HN2 (1 μM)-treated adherent cells, and HN2 (1 μM)-treated floating cells
p53 has been shown to be required for apoptotic cell death induced by various DNA damage.19 To investigate whether the tetraploid G1 apoptosis triggered by gross chromosomal abnormalities resulting from ICL is p53-dependent, we examined the apoptotic profiles of AA8 cells transfected with dominant-negative p53 (dn-p53)30 following HN2 treatment. Overexpression of dn-p53 did not affect the percentage of apoptotic and dead cells, as determined by Annexin-V (Figure 5b, left) and sub-G1 (Figure 5b, right) analysis. Because overexpression of dn-p53 can inhibit p53-family members (p53, p63, p73),31 these results suggest that the ICL-induced tetraploid G1 cell death observed in AA8 cells is independent of p53.
To investigate whether ICL induces giant cell death in a human cancer cell, we analysed HN1- and HN2-treated HeLa cells. The HeLa cells were sensitive to HN2, with an approximate 100-fold increased sensitivity over monofunctional HN1 (Figure 5c). Both HN1 and HN2 induced typical apoptosis in HeLa cells following exposure to equitoxic concentrations (90% growth inhibition, GI90) of these agents, as determined by fluorescence video microscopy and sub-G1 analysis (Supplementary Figure S5 and Supplementary Figure S6). Lower concentrations (GI50) of HN2-treated, H2B-GFP-expressing HeLa cells showed the formation of giant cells with enlarged nuclei and, consequently, giant cell death (Figure 5d). Cell-cycle histograms showed increasing sub-G1 peaks from 24 h after HN2 (GI50) treatment following tetraploid (4n) and octaploid (8n) states (Figure 5e).
ICL repair increases G2 arrest and chromosomal abnormalities following HN2 treatment
We investigated whether failure of ICL repair contributes to prolongation of G2 arrest, gross chromosomal abnormalities, and cell death. Equitoxic doses (LD80) of HN2 were determined to be 10, 0.8, and 0.3 μM for XPG (UV135)-, ERCC1 (UV96)-, and XRCC3 (irs1SF)-deficient cells, respectively (Figure 6a). When LD50 values were compared, XRCC3- and ERCC1-deficient cells were, respectively, 63- and 16-fold more sensitive to HN2 than their isogenic parent (AA8) cells, indicating the crucial role of HR and ERCC1 in the repair of HN2-induced DNA damage as shown previously.2, 13 By contrast, the XPG-deficient cells showed similar sensitivity to AA8 cells, suggesting that this NER protein does not have a significant role in the repair of HN2-induced ICL lesions.

Cell death associated with decreased chromosomal abnormalities and less prolongation of G2 arrest in ICL-repair-deficient cells following equitoxic HN2 treatment. (a) Hypersensitivity of ERCC1- and XRCC3-defective cells to HN2. Percentage cell survival of wild-type (AA8), XPG-deficient (UV135), ERCC1-deficient (UV96), and XRCC3-deficient (irs1SF) cell lines exposed to HN2 as determined by clonogenic assay. The data are presented as the mean±S.E.M. (b) Induction of cell death associated with decrease in G2 arrest in ERCC1- and XRCC3-deficient cells following equitoxic HN2 treatment. The sub-G1 cell-cycle profiles of untreated and HN2-treated ERCC1-deficient (0.8 μM) and XRCC3-deficient cells (0.3 μM). Floating cells were included in this analysis. (c) HN2-treated XRCC3-deficient cells induced cell death following cell division without enlargement of cells. Phase-contrast and/or epifluorescence images of H2B-GFP-expressing irs1SF cells treated with HN2 (LD80=0.3 μM) showing interphase at 17 h 35 min, prophase at 18 h 5 min, pro-metaphase at 18 h 10 min, a metaphase-like stage at 18 h 15 min, anaphase to telophase at 18 h 40 min to 18 h 45 min, interphase at 19 h 10 min, multinucleated G1 (2n) stage at 21 h 50 min to 37 h 30 min, and induction of cell death at 40 h 50 min to 46 h 40 min as observed by fluorescence video microscopy. (d) The amount of giant interphase nucleus and abnormal metaphase chromosomes observed in HN2-treated irs1SF cells decreased following equitoxic HN2 treatment. DAPI-stained inverted fluorescence images of interphase nucleus and metaphase chromosomes of untreated and HN2 (0.3 μM)-treated irs1SF cells at 48 and 72 h. Images were captured at the same magnification (objective, 100 × 10)
To determine the mechanism of ICL-induced cell death in the repair-deficient cells, we performed membrane and organelle analyses in XPG-, ERCC1-, and XRCC3-deficient, HN2-treated cells. We observed HN2-induced apoptosis between 24 and 72 h in these cell lines, along with increased PS membrane externalisation and mitochondrial permeablisation (Supplementary Figure S7). We then simultaneously examined apoptosis and cell-cycle arrest in ERCC1- and XRCC3-deficient cells following exposure to equitoxic concentrations of HN2 by sub-G1 analysis. In agreement with the membrane and organelle analyses, both ERCC1- and XRCC3-deficient cells showed an increased number of sub-G1 cells between 24 and 72 h (Figure 6b); however, G2 arrest was only transiently induced and did not persist in these cells (Supplementary Figure S8), in contrast to the wild-type cells (Figure 4e).
To investigate the fate of ICL-repair-deficient cells, we generated H2B-GFP-expressing irs1SF cells and observed them for 72 h by fluorescence video microscopy following equitoxic HN2 treatment. XRCC3-deficient cells did not show giant cell death, but cell death after cell division, followed by relatively normal chromosomal segregation compared with AA8 cells in response to equitoxic HN2 treatment (Figure 6c). Incomplete cytokinesis, which was a critical step in the formation of ICL-induced, tetraploid, multinucleated giant cells in wild-type cells (Figure 2), was attenuated in XRCC3-deficient cells. The percentage of incomplete cytokinesis relative to the number of normal cell divisions was significantly decreased in XRCC3-deficient cells as compared with the wild-type cells, as determined by a cell-tracking system between 48 and 72 h following equitoxic HN2 treatment (Supplementary Table S1 and Supplementary Figure S9).
Although both wild-type and XRCC3-deficient cells underwent apoptosis at similar time points (Figure 3 and Supplementary Figure S7), XRCC3-deficient cells showed less incomplete cytokinesis, with relatively normal cell division. We therefore investigated the chromosomal status in HN2-treated, XRCC3-deficient cells. Giant interphase nuclei were not observed in XRCC3-deficient cells at 48 and 72 h following HN2 treatment (Figure 6d). Moreover, chromosomal abnormalities were significantly decreased in XRCC3-deficient cells as compared with wild-type cells (Figures 5a and 6d). Therefore, although both cell types undergo the same amount of cell death through apoptosis and at similar time points, less giant nuclei, chromosomal abnormalities, and amplifications were observed in XRCC3-deficient cells as compared with wild-type cells.
Discussion
DNA ICL agents are widely used in cancer chemotherapy; however, an emergence of acquired drug resistance, due at least partly to ICL repair, is a major clinical problem.6, 7 In this study, we have investigated cell death resulting from ICL in mammalian cells, and a connection between cell death and ICL repair associated with prolonged G2 arrest and incomplete cytokinesis has been indicated. Cellular fate following ICL damage was revealed by using time-lapse video microscopy, and the mechanism of ICL-induced cell death was confirmed as p53-independent apoptosis. The timing of cell death suggests that it is induced after an attempt to repair the ICL damage. Moreover, the trigger of cell death was initiated after a failure of cytokinesis, associated with chromosome abnormalities resulting from incomplete or inappropriate ICL repair. By contrast, an HR repair-deficient cell line showed diminished G2 arrest, reduced giant cell formation, and a lower frequency of incomplete cytokinesis. Collectively, our results illustrate the coordination of ICL-induced cellular responses, including cell-cycle arrest, DNA damage repair, and cell death.
An investigation of the relationship between repair of DNA ICL damage, cell-cycle response, and cell death enabled the identification of a link between HR repair and ICL-induced G2 arrest in mammalian cells. ICLs are most critical in the S-phase of proliferating cells32 and are processed through an unhooking of ICLs and subsequent HR in the late S-to-G2 phases.2, 17 In agreement, our results demonstrated the hypersensitivity of ERCC1- and XRCC3-deficient cells to HN2 as determined by a colony formation assay. Moreover, the clonogenic cell-cycle analyses demonstrated ICL dose-dependent cytotoxicity accompanied by induction of G2 arrest, suggesting a connection between G2 arrest and induction of cell death. Further investigation revealed that the persistence of ICLs, rather than the initial amount formed, activated and maintained G2 arrest for more than 48 h in wild-type cells (Figure 1b).33 Importantly, XRCC3-deficient cells were severely impaired in the maintenance of G2 arrest when compared with the wild-type cells, suggesting that HR repair contributes to the maintenance of G2 arrest induced by DNA-cross-linking drugs.
The fate of the ICL damage following a prolonged G2 arrest is characterised as a multinucleated, tetraploid G1 cell death associated with incomplete cytokinesis. Activation of G2 arrest occurs as part of the repair process of ICL damage in surviving cells,19 and also occurs as part of the process that leads to cell death. Cell death following damage-induced G2 arrest has been reported to occur in the G2,20 M,21 or the subsequent G1/S phases.12 In the current study, ICL-damaged cells showed transient mitosis and a novel giant cell death in the tetraploid G1 phase following the prolonged G2 arrest. This observation highlights the importance of real-time observation using time-lapse video microscopy rather than just a single time point analysis, which can often result in the misinterpretation of cell-cycle transition events. Interestingly, this ‘giant’ cell death was not observed in XRCC3-defective cells, suggesting that ICL repair, especially by HR, participates not only in the maintenance of G2 arrest, but also in ICL-induced ‘giant’ cell death.
A comprehensive investigation into the mechanism of ICL-induced cell death was conducted. Cell death can occur as a result of multiple processes, including apoptosis, necrosis, mitotic catastrophe, and apoptotic-like cell death,20, 21 and can be determined by morphological and biochemical changes. Previous studies in mouse embryonic fibroblasts have indicated that alkylation DNA damage (both monofunctional and bifunctional) stimulates a regulated form of necrotic cell death independent of p53.34 The ICL-induced tetraploid G1 cell death observed in the current study is associated with several hallmarks of apoptotic cell death determined by four independent criteria: membrane PS externalisation, mitochondrial permeability, activation of caspases, and elevated levels of a sub-G1 phase fraction. HN2-induced ICLs form rapidly in cells, reaching a peak by 1 h.2 The timing of cell death after drug treatment was 24 h onwards and occurs after G2 arrest, suggesting that cell death is initiated after an attempt at ICL damage repair. Whereas wild-type cells showed an induction of apoptosis concomitant with prolonged G2 arrest in tetraploid (4n) cells, XRCC3-deficient cells showed an induction of apoptosis after an impaired G2 arrest in diploid (2n) cells. It is noteworthy that this type of cell death following prolonged G2 arrest was only induced by the bifunctional alkylating agent HN2 and not by the monofunctional agent HN1 at equitoxic drug concentrations, suggesting that DNA ICLs and their processing are critical for this novel apoptotic cell death.
Wild-type cells following cross-linking drug treatment possess ‘giant’ nuclei with gross chromosomal abnormalities. By contrast, XRCC3-deficient cells at equitoxic doses showed diminished formation of ‘giant’ nuclei, with fewer chromosomal abnormalities. It is therefore likely that the abnormal chromosomes that triggered ‘giant’ cell death resulted from aberrant repair of ICL damage (e.g. over-processing of ICL damage repair and/or an excess of recombination events). In CHO cells, the ICL-induced tetraploid G1 apoptosis was shown to be independent of p53-family proteins. It is notable that histone-H2B-GFP-expressing HeLa cells also demonstrated tetra/octaploid apoptosis following a prolonged cell-cycle arrest, indicating the occurrence of ‘giant’ cell death after an attempt to repair the ICL damage in human cancer cells. In fact, the multinucleated ‘giant’ cell formation and polyploid cell death could be a general hallmark of cancer,35 as well as a cellular response to various genotoxic stimuli such as UVA exposure,36 ionising radiation,37 and chemotherapy,38 and often associated with mutant p53.39 Overall, our data suggest a mechanism of ICL-induced cell death in mammalian cells (Figure 7).

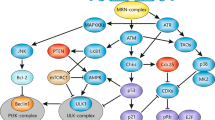
Mechanism of cell death resulting from DNA ICLs in mammalian cells. ICL-damaged cells undergo cell death through apoptosis as a result of persistence of ICLs in G1, collapse of the stalled replication fork in S, inefficient or excess ICL repair in G2, mitotic catastrophe or incomplete cytokinesis in M, and multi-nucleation in the tetraploid G1 phase. By contrast, ICL-damaged cells may survive after ICL removal in G1, translesion synthesis (TLS) in S, efficient ICL repair involving HR in G2, mitotic slippage in M, polyploidy in the tetraploid G1 phase, and after endoreduplication. The pathway indicates by arrows is the one elucidated in this study, and the pathways indicated by dotted arrows are the ones proposed in previous studies
In conclusion, the results provided here describe for the first time the occurrence of a novel mechanism of cell death following an attempt to repair ICL damage. Further investigation regarding the mechanism underlying cell death with respect to (1) ICL-induced damage-repair pathways in the regulation of the cell cycle; (2) induction of cell death following repair of ICL-induced damage; and (3) chromosomal dynamics during incomplete cytokinesis will further our understanding of the ICL-induced apoptotic cell death of cancer cells following chemotherapy. This may lead to strategies to eliminate cells that are tolerant or acquire resistance to conventional chemotherapy, and may enhance the therapeutic index in combination with newly developed antitumor agents and ultimately lead to new therapeutic strategies for the treatment of cancer.
Materials and Methods
Cell lines and culture conditions
Cells were obtained from the departmental cell bank, unless otherwise stated. Wild-type, repair-proficient (AA8), XPG-deficient (UV135), ERCC1-deficient (UV96), XRCC3-deficient (irs1SF) CHO cell lines were obtained from the departmental cell bank (Cancer Research UK Drug-DNA Interactions Research Group, London, UK). HeLa, the human cervix cancer cell line, was purchased from Cancer Research UK cell culture department (London, UK). The histone-H2B-GFP HeLa cell line was obtained from G Wahl (Salk Institute, La Jolla, CA, USA). Histone-H2B-GFP AA8, histone-H2B-GFP irs1SF and dn-p53 AA8 were generated for this study. The CHO cell lines were grown in Ham F12 nutrient medium (Sigma, Poole, UK). HeLa cells were grown in DMEM high-glucose medium (Autogen Bioclear, Calne, UK). These media were supplemented with 10% foetal bovine serum (FBS), 2 mM glutamine, and penicillin/streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. In addition, DMEM medium for culturing the H2B-GFP HeLa cells was supplemented with blasticidin (Invitrogen-Autogen Bioclear, Calne, UK).
Colony-based survival assay
Three hundred cells were plated onto petri dishes (100 mm) 24 h prior to HN2 treatment. Drug dilutions prepared fresh prior to the treatment and cells were treated in serum-free medium for 1 h then replaced in full medium. After 10–14 days, colonies were stained with 1% methylene blue and dried overnight. Colonies containing more than approximately 50 cells were counted.
Cytotoxicity assay
Cell proliferation was assessed by using the sulphorhodamine-B (SRB) assay as described previously.2
Cell-cycle analysis
Cells were harvested by 5 ml of trypsin EDTA (Autogen Bioclear) after HN2 drug treatment and washed in PBS (Sigma), then fixed in ice-cold 70% ethanol with gentle vortexing. After a minimum of 30 min at 4 °C, the cells were washed twice in PBS and resuspended in 100 μl of 100 μg/ml ribonuclease (Sigma) and 400 μl propidium iodide (PI) (50 μg/ml) (Sigma). After incubation for 30 min, samples were analysed by using a FACSCalibur (BD BioSciences, San Jose, CA, USA), with PI fluorescence being measured above 670 nm. Cell doublets and apoptotic cells were excluded from analysis.
BrdU cell-cycle analysis
Prior to drug treatment, cells were treated with 10 μM BrdU (Sigma) for 30 min in serum-containing medium. The cells were washed twice with free medium followed by drug treatment for 1 h in serum-free medium. The cells were grown in non-drug-containing medium until an appropriate time course. After harvesting, the cells were fixed in ice-cold 70% ethanol and kept at 4 °C for at least 30 min before further processing. After washing, the cells were treated with 1.5 ml of 2 M hydrochloric acid and incubated 30 min at room temperature. A 0.5-ml volume of PBS was added and acid was spun off at 2000 r.p.m. followed by washing in 0.5 ml of PBS and in 0.5 ml of PBS-T (PBS, 0.1% BSA, 0.2% Tween-20, pH 7.4). A 2-μl volume of anti-BrdU antibody (BD BioSciences, Oxford, UK) was added directly to the cell pellet and incubated for 15 min at room temperature and samples were washed twice in PBS-T. Then the samples were stained with 50 μl of FITC-conjugated rabbit anti-mouse secondary antibody (Dako, Ely, UK) at a 1 in 100 dilution for 15 min at room temperature and washed in PBS. A 50-μl volume of RNase (100 μg/ml) (Sigma) and 300–400 μl of PI (50 μg/ml) were added just before analysis. Samples were analysed on a FACSCalibur (BD BioSciences). Cell doublets and debris were excluded and FITC fluorescence was measured between 515 and 545 nm, and PI fluorescence above 670 nm.
Cell sorting and survival assay
Cell sorting was used to select cells for the clonogenic survival assay. Cells were harvested by 5 ml of trypsin EDTA while in exponential growth. To stain cellular DNA, 10 μg/ml Hoechst-33342 (bis-benzamide; Sigma) was added and incubated at 37 °C for 1 h together with different concentrations of HN2. After the treatment, the cells were sorted depending on the cell-cycle population (G1 and G2) into culture medium based on cell integrity (forward versus side scatter) and DNA profile (Hoechst-33342 staining) by using a FACS Vantage (BD BioSciences) cell sorter. For clonogenic assays, 300–5000 cells were sorted per T25 flask depending on the expected surviving fraction after HN2 treatment to keep the number of colonies per flask at approximately 200. Three replicates were used at each dose in each experiment so at least 900 cells were assessed at each dose point. After 7–10 days of incubation at 37 °C, the flasks were stained with crystal violet (1%) and colonies of greater than 50 cells were counted manually.
Phase-contrast, time-lapse video microscopy
Cells were plated on 35 mm petri dishes 24 h prior to drug treatment. The drug-containing dishes were washed and replaced with full medium and placed onto the video camera chamber in a humidified atmosphere supplied with 5% CO2 at 37 °C. Images were captured every 5–10 min using × 10 to × 20 objective lenses of an Olympus inverted microscope. Phase-contrast images were acquired by using the Kinetic Imaging software connected to a Sony CCD-IRS camera, which also controlled the shutters and the filter wheels to limit light exposure.
Fluorescence video microscopy
For monitoring living cell cultures, 5 × 104 cells were plated onto 35 mm glass-bottom culture dishes (MatTek Corporation, Ashland, MA, USA) 24 h prior to any drug treatment. Histone-H2B-GFP-expressing AA8 and irs1SF cells were treated with HN2 for 1 h in a phenol red-free and serum-free HAM F12 medium (Cancer Research UK cell culture department). After the treatment, the drug-containing media were discarded and cells were washed with a phenol red-free, serum-free HAM F12 medium, and then the cells were incubated in a phenol red-free HAM F12 medium supplemented with 10% FBS and 2 mM glutamine. For image acquisition, the culture dishes were kept in a special chamber, which was kept at 37 °C in a humidified atmosphere containing 5% CO2. Phase-contrast images of cells and/or epifluorescence images of cell nuclei were acquired on an Axiovert TV 135 microscope (Carl Zeiss, Maple Grove, MN, USA) equipped with a × 63 NA 1.4 objective lens and an Orca ER CCD camera (Hamamatsu, Photonics K.K., Hamamatsu, Japan) by using Acquisition Manager (Kinetic Imaging, Liverpool, UK).
Cell membrane analysis (Annexin-V versus PI)
Cells were treated with drug for 1 h in a serum-free medium. After the drug treatment, the cells were washed twice with serum-free medium. Thereafter, the cells were grown in non-drug-containing medium for the appropriate time period. After harvesting, both floating cells and adherent cells were collected and resuspended in 0.5 ml of Annexin binding buffer (10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2) (Pharmingen, San Diego, CA, USA) and 3 μl of Annexin-V–FITC were added. Samples were incubated in the dark at room temperature for 20 min and 50 μl of PI (50 μg/ml) were added just before analysis. The samples were analysed by using a FACSCalibur (BD BioSciences), with FITC fluorescence being measured between 515 and 545 nm, and PI fluorescence being measured above 670 nm.
Mitochondrial permeabilisation assay
During apoptosis there is often a collapse of mitochondrial membrane potential. Laser Dye Styryl-751 (LDS-751) stains activate mitochondria and can be detected by flow cytometry.40 Cells were treated with drug for 1 h in serum-free medium. After the drug treatment, the cells were washed twice with serum-free medium. Thereafter, the cells were grown in non-drug-containing medium for the appropriate time course. After harvesting, both floating and adherent cells were collected and resuspended in 0.5 ml of PBS and LDS-751 (Exciton, Dayton, OH, USA) to a final concentration of 100 nM and incubated at room temperature for 20 min, and before analysis 10 μl of TO-PRO-3 iodide (1 nM; Molecular Probes, Eugene, OR, USA) were added. Samples were analysed on a FACSCalibur (BD BioSciences), with LDS-751 being excited by a 488-nm laser and the emitted fluorescence measured between 560 and 600 nm. TO-PRO-3 was excited by a 633-nm laser and the emitted fluorescence was collected with a 660/16-nm band-pass filter.
Sub-G1 analysis
Cells were treated with drug for 1 h in serum-free medium. After the drug treatment, the cells were washed twice with serum-free medium. Thereafter, the cells were grown in non-drug-containing medium for the appropriate time course. After harvesting, both floating and adhered cells were collected and fixed in ice-cold 70% ethanol with gentle vortexing and kept for a minimum of 30 min at 4 °C. Samples were washed twice in phosphate-citrate buffer (0.2 M Na2HPO4, 0.1 M citric acid, pH 7.8) and resuspended in 100 μl of RNase (100 μg/ml) and 400 μl of PI (50 μg/ml). After incubation for 30 min, the samples were analysed by using a FACSCalibur (BD BioSciences), with PI fluorescence being measured above 670 nm. Cell doublets and aggregates were excluded from analysis.
Activation of caspase-3 determined by western blotting
Cell samples were prepared as described previously.27 Briefly, Cells were washed with PBS and lysed in ice-cold NP-40 blase buffer (2% NP-40, 2 mM NaO4Va, 50 mM NaF, 20 μg/ml Apro, 20 μg/ml leupeptine, 20 μg/ml bensamidine). Total cellular proteins (10–20 μg) were boiled in 4 × sample buffer (250 mM Tris-HCl (pH 6.8), 8% SDS, 40% glycerol, 20% β-mercaptoethanol, 1 mg/10 ml bromophenol blue) and separated on 12–15% SDS polyacrylamide gels and blotted onto nitrocellulose overnight. After the transfer, the membranes were washed in PBS and PBS-T, and incubated with a cleaved caspase-3 rabbit monoclonal antibody (Cell Signaling Technology, Beverly, MA, USA), followed by subsequent incubation with a horseradish peroxidase-conjugated anti-rabbit secondary antibody (Amersham, Little Chalfont, UK). Immunoreactive bands were visualised by enhanced chemiluminescence (Amersham).
Metaphase chromosome and interphase nucleus preparation
After 1 h of HN2 treatment, cells were incubated in medium containing 0.05 mg/ml colcemid (Gibco, Grand Island, NY, USA) at 37 °C for 1 h. The cells were treated with a hypotonic solution of 0.075 M KCl for 15 min at room temperature. The cells were then fixed three times with methanol/acetic acid (3 : 1) and metaphase preparations were made. The slides were mounted in a DAPI/Antifade mounting medium (Abbott Laboratories, Kent, UK). Spectral images were captured by using a CCD camera attached to AX10 plan microscope (Zeiss, Birmingham, UK).
Production of histone-H2B-GFP and dn-p53-expressing cells
H2B-GFP expression pCLNR-H2BG41 and a dn-p53, pBabepuro-p53175 plasmids were transfected together with a packaging pHIT60 carrying MLV gag-pol, and an envelope glycoprotein expression plasmid, pMD-G, encodes the vesicular stomatitis virus G-protein (VSV-G) under the control of the hCMV promoter. Transfection of 293T cells was conducted by using Lipofectamine (Invitrogen, Paisley, UK) as described in the manufacturer's instructions. CHO cells were plated at 1 × 105 cells per well in 24-well plates 24 h before infection. On the day of infection, the filtered supernatant was serially diluted in OptiMEM and added to the wells in the presence of 1 × 105 μg/ml polybrene (hexadimethrine bromide, Sigma). The cells were incubated overnight at 37 °C. The virus was then washed off the cells and replaced with HAM F12 containing 10% FCS. Cells expressing H2B-GFP were sorted by using a FACS Vantage (BD BioSciences). The cells were then cultured and maintained in HAM F12 medium. The dn-p53-expressing cells were selected by using a HAM F12 medium containing 10 μg/ml puromycin for 2 weeks.
Abbreviations
- CHO:
-
Chinese hamster ovary
- FA:
-
Fanconi anaemia
- GI50:
-
growth inhibition by 50%
- GI90:
-
growth inhibition by 90%
- HN1:
-
monofunctional version of mechlorethamine
- HN2:
-
mechlorethamine
- ICL:
-
interstrand cross-link
- LD80:
-
lethal dose 50%
- LD50:
-
lethal dose 80%
- PI:
-
propidium iodide
- PS:
-
phosphatidylserine
References
Hartley JA . Alkylating agents In:Souhami RL, Tannock I, Hohenberger P, Horiot J-C (eds) Oxford Textbook of Oncology. Oxford University Press: Oxford, UK, 2001, pp 639–654.
De Silva IU, McHugh PJ, Clingen PH, Hartley JA . Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 2000; 20: 7980–7990.
McHugh PJ, Spanswick VJ, Hartley JA . Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol 2001; 2: 483–490.
O’Connor PM, Kohn KW . Comparative pharmacokinetics of DNA lesion formation and removal following treatment of L1210 cells with nitrogen mustards. Cancer Commun 1990; 2: 387–394.
Sunters A, Springer CJ, Bagshawe KD, Souhami RL, Hartley JA . The cytotoxicity, DNA crosslinking ability and DNA sequence selectivity of the aniline mustards melphalan, chlorambucil and 4-[bis(2-chloroethyl)amino] benzoic acid. Biochem Pharmacol 1992; 44: 59–64.
Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG et al. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 2002; 100: 224–229.
Wynne P, Newton C, Ledermann JA, Olaitan A, Mould TA, Hartley JA . Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. Br J Cancer 2007; 97: 927–933.
Ledermann JA, Gabra H, Jayson GC, Spanswick VJ, Rustin GJS, Jitlal M et al. Inhibition of carboplatin-induced DNA interstrand crosslink repair by gemcitabine in patients receiving these drugs for platinum-resistant ovarian cancer. Clinical Cancer Res 2010; 16: 4899–4905.
Dronkert ML, Kanaar R . Repair of DNA interstrand cross-links. Mutat Res 2001; 486: 217–247.
Gottesman MM . Mechanisms of cancer drug resistance. Annu Rev Med 2002; 53: 615–627.
Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol 2004; 24: 5776–5787.
McHugh PJ, Sarkar S . DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle 2006; 5: 1044–1047.
Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1998; 1: 783–793.
Yu VP, Koehler M, Steinlein C, Schmid M, Hanakahi LA, van Gool AJ et al. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev 2000; 14: 1400–1406.
Grompe M, D’Andrea A . Fanconi anemia and DNA repair. Hum Mol Genet 2001; 10: 2253–2259.
Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008; 134: 969–980.
Hoeijmakers JH . Genome maintenance mechanisms for preventing cancer. Nature 2001; 411: 366–374.
Ben Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J . Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell 2009; 35: 704–715.
Zhou BB, Elledge SJ . The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–439.
Chen QM, Merrett JB, Dilley T, Purdom S . Downregulation of p53 with HPV E6 delays and modifies cell death in oxidant response of human diploid fibroblasts: an apoptosis-like cell death associated with mitosis. Oncogene 2002; 21: 5313–5324.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G . Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–2837.
Thompson CB . Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267: 1456–1462.
Green DR, Reed JC . Mitochondria and apoptosis. Science 1998; 281: 1309–1312.
Andreassen PR, Lohez OD, Lacroix FB, Margolis RL . Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12: 1315–1328.
Ivanov A, Cragg MS, Erenpreisa J, Emzinsh D, Lukman H, Illidge TM . Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J Cell Sci 2003; 116: 4095–4106.
Hartley JM, Spanswick VJ, Gander M, Giacomini G, Whelan J, Souhami RL et al. Measurement of DNA cross-linking in patients on ifosfamide therapy using the single cell gel electrophoresis (comet) assay. Clin Cancer Res 1999; 5: 507–512.
Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM . Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem 1999; 274: 5053–5060.
Chu K, Leonhardt EA, Trinh M, Prieur-Carrillo G, Lindqvist J, Albright N et al. Computerized video time-lapse (CVTL) analysis of cell death kinetics in human bladder carcinoma cells (EJ30) X-irradiated in different phases of the cell cycle. Radiat Res 2002; 158: 667–677.
Huang H, Fletcher L, Beeharry N, Daniel R, Kao G, Yen TJ et al. Abnormal cytokinesis after X-irradiation in tumor cells that override the G2 DNA damage checkpoint. Cancer Res 2008; 68: 3724–3732.
Tang DG, Tokumoto YM, Apperly JA, Lloyd AC, Raff MC . Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 2001; 291: 868–871.
Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G et al. Roles for p53 and p73 during oligodendrocyte development. Development 2004; 131: 1211–1220.
McHugh PJ, Sones WR, Hartley JA . Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol 2000; 20: 3425–3433.
Brozovic A, Damrot J, Tsaryk R, Helbig L, Nikolova T, Hartig C et al. Cisplatin sensitivity is related to late DNA damage processing and checkpoint control rather than to the early DNA damage response. Mutat Res 2009; 670: 32–41.
Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB . Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004; 18: 1272–1282.
Ganem NJ, Storchova Z, Pellman D . Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev 2007; 17: 157–162.
Brathen M, Banrud H, Berg K, Moan J . Induction of multinucleated cells caused by UVA exposure in different stages of the cell cycle. Photochem Photobiol 2000; 71: 620–626.
Illidge TM, Cragg MS, Fringes B, Olive P, Erenpreisa JA . Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int 2000; 24: 621–633.
Sneige N, Kemp B, Pusztai L, Asmar L, Hortobagyi GN . Chemotherapy-induced histologic changes in mastectomy specimens and their potential significance. Breast 2001; 10: 492–500.
Kasiappan R, Shih HJ, Chu KL, Chen WT, Liu HP, Huang SF et al. Loss of p53 and MCT-1 overexpression synergistically promote chromosome instability and tumorigenicity. Mol Cancer Res 2009; 7: 536–548.
Frey T . Nucleic acid dyes for detection of apoptosis in live cells. Cytometry 1995; 21: 265–274.
Kanda T, Sullivan KF, Wahl GM . Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 1998; 8: 377–385.
Acknowledgements
We thank Professor Geoffrey M Wahl for providing the pCLNR-H2BG plasmid and the H2B-GFP-HeLa cell line, and Dr. Alison C Lloyd for providing the dn-p53, pBabepuro-p53175 plasmid. We also thank Professor Denise Sheer and Ms Radost Vatcheva for support in chromosomal experiments; Ms Deborah Aubyn, Dr. Colin Gray, and Dr. Daniel Zicha for time-lapse video-microscopy experiments and analyses; Professor Parmjit S Jat for help in bacterial transformation and plasmid constructions; Professor Gerry Cohen and Dr. Satoshi Inoue, and Dr. Aya Takesono for advices in apoptotic assays; Mr Michael Woodcook for help in cell sorting clonogenic experiments and analyses; and Dr. Yasuhiro Takeuchi and Dr. Yasuhiro Ikeda for providing the pHIT60 and pMD-G plasmids and retroviral experiments. Finally, we thank Dr. Tim Hunt and Professor Martin C Raff for helpful discussions and advice. This work was supported by Cancer Research UK (C2259/A9994 to JAH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies the paper on Cell Death and Disease website )
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Osawa, T., Davies, D. & Hartley, J. Mechanism of cell death resulting from DNA interstrand cross-linking in mammalian cells. Cell Death Dis 2, e187 (2011). https://doi.org/10.1038/cddis.2011.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.70
Keywords
This article is cited by
-
PIK3R3 inhibits cell senescence through p53/p21 signaling
Cell Death & Disease (2020)
-
Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo
Cell Death & Disease (2016)
-
Senescence and aging: the critical roles of p53
Oncogene (2013)
-
Analysis of apoptosis methods recently used in Cancer Research and Cell Death & Disease publications
Cell Death & Disease (2012)
