
Abstract
After axotomy of embryonic hippocampal neurons in vitro, some of the axotomized axons lose their identity, and new axons arise and grow. This axotomy-induced axonogenesis requires importin, suggesting that some injury-induced signals are transported via axons to elicit axonogenesis after axotomy. In this study, we show that STAT3 is activated in response to axotomy. Because STAT3 was co-immunoprecipitated with importin β in the axotomized neurons, we suggest that STAT3 is retrogradely transported as molecular cargo of importin α/β heterodimers. Indeed, inhibition of importin α binding with STAT3 resulted in the attenuation of axonogenesis. Silencing STAT3 blocked the axonogenesis, demonstrating that STAT3 is necessary for axotomy-induced axonogenesis. Furthermore, the overexpression of STAT3 enhanced axotomy-induced axonogenesis. Taken together, these results demonstrate that activation and retrograde transport of STAT3 in injured axons have key roles in the axotomy-induced axonogenesis of hippocampal neurons.
Similar content being viewed by others

Main
The cell body of an injured neuron must receive accurate and timely information about axonal damage to reproduce polarization. It has been widely recognized that a dendrite is transformed into a new axon and that an injured axon regrows after axonal injury.1, 2 In addition to examining these responses, we have focused on the previously unrecognized phenomenon of a new neurite arising from a cell body after axotomy and becoming an axon.3 In approximately one-tenth of cultured rat hippocampal neurons on embryonic day (E) 18–19, new neurites arise from cell bodies and grow after axotomy (axonogenesis).3 These neurites become Tau-1 positive, and the injured axons lose immunoreactivity for Tau-1. This axotomy-induced axonogenesis is the third form of response to axonal injury and may be important for plasticity induced by axonal injury. The mechanism underlying this phenomenon has remained a significant question, however.
Because the cell bodies of injured neurons must receive the signals for axonal damage to produce new axons, we reasoned that an injury signal would be transported retrogradely to the cell body by the dynein–dynactin complex. Indeed, axonogenesis can be delayed in these neurons by inhibiting the dynein–dynactin complex through overexpression of p50.3 Importin β, which is locally translated after axotomy, associates with dynein and is required for axotomy-induced axonogenesis in hippocampal neurons.3 Importin α protein is constitutively complexed with the retrograde motor dynein; after lesioning, importin β1 mRNA localizes in the axoplasm and is rapidly translated into importin β1 protein, leading to the formation of importin α/β1 heterodimers bound to the retrograde motor dynein.4 Thus, the axoplasmic importin–dynein complex enables retrograde injury signaling in injured axons. The identification of the molecules that are retrogradely transported is expected to elucidate the mechanism of this interesting phenomenon.
Other studies have identified a number of transcriptional factors in peripheral nerve axons that might be implicated in retrograde signaling,5 some of which have been reported to interact with importins and their associated molecules for nucleocytoplasmic transport. It is tempting, therefore, to speculate that transcriptional factors interact in axons via importin–dynein-mediated transport as well. Our study showed that STAT3 was activated in response to axotomy and was required for axotomy-induced axonogenesis.
Results
STAT3 inhibitor attenuates axotomy-induced axonogenesis
We cultured hippocampal rat neurons (E18–19) at low density for 3 days and cut the axons of the stage 3 polarized neurons. We then observed morphological changes in these neurons for 12 h using time-lapse imaging. Among 65 axotomized neurons (excluding 10 neurons that died within 3 h of axotomy), axotomized axons regrew in 35 neurons. The remaining dendrites became new axons in 7 neurons, and new neurites arose from the cell bodies after axotomy, grew, and became Tau-1 positive (axonogenesis) in 8 neurons.3 This axonogenesis was specific to the axotomized neurons, because it was never observed in 62 non-axotomized neurons at the same stage.3 We assumed that some injury-induced signals would be transported with importin α/β heterodimers and dynein complex from the injury site to the cell body. Therefore, we sought to determine the signal that is transported in the axotomized neurons. STAT3,6, 7, 8, 9, 10, 11, 12 NF-κB,13, 14, 15 nuclear factor of activated T cells (NFAT),13, 16, 17, 18 and ERK19, 20 are known molecular cargoes of importin, and they also have roles in axon outgrowth and neuronal survival.
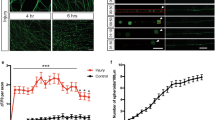
We treated axotomized neurons with membrane-permeable inhibitors AG490 (a STAT3 inhibitor), 6-amino-4-(4-phenoxyphenylethylamino) quinazoline (an inhibitor of NF-κB), U0126 (an inhibitor of ERK), and 11R-vivit (a NFAT inhibitor). AG490 efficiently inhibited axonogenesis but did not modulate the regrowth response (Figure 1a). Although the NF-κB and ERK inhibitors prevented axonogenesis, they also blocked the regrowth response (Figures 1b and c), suggesting that these agents suppress the outgrowth of processes nonspecifically. The NFAT inhibitor did not modulate axotomy-induced axonogenesis (Figure 1d). These results prompted us to focus on the role of STAT3 in axotomy-induced axonogenesis.

Inhibitor study on axotomy-induced axonogenesis Effects of signal transduction inhibitors on the responses of axotomized hippocampal neurons at 3 DIV. The graphs show the number of neurons with the indicated morphological changes. After axotomy, axonogenesis, but not regrowth of the injured axons, was suppressed by AG490 (a; n=74). Regrowth as well as axonogenesis was inhibited by treatment with NFκB inhibitor (b; n=35) or U0126 (c; n=13). No significant effect was observed with NFAT inhibitor (d; n=10)
STAT3 is activated after axotomy
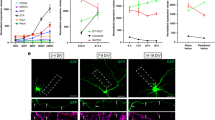
We tested whether STAT3 was activated after axotomy. The axons of the polarized neurons at 3 days in vitro (DIV) were cut and immunostained for phosphorylated STAT3 (pSTAT3) at 10 min, 1, and 3 h after axotomy. The level of pSTAT3 in the nucleus of the axotomized neurons increased significantly at 1 h after axotomy compared with that of non-axotomized neurons (Figures 2a–c). When p50 was transfected with the neurons, however, upregulation of axotomy-induced pSTAT3 in the cell bodies disappeared (Figure 2c).

STAT3 is required for axotomy-induced axonogenesis (a) Immunocytochemistry for phosphorylated STAT3 (pSTAT3) before and at 1 h after axotomy. pSTAT3 was increased in the nuclei of axotomized hippocampal neurons (arrowhead) compared with the non-axotomized neurons (asterisks). Arrows indicate the injury site, and arrowheads indicate the cell bodies of axotomized neurons. Scale bar, 50 μm. (b) The hippocampal neurons were double-immunostained for pSTAT3 (red) and DAPI (blue). pSTAT3 was expressed in the nucleus of the neurons after axotomy. Scale bar, 20 μm. (c) Relative level of pSTAT3 in the nucleus. The level of pSTAT3 in the nucleus of axotomized neurons increased significantly at 1 h after axotomy but returned to the control level 3 h after axotomy. In cells with p50 overexpression, the level of pSTAT3 in the nucleus did not increase at 1 or 3 h after axotomy (each group, n=13–15). The ratio of pSTAT3/STAT3 was calculated from the fluorescent intensities in immunocytochemistry. Data are represented as mean±S.E.M. *P<0.05. (d) Procedure for transfection of neurites from the explant. (e) Western blot analysis of lysates from axotomized hippocampal explants from 0 min to 1 h after axotomy. Lysates were obtained from the cell bodies that included the proximal axons as well as from the distal axons. (f and g) Relative changes in the level of pSTAT3. The level of pSTAT3 increased at 5 and 10 min after axotomy in both tissues. The ratio was obtained by measuring the band intensities. In the cell bodies and proximal axons (f), the increase was gradual and remained high 60 min after axotomy. In the distal axons (g), the increase peaked at 10 min after axotomy and decreased dramatically thereafter. Data are represented as the mean±S.E.M. of three independent experiments. **P<0.01; *P<0.05 compared with control
Next, we evaluated the amount of pSTAT3 in the axotomized neurons. To obtain enough protein for Western blotting, we used an explant culture of the hippocampus from E18 rats. We cut the axons of the hippocampal explants with a blade (Figure 2d) and collected the cell bodies, including the proximal axons and the distal axons (Figure 2e) at 0, 5, 10 min, and 1 h after axotomy. The phosphorylation level of STAT3 was increased after axotomy in both the cell bodies and distal axons (Figures 2e–g). The phosphorylation levels of STAT3 increased gradually up to 10 min after axotomy and remained high even at 1 h after axotomy in the cell bodies and injured the proximal axons (Figures 2e and f). In the distal axons, the level of pSTAT3 peaked 10 min after axotomy and decreased below the baseline 1 h after axotomy (Figures 2e and g). These results demonstrate that pSTAT3 increased in the axons in response to axotomy.
STAT3 is transported through injured axons with importin β
We examined the interaction between pSTAT3 and importin β in axotomized hippocampal neurons. Coimmunoprecipitation analysis revealed that pSTAT3 was associated with importin β1, and this association was increased at 1 h after axotomy (Figure 3a). We have reported that importin β increases in axons in response to axotomy and that they interact with the dynein motor complex for retrograde transport.3 These results suggest that pSTAT3 interacts with dynein–importin α/β heterodimers for retrograde transport.

STAT3 binding to importin is required for axotomy-induced axonogenesis (a) Coimmunoprecipitation of importin β1 with pSTAT3. Lysates were prepared from hippocampal explants at 1 h after axotomy. (b) Immunostaining of a hippocampal neuron at 3 DIV reveals expression of importin α5. Scale bar, 50 μm. (c) Fluorescein-TAT-protein transduction domain-STAT3 (FLC-TAT-PTD-STAT3) (210–219) was efficiently transduced in the hippocampal neurons. Scale bar, 50 μm. (d) The hippocampal neurons were double-immunostained for pSTAT3 (red) and DAPI (blue). pSTAT3 was expressed in the cytoplasm, but not in the nucleus in the neurons transduced with FLC-TAT-PTD-STAT3 (210–219). Scale bar, 30 μm. (e) Number of neurons with the indicated morphological changes. No axonogenesis occurred after axotomy in the neurons transduced with FLC-TAT-PTD-STAT3 (210–219) (n=28). TAT-PTD control neurons, n=16. (f) Number of neurons with and without axonogenesis. Axonogenesis was attenuated in neurons transduced with TAT-PTD STAT3 (210–219), but not in those transduced with TAT-PTD control. **P<0.05
We then attempted to assess whether the binding of STAT3 with importin α/β heterodimers was required for axotomy-induced axonogenesis. Because Arg214/215 of STAT3 is the binding site for importin α5,7, 21 we intended to block the binding of STAT3 with importin α5 specifically by using STAT3, a 10-amino-acid residue peptide (210–219) that includes the binding site of STAT3 with importin α5. This peptide was fused with the amino (N)-terminal protein transduction domain (11 amino acids) from the HIV protein TAT-protein transduction domain (TAT-PTD) and conjugated with fluorescein (FLC) to produce FLC-TAT-PTD-STAT3 (210–219). We used immunocytochemistry to confirm that importin α5 was expressed in rat hippocampal neurons (Figure 3b). In the dissociated culture of the hippocampal neurons, we transduced FLC-TAT-PTD-STAT3 (210–219) (Figure 3c). Immunocytochemical data revealed that the signal for pSTAT3 was absent in the nuclei of neurons transduced with FLC-TAT-PTD-STAT3 (210–219), demonstrating that the binding of STAT3 with importin α5 was blocked successfully by transduction of FLC-TAT-PTD-STAT3 (210–219) (Figure 3d). By transducing this peptide, we attenuated injury-induced axonogenesis completely (Figures 3e and f). Thus, the binding of STAT3 with importin α5 is required for axotomy-induced axonogenesis.
STAT3 is required for the axotomy-induced axonogenesis
We next investigated the role of STAT3 in axotomy-induced axonogenesis by knocking down its endogenous expression in cultured hippocampal neurons. Nucleofection with the STAT3 siRNA construct, but not with the control siRNA construct, decreased STAT3 expression in the hippocampal neurons at 3 DIV (Figure 4a). We performed nucleofection of the STAT3 siRNA construct in combination with the GFP construct to visualize the transfected neurons (Figure 4b). We transected the axons of the transfected neurons and found that neurons transfected with STAT3 siRNA did not have a significantly different number of processes (Figure 4c) or axonal length (Figure 4d) compared with those transfected with control siRNA. Thus, STAT3 is not involved in the neuritogenesis or the elongation of the processes of uninjured neurons. We observed no axonogenesis after axotomy in the 38 STAT3 siRNA-transfected neurons, however (Figures 4e and f). Thus, STAT3 appears to be necessary for axotomy-induced axonogenesis.

STAT3 is required for axotomy-induced axonogenesis. (a) Western blot analysis for the detection of STAT3. The hippocampal neurons at 3DIV were cotransfected with STAT3 siRNA or control siRNA in combination with GFP. (b) Immunostaining for STAT3 shows that the expression of STAT3 decreased in the STAT3 siRNA-transfected neurons compared to non-transfected neighboring neurons. Scale bar, 50 μm. (c) Number of neurites. Cultured hippocampal neurons were transfected with STAT3 siRNA (n=23) or control siRNA (n=20). Data are represented as mean±S.E.M. (d) Axonal length. Data are represented as mean±S.E.M. (e) Number of neurons with the indicated morphological changes. No axonogenesis occurred after axotomy in the neurons transfected with STAT3 siRNA (n=38). Control siRNA, n=17. (f) Number of neurons with and without axonogenesis. Knockdown of STAT3 inhibited axonogenesis. **P<0.01
Overexpression of STAT3 promotes axotomy-induced axonogenesis
We transfected Flag-tagged STAT3 construct in combination with GFP into the cultured hippocampal neurons. We verified that almost all of the GFP-expressing neurons also expressed Flag-tagged STAT3 (Figure 5a). Neurons overexpressing STAT3 showed no significant difference in the number of processes (Figure 5b) or in axonal length (Figure 5c) compared with those of neurons expressing GFP alone. Interestingly, axonogenesis after axotomy occurred in 12 out of 28 STAT3-overexpressed neurons, whereas the same occurred in 9 out of 52 neurons expressing GFP alone (Figures 5d and e), which indicates that axotomy-induced axonogenesis is enhanced by the overexpression of STAT3. These results demonstrate that STAT3 accelerates axotomy-induced axonogenesis.

Overexpression of STAT3 enhances axotomy-induced axonogenesis. (a) Cultured hippocampal neurons at 3 DIV were transfected with Flag-STAT3 and GFP. Scale bar, 50 μm. (b) Number of neurites. Cultured hippocampal neurons were transfected with GFP (n=25) or GFP plus Flag-STAT3 (n=28). Data are represented as mean±S.E.M. (c) Axonal length. Data are represented as mean±S.E.M. (d and e) Axonogenesis after axotomy was increased in the neurons expressing GFP plus Flag-STAT3 (n=28). GFP, n=52. *P<0.05
Discussion
In this study, we have shown that STAT3 is involved in axonogenesis after axotomy. STAT3 is activated in response to axotomy and is transported with importin β through the injured axons. STAT3 is required specifically for the axotomy-induced axonogenesis, and overexpression of STAT3 promotes this event.
Accumulated evidence suggests that STAT3 participates in the responses of injured neurons in vitro and in vivo. It has been reported previously that axotomy increases pSTAT3 in the sciatic nerve.9, 22, 23 Moreover, pSTAT3 has been shown to be transported retrogradely from an injury site to the cell body after sciatic nerve, facial nerve, and hypoglossal nerve lesioning.24 STAT3 is activated in injured sensory neurons, and the application of a JAK inhibitor attenuates the injury response in vitro.25 Further, in vivo peripheral nerve lesioning has been demonstrated to lead to a very rapid activation of STAT3 in sciatic nerve axons at the lesion site.22 This response increases during the first 24 h after injury and extends back to cell bodies over a time course consistent with that required for the retrograde transport of activated STAT3 from injury site to cell body.22 The phosphorylation of STAT3 and its translocation from cytoplasm to the nuclei of DRG neurons and motor neurons are increased after injury,9, 24 and STAT3 has been reported to interact with a number of importins.6, 8 Taken together, these findings suggest that STAT3 is retrogradely transported to elicit responses to axonal lesioning. Although STAT3 regulates a wide variety of responses in neurons, the present study is the first to demonstrate that it is involved in axonogenesis. Intriguingly, the data suggest that STAT3 is not a global regulator of axonogenesis but is specifically associated with injury-induced axonogenesis.
Axonogenesis after axotomy was specifically suppressed in neurons transfected with STAT3 siRNA, as the siRNA transfection had no effect on the number of processes or the axonal elongation of uninjured cells. STAT3 is activated in response to growth factors, cytokines, and hormones known to have a protective role after cerebral ischemia and nerve injury.26, 27, 28 Schweizer et al12 used a tissue-specific gene ablation of STAT3 in motor neurons to study the role of neural STAT3 in nerve injury in adults and reported that STAT3 contributes to motor neuron protection after injury-induced neuronal death. Naturally occurring motor neuron death during the embryonic period was not enhanced in neurofilament light chain promoter–Cre STAT3-knockout mice. Facial motor neuron survival was significantly reduced in these mice after nerve lesioning in adults, however. This result suggests that STAT3 has an important role in promoting neuronal survival after injury. Consistent with this observation, knockdown of STAT3 increased neuronal death after axotomy in our study. Because more than half of the STAT3 siRNA-transfected neurons died within 6 h after axotomy, STAT3 appears to be necessary for cells to survive axotomy. Hence, STAT3 contributes to survival and axonogenesis in axotomized neurons.
The neuropoietic cytokines IL-6, leukemia inhibitory factor (LIF), and ciliary neurotrophic factor (CNTF), which are known to be induced after ischemia and nerve injury and which exert neuroprotective and regenerative effects,27, 29, 30, 31, 32, 33 share the signaling receptor gp130. In peripheral nerves, CNTF is stored in the cytoplasm of Schwann cells and may be released by injury.34, 35, 36, 37, 38 LIF and IL-6 are present at very low levels in the nerve as well.39, 40, 41, 42 After experimental axonal injury, IL-6 was synthesized in cortical, thalamic, and hippocampal neurons.43 Although STAT3 was phosphorylated in axotomized neurons, it remains unknown which ligand phosphorylated STAT3 after axonal injury. Because we used dissociated cultures of hippocampal neurons in this study, we assumed that the injured neurons themselves produced some ligand, such as IL-6, CNTF, or LIF, that activated STAT3 or that they induced STAT3 activation independent of any ligand.
The in vivo relevance of our observations is an issue to be addressed in the future. In vivo synaptic plasticity in the formation of new circuits through collateral sprouting of injured axons is an important component of the functional recovery process in patients with central nervous system injury.44, 45 These reorganization processes possibly occur in cortical and subcortical motor areas. Whether axonogenesis occurs and has a role in recovery after central nervous system injury, such as traumatic brain injury and spinal cord injury is unknown, however. The next challenge will be to explore whether new axons arise in vivo from cell bodies of neurons after these injuries.
Materials and Methods
Dissociated cell culture
All experimental procedures were approved by the Institutional Ethics Committee of Osaka University. Hippocampal neurons obtained from Wistar rat pups on E18–19 were dissociated using trypsinization (treatment with 0.25% trypsin in PBS for 15 min at 37 °C) and then resuspended in DMEM/F12 (Invitrogen, Carlsbad, CA, USA) containing 10% FBS and triturated. Subsequently, the neurons were washed three times. The cells were suspended in DMEM/F12 containing 10% FBS, plated on poly-L-lysine- and laminin-coated dishes, and maintained at 37 °C in 5% CO2. The culture medium was replaced with serum-free DMEM/F12 supplemented with B27 (Invitrogen) at 12 h after plating, by when the cells had attached. For the immunocytochemistry experiments to detect pSTAT3, cells were suspended in DMEM/F12 containing 3% FBS, and the culture medium was replaced with serum-free DMEM/F12 supplemented with B27 at 12 h after plating because the level of pSTAT3 in hippocampal neurons is increased by serum. Where indicated, the following reagents were used at the indicated concentrations: 50 μM AG490 (a STAT3 inhibitor; Calbiochem, San Diego, CA, USA), 50 nM 6-amino-4-(4-phenoxyphenylethylamino) quinazoline (an inhibitor of nuclear factor-κB [NF-κB]; Calbiochem), 10 μM U0126 (an inhibitor of ERK; Calbiochem), 1 μM 11R-vivit (RRRRRRRRRRR-GGG-MAGPHPVIVITGPHEE, an inhibitor of NFAT; Calbiochem). Cells were pretreated with each inhibitor for 30 min before axotomy.
Explant culture
The hippocampus was removed from Wistar rat pups on E18 according to a previously reported method, with slight modification.46 The hippocampus was chopped into 300- to 600-μm pieces using fine tweezers. These pieces were placed in a 3.5-cm tissue culture dish containing DMEM/F12, 1.5 ml and supplemented with 10% FBS. After 2 days of incubation at 37 °C in a 5% CO2 incubator, the medium was replaced with DMEM/F12 supplemented with 2% B27. At 10 DIV, the extended neurites were transected with a blade, according to a previously reported method.47
Nucleofection procedure
For each transfection experiment, 4.0–5.0 × 106 cells were used with the Nucleofector II device (Amaxa Biosystems, Köln, Germany). Dissociated hippocampal neurons were spun at 800 r.p.m. for 3 min, and the medium was removed. Cells were then resuspended in 100 μl of rat neuron Nucleofector solution (Amaxa Biosystems) at RT, and 5 μg of plasmids were added, including GFP-expression vector (Amaxa Biosystems) and Flag-STAT3-C Flag pRc/CMV (Addgene, Cambridge, MA, USA) at the ratio of 1 : 10. The mixture of neurons, solution, and plasmids was transferred to a 2-mm electroporation cuvette (Amaxa Biosystems), inserted into the Nucleofector, and processed with program O-03. Immediately after transfection, 1 mL of DMEM/F12 supplemented with 10% FBS was added to the hippocampal neurons to reduce damage, and the cells were plated on poly-L-lysine- and laminin-coated dishes. The culture medium was replaced with serum-free DMEM/F12, supplemented with B27 at 3 h after plating to reduce damage to the cells. To knockdown the expression of STAT3, the neurons were cotransfected with 500 pmol of rat STAT3 siRNA (5′-CCACUCUGGUGUUUCAUAACCUCUU-3′) (Stealth Select RNAi; Invitrogen) or control siRNA (Stealth RNAi Negative Control Low GC Duplex; Invitrogen) in combination with 2.5 μg of GFP-plasmid (Amaxa Biosystems). The number of processes and axonal length of the neurons were measured at 48 h after nucleofection.
Axotomy and time-lapse imaging
The culture dish was secured in a chamber that was supplied continuously with 5% CO2 in air. The chamber was placed on an Olympus IX81 inverted phase-contrast microscope equipped with a heated stage apparatus (model MI-IBC-IF; Olympus, Tokyo, Japan). We chose 10–15 polarized hippocampal neurons in each dish at 3 DIV and cut the axons of the neurons using a 30-G needle through the microscope. Images of the axotomized neurons were acquired every 3 min for 12 h using a × 40 objective lens with a charge-coupled device video camera (Cooke, Kelheim, Germany). Images were combined into a time-lapse sequence using the MetaMorph software (Molecular Devices, Sunnyvale, CA, USA).
Transduction of TAT–PTD fusion protein
FLC—TAT–PTD (YGRKKRRQRRR)-STAT3 210–219 (LDQMRRSIVS) fusion peptide and FLC–TAT–PTD (YGRKKRRQRRR)–(SRQLVMRDIS) fusion peptide (control) were synthesized by Sigma Genosys (Ishikari, Japan). The peptides, which were purified using preparative reverse-phase HPLC, displayed >95% purity. Hippocampal neurons were incubated with 5 μmol FLC–TAT–PTD-STAT3 210–219 or FLC–TAT–PTD in serum-free DMEM/F12 supplemented with B27 for 3 h at 3 DIV. Subsequently, the neurons were washed with PBS, and the culture medium was replaced with serum-free DMEM/F12 supplemented with B27. The treated neurons were observed for 12 h after axotomy using time-lapse imaging.
Fluorescence immunostaining
Cells were fixed in 2% paraformaldehyde and 2% sucrose in 0.1 mol phosphate buffer for 20 min at RT. They were incubated with a blocking solution containing 5% BSA and 0.1% Triton-X in PBS for 1 h and overnight at 4 °C with anti-importin α5 (diluted 1 : 100 in the blocking solution; Sigma-Aldrich, St. Louis, MO, USA), anti-phospho STAT3 (Ty705) antibody (diluted 1 : 500 in the blocking solution; Cell Signaling Technology, Danvers, MA, USA), anti-STAT3 (124H6) mouse monoclonal antibody (diluted 1 : 500 in the blocking solution; Cell Signaling Technology), and anti-FLAG antibody (diluted 1 : 1000 in the blocking solution; Sigma-Aldrich). After primary antibody incubation, sections were washed three times with PBS and incubated with 1 μg/ml DAPI and fluorescent dye Alexa 488-conjugated anti-rabbit IgG or 568-conjugated anti-mouse IgG (diluted 1 : 1000 in 5% BSA in PBS; Molecular Probes/Invitrogen) at RT for 1 h. They were then observed with an Olympus IX81 inverted phase-contrast microscope.
Western blotting
Cells were lysed using a mixture containing 50 mmol Tris-HCl (pH 7.4), 150 mM NaCl, 1% Np-40, 0.1% SDS, 2 mmol EDTA, 1 mmol Na3VO4, 1 mmol NaF, and a protease inhibitor (Roche Diagnostics, Indianapolis, IN, USA). The homogenate was centrifuged at 15 000 r.p.m. for 10 min, and the supernatant was stored at −20 °C. The protein concentration was measured using a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher Scientific, Rockford, IL, USA). An equal amount of the protein was loaded into each lane, run on SDS-PAGE, and transferred to a PVDF membrane (Millipore, Billerica, MA, USA). The protein samples were boiled in sample buffer for 5 min, run on SDS-PAGE, and transferred to PVDF membranes (Millipore). The membranes were blocked for 1 h at RT with 5% BSA and incubated for 2 h at RT with anti-phospho STAT3(Ty705) antibody (diluted 1 : 1000; Cell Signaling Technology) or anti-STAT3(124H6) antibody (diluted 1 : 500; Cell Signaling Technology). HRP-conjugated secondary antibodies (diluted 1 : 1000; Cell Signaling Technology) and ECL Plus reagents (GE Healthcare, London, UK) were used for detection. The membrane was exposed to X-ray film or the LAS-3000 image system (Fujifilm, Tokyo, Japan), according to the manufacturer's specifications.
Coimmunoprecipitation assay
Rat hippocampal explants were lysed in 50 mol Tris-HCl (pH 7.5), 150 mmol NaCl, 10% glycerol, and 1% NP-40 supplemented with protease inhibitor cocktail tablets (Roche Diagnostics). The lysates were incubated on a rocking platform at 4 °C for 20 min and clarified by centrifugation at 13 000 × g at 4 °C for 10 min. The supernatants were precleared for 30 min by incubating with 60 μl of protein-G Sepharose beads (GE Healthcare). After a brief centrifugation to remove the precleared beads, the cell lysates were incubated overnight (for coimmunoprecipitation with rat hippocampal explants extracts) at 4 °C with anti-importin β antibody (Abcam, Cambridge, MA, USA). The immunocomplexes were collected for 1 h at 4 °C with protein-G Sepharose beads coated with 0.1% BSA in PBS. The beads were washed four times with lysis buffer. The bound proteins were solubilized with × 2 sample buffer and subjected to SDS-PAGE followed by immunoblotting.
Morphometric analysis
We classified the morphological changes of axotomized neurons into five groups as follows: axonogenesis, new neurites that arose from cell bodies after axotomy and grew; dendrites that changed into axons, dendrites that grew and became axons instead of axotomized axons; regrowth, axotomized axons that regrew; no change, axotomized axons that retracted or showed no morphological change; and death, neurons died within 3 h of axotomy. We performed immunocytochemistry for Tau-1 in the first set of experiments, and confirmed that all the new neurites that arose from cell bodies after axotomy and grew were axons.3
For the quantification of neurite outgrowth, images were captured on an Olympus IX81 inverted phase-contrast microscope mounted with a charge-coupled device video camera (Cooke, Leicester, UK) using a × 20 objective lens. Analysis was performed using MetaMorph software (Molecular Devices). Twenty to twenty-eight neurons per well were analyzed. Processes were defined as neurites that emerged from the cell body, and an axonal length was defined as the length of the longest neurite.
Statistical analysis
Significant differences in the data for axonogenesis after axotomy (Figures 3f, 4f and 5e) were determined using the χ2-test. Significant differences in other data were determined using Scheffe's multiple comparison tests. A P-value<0.05 was considered significant.
Abbreviations
- E:
-
embryonic day
- NFκB:
-
nuclear factor-κB
- NFAT:
-
nuclear factor of activated T cells
- DIV:
-
days in vitro
- pSTAT3:
-
phosphorylated STAT3
- TAT-PTD:
-
TAT protein transduction domain
- FLC:
-
fluorescein
- LIF:
-
leukemia inhibitory factor
- CNTF:
-
ciliary neurotrophic factor
References
Dotti CG, Banker GA . Experimentally induced alteration in the polarity of developing neurons. Nature 1987; 330: 254–256.
Gomis-Ruth S, Wierenga CJ, Bradke F . Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol 2008; 18: 992–1000.
Ohara R, Hata K, Yasuhara N, Mehmood R, Yoneda Y, Nakagawa M et al. Axotomy induces axonogenesis in hippocampal neurons by a mechanism dependent on importin β. Biochem Biophys Res Commun 2011; 405: 697–702.
Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ et al. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 2003; 40: 1095–1104.
Abe N, Cavalli V . Nerve injury signaling. Curr Opin Neurobiol 2008; 18: 276–283.
Liu L, McBride KM, Reich NC . STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA 2005; 102: 8150–8155.
Ma J, Cao X . Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell Signal 2006; 18: 1117–1126.
Ushijima R, Sakaguchi N, Kano A, Maruyama A, Miyamoto Y, Sekimoto T et al. Extracellular signal-dependent nuclear import of STAT3 is mediated by various importin alphas. Biochem Biophys Res Commun 2005; 330: 880–886.
Qiu J, Cafferty WB, McMahon SB, Thompson SW . Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci 2005; 25: 1645–1653.
Dziennis S, Jia T, Ronnekleiv OK, Hurn PD, Alkayed NJ . Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J Neurosci 2007; 27: 7268–7274.
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 2006; 12: 829–834.
Schweizer U, Gunnersen J, Karch C, Wiese S, Holtmann B, Takeda K et al. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J Cell Biol 2002; 156: 287–297.
Torgerson TR, Colosia AD, Donahue JP, Lin YZ, Hawiger J . Regulation of NF-kappa B, AP-1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-kappa B p50. J Immunol 1998; 161: 6084–6092.
Mattson MP, Culmsee C, Yu Z, Camandola S . Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem 2000; 74: 443–456.
Fagerlund R, Kinnunen L, Kohler M, Julkunen I, Melen K . NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J Biol Chem 2005; 280: 15942–15951.
Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW et al. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 1999; 401: 703–708.
Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P et al. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005; 309: 1570–1573.
Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A et al. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem 2005; 280: 2818–2825.
Whitehurst AW, Wilsbacher JL, You Y, Luby-Phelps K, Moore MS, Cobb MH . ERK2 enters the nucleus by a carrier-independent mechanism. Proc Natl Acad Sci USA 2002; 99: 7496–7501.
Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M . Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 2005; 45: 715–726.
Sato N, Tsuruma R, Imoto S, Sekine Y, Muromoto R, Sugiyama K et al. Nuclear retention of STAT3 through the coiled-coil domain regulates its activity. Biochem Biophys Res Commun 2005; 336: 617–624.
Lee N, Neitzel KL, Devlin BK, MacLennan AJ . STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J Comp Neurol 2004; 474: 535–545.
Sheu JY, Kulhanek DJ, Eckenstein FP . Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol 2000; 166: 392–402.
Schwaiger FW, Hager G, Schmitt AB, Horvat A, Hager G, Streif R et al. Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur J Neurosci 2000; 12: 1165–1176.
Liu RY, Snider WD . Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci 2001; 21: RC164.
Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T et al. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem 2005; 94: 459–468.
Leibinger M, Muller A, Andreadaki A, Hauk TG, Kirsch M, Fischer D . Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. J Neurosci 2009; 29: 14334–14341.
Komine-Kobayashi M, Zhang N, Liu M, Tanaka R, Hara H, Osaka A et al. Neuroprotective effect of recombinant human granulocyte colony-stimulating factor in transient focal ischemia of mice. J Cereb Blood Flow Metab 2006; 26: 402–413.
Lin TN, Wang PY, Chi SI, Kuo JS . Differential regulation of ciliary neurotrophic factor (CNTF) and CNTF receptor alpha (CNTFR alpha) expression following focal cerebral ischemia. Brain Res Mol Brain Res 1998; 55: 71–80.
Loddick SA, Turnbull AV, Rothwell NJ . Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18: 176–179.
Suzuki S, Tanaka K, Nogawa S, Nagata E, Ito D, Dembo T et al. Temporal profile and cellular localization of interleukin-6 protein after focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1999; 19: 1256–1262.
Herrmann O, Tarabin V, Suzuki S, Attigah N, Coserea I, Schneider A et al. Regulation of body temperature and neuroprotection by endogenous interleukin-6 in cerebral ischemia. J Cereb Blood Flow Metab 2003; 23: 406–415.
MacLaren RE, Buch PK, Smith AJ, Balaggan KS, MacNeil A, Taylor JS et al. CNTF gene transfer protects ganglion cells in rat retinae undergoing focal injury and branch vessel occlusion. Exp Eye Res 2006; 83: 1118–1127.
Dobrea GM, Unnerstall JR, Rao MS . The expression of CNTF message and immunoreactivity in the central and peripheral nervous system of the rat. Brain Res Dev Brain Res 1992; 66: 209–219.
Friedman B, Scherer SS, Rudge JS, Helgren M, Morrisey D, McClain J et al. Regulation of ciliary neurotrophic factor expression in myelin-related Schwann cells in vivo. Neuron 1992; 9: 295–305.
Rende M, Muir D, Ruoslahti E, Hagg T, Varon S, Manthorpe M . Immunolocalization of ciliary neuronotrophic factor in adult rat sciatic nerve. Glia 1992; 5: 25–32.
Sendtner M, Stockli KA, Thoenen H . Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J Cell Biol 1992; 118: 139–148.
Sendtner M, Gotz R, Holtmann B, Thoenen H . Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J Neurosci 1997; 17: 6999–7006.
Curtis R, Scherer SS, Somogyi R, Adryan KM, Ip NY, Zhu Y et al. Retrograde axonal transport of LIF is increased by peripheral nerve injury: correlation with increased LIF expression in distal nerve. Neuron 1994; 12: 191–204.
Bourde O, Kiefer R, Toyka KV, Hartung HP . Quantification of interleukin-6 mRNA in wallerian degeneration by competitive reverse transcription polymerase chain reaction. J Neuroimmunol 1996; 69: 135–140.
Marz P, Cheng JG, Gadient RA, Patterson PH, Stoyan T, Otten U et al. Sympathetic neurons can produce and respond to interleukin 6. Proc Natl Acad Sci USA 1998; 95: 3251–3256.
Reichert F, Levitzky R, Rotshenker S . Interleukin 6 in intact and injured mouse peripheral nerves. Eur J Neurosci 1996; 8: 530–535.
Hans VH, Kossmann T, Lenzlinger PM, Probstmeier R, Imhof HG, Trentz O et al. Experimental axonal injury triggers interleukin-6 mRNA, protein synthesis and release into cerebrospinal fluid. J Cereb Blood Flow Metab 1999; 19: 184–194.
Raineteau O, Schwab ME . Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2001; 2: 263–273.
Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME . The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci 2004; 7: 269–277.
Yamagishi S, Fujitani M, Hata K, Kitajo K, Mimura F, Abe H et al. Wallerian degeneration involves Rho/Rho-kinase signaling. J Biol Chem 2005; 280: 20384–20388.
Tanaka T, Ueno M, Yamashita T . Engulfment of axon debris by microglia requires p38 MAPK activity. J Biol Chem 2009; 284: 21626–21636.
Acknowledgements
This study was supported by a Grant-in-Aid for Young Scientists (S) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ohara, R., Fujita, Y., Hata, K. et al. Axotomy induces axonogenesis in hippocampal neurons through STAT3. Cell Death Dis 2, e175 (2011). https://doi.org/10.1038/cddis.2011.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.59
Keywords
This article is cited by
-
Sorting nexin 3 induces heart failure via promoting retromer-dependent nuclear trafficking of STAT3
Cell Death & Differentiation (2021)
