
Abstract
The formation of cerebral senile plaques composed of amyloid β peptide (Aβ) is a fundamental feature of Alzheimer's disease (AD). Glial cells and more specifically microglia become reactive in the presence of Aβ. In a triple transgenic model of AD (3 × Tg-AD), we found a significant increase in activated microglia at 12 (by 111%) and 18 (by 88%) months of age when compared with non-transgenic (non-Tg) controls. This microglial activation correlated with Aβ plaque formation, and the activation in microglia was closely associated with Aβ plaques and smaller Aβ deposits. We also found a significant increase in the area density of resting microglia in 3 × Tg-AD animals both at plaque-free stage (at 9 months by 105%) and after the development of A plaques (at 12 months by 54% and at 18 months by 131%). Our results show for the first time that the increase in the density of resting microglia precedes both plaque formation and activation of microglia by extracellular Aβ accumulation. We suggest that AD pathology triggers a complex microglial reaction: at the initial stages of the disease the number of resting microglia increases, as if in preparation for the ensuing activation in an attempt to fight the extracellular Aβ load that is characteristic of the terminal stages of the disease.
Similar content being viewed by others

Main
Microglial cells, which were discovered by Pio Del Rio Hortega,1 are the resident macrophages of the CNS. These cells of myeloid origin2 enter the CNS shortly after birth, and disseminate over the brain and the spinal cord, where they rapidly transform into the resting microglia with a peculiar morphological appearance.3 The resting microglial cells have small somata and multiple fine processes, with each cell occupying the defined domain that does not overlap with the neighbouring microglia. In the healthy CNS, microglial processes are on a constant move, scanning the microenvironment in their territorial domains.4, 5 Insults to the nervous system trigger a complex and multi-stage activation of microglia,6 which results in both phenotypic and functional changes.
Being the intrinsic CNS defence system, microglia are intimately involved in all forms of neuropathology,7 including various types of neurodegeneration. Alzheimer's disease (AD8) is the most common form of neurodegeneration, which results in severe and irreversible cognitive decline and dementia.9 The loss of neurones and synaptic connectivity in AD has been specifically linked to the extracellular deposition of amyloid β-peptide (Aβ), with its progressive aggregation to form insoluble amyloid plaques as well as intracellular Tau neuro-fibrillary tangles.10, 11 Microglial activation in AD has been discovered two decades ago,12 and activated microglia are generally believed to be involved in AD-associated inflammatory response,13 although the precise role of microglial cells in AD pathogenesis remains to be elucidated and characterised14, 15
Some studies have shown that activated microglia contribute to neurodegeneration in AD, either via the chronic secretion of pro-inflammatory peptides16 or by being involved in the formation of Aβ plaques.17 However, other reports have suggested a neuroprotective role of microglia in AD through, for example, Aβ clearance.15, 18 At any rate the concentration of activated microglia in the vicinity of Aβ plaques is routinely observed,19, 20, 21, 22 although the changes in the resting microglia population in AD remain unknown.
The aim of the present study was to quantify the density of resting and activated microglia in the CA1 subfield of the hippocampus in a triple transgenic mouse model (3 × Tg-AD) of AD. Developed by Oddo et al.,23, 24 3 × Tg-AD mice express mutant versions of the human presenilin 1 (PS1M146V) and tau (P301L) genes on a Tg2576 (amyloid precursor protein (APP)Swe mutant) background, resulting in the formation of both Aβ plaques and neurofibrillary tangles with a similar spatial and temporal distribution to that observed in human AD patients. We provide further evidence for the localisation of reactive microglia to amyloid plaques within CA1; in addition, we discovered a significant increase in the density of resting microglial cells, which precedes the massive activation of microglia.
Results
The populations of resting and activated microglia were analysed in the hippocampi of 3 × Tg-AD mice at three different ages, at 9, 12 and 18 months. Importantly, at 9 months of age the hippocampal tissue of these animals is virtually plaque-free, whereas the plaque load becomes substantial at 12 and 18 months.23, 25, 26 The resting microglial cells were identified by specific staining with tomato lectin,27 whereas activated microglia were stained with Mac-1 antibody raised against CD11b.28
In the dorsal hippocampus, and more specifically within the CA1 of both non-Tg and 3 × Tg-AD mice, we observed two different phenotypes of microglial cells (Figures 1 and 2). The resting, tomato-lectin immunoreactive (TL-IR) cells were characterised by a small cell body equipped with thin-to-medium ramified processes extending to the surrounding neuropil (Figure 1b and c), typical of resting microglia. Occasionally, resting microglia were also observed in the vicinity of dense deposits that were likely to be potential Aβ aggregates (data not shown). The other type of microglial cells, which were reactive for Mac-1, showed enlarged cell bodies from which processes with an enlarged and thicker appearance emanated (Figure 2d and e), thus being consistent with a reactive phenotype. Reactive microglia were predominant in 3 × Tg-AD mice (Figure 2a–c).

Visualisation and quantification of resting microglia in the hippocampi of 3 × Tg-AD animals. (a) Bar graph showing the area density of resting microglia (Sv; number/mm) in the hippocampal CA1 of 3 × Tg-AD mice and non-Tg control mice. (b, c) Brightfield micrographs showing the characteristic morphology of resting TL-IR microglia with small cell body equipped with thin to medium ramified processes extending to the surrounding neuropil in the CA1 subfield of the hippocampus of 9- and 18-month-old 3 × Tg-AD mouse, respectively, which is not modified either by age or by Aβ amyloid plaques. Scale bar=5 μm

Microglial activation in plaque-infested hippocampal tissue. (a) Bar graph showing the effect of age-related changes on MAC-1-IR reactive microglia within the CA1 subfield of the hippocampus at 9, 12 and 18 months between non-Tg control and 3 × Tg-AD mice. (b, c) Brightfield micrographs showing the increase in MAC-1-IR reactive microglia (asterisk) in 3 × Tg-AD (c) versus non-Tg control mice (b) in the hippocampal CA1. (d, e) High-magnification micrographs illustrating the characteristic morphology of reactive microglia within the CA1 subfield of the hippocampus of an 18-month-old 3 × Tg-AD mouse. Reactive microglia appear with most enlarged cell bodies from which a greater number of numerous processes emanated, but with an enlarged and thicker (arrows) appearance (f, g). Confocal images showing recruitment of MAC-1-IR microglia (green) in the vicinity of Aβ amyloid plaques (red; f, g) and aggregates (h) in the CA1 subfield of the hippocampus of an 18-month-old 3 × Tg-AD mouse. In (f) we can also observe this reactive microglia surrounding a blood vessel (BV) attaint of Aβ deposits. Scale bars: (b, c) 100 μm, (d, e) 5 μm and (f) 20 μm
Resting microglia in AD
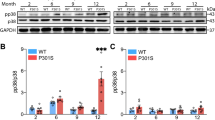
There was no difference between the morphological characteristics of TL-IR resting microglia in either 3 × Tg-AD or the respective non-Tg controls, independent of the age (Figure 1b and c). The area density (Sv, number/mm2) of resting microglia was significantly larger in 3 × TG-AD animals when compared with the control at all ages (P<0.01; Figure 1a). When the Sv was compared between 3 × Tg-AD and non-Tg controls, it was significantly larger at 9 (105%, P=0.058), 12 (54%, P=0.0198) and 18 (131%, P=0.0116; Figure 1a) months. In non-Tg animals, ageing (9–18 months) did not affect the Sv of resting microglia, whereas in 3 × Tg-AD animals we found a clear age-dependent increase in Sv between 9 and 18 months (F2,13=7.078; P=0.0106).
Activated microglia in AD
We found no change in the typical morphology of Mac-1-IR reactive microglia in all age groups in both control and 3 × Tg-AD animals. In control animals the area density of Mac-1-IR was stable at all ages, showing no significant changes at 9, 12 and 18 months (Figure 2a). In contrast, in the 3 × Tg-AD mice the area density of Mac-1-IR (total activated microglial cells) was significantly increased throughout the whole extension of CA1 (Figure 2b and c) at 12 months (111%; P=0.0311) and 18 months (88%; P=0.0330), but showed only a non-significant rise at 9 months (55.44%, P=0.1163). The Mac-1 activated microglia were almost exclusively localised in close proximity to Aβ plaques and/or aggregates (Figure 2f–h). The cell bodies of these reactive microglial cells were distributed in a circular shape around the plaque periphery (Figure 2f–h). The Mac-1-IR activated microglial cells were normally present in multiple clusters, revealing an active phagocytic activity as shown by a high degree of co-localisation with Aβ (Figure 2f, g). This phagocytic function was effective in removing the Aβ load, as shown by the presence of small packs of broken Aβ aggregates surrounded by hypertrophic and multiprocess MAC-1-IR cells (Figure 2h). Reactive microglia were also present around vascular elements that accumulated Aβ, which is typical of AD-associated angiopathy (Figure 2f).
Discussion
Although it has long been known that activated microglia are associated with compact Aβ plaques,29 their role in the development of AD is not completely understood.2, 14 Although numerous studies have attempted to characterise microglial function in AD, few have aimed to quantify the changes in the two types of microglia population (resting and reactive). In the present study, we investigated the changes in the density of microglial cells in 3 × Tg-AD mice, with particular focus on the CA1 subfield of the hippocampus. The CA1 area was shown to be highly susceptible to Aβ pathology in both human AD patients and 3 × Tg-AD mice, which is also important in the context of memory impairment. Within the hippocampus the CA1 is the major component of the Schaffer collateral pathway, acting as a neural conduit between the hippocampal subfields, subiculum and entorhinal cortex.30 As such, neuronal activity in CA1 has been strongly linked to memory formation, an aspect of cognitive function, which is typically compromised in AD.9
We found that the reactive microglia was densely populated with regions of high Aβ deposition, with the processes of many of these cells appearing to be in contact with Aβ aggregates. Some microglial cells contained an intracellular deposition of Aβ, thus indicating active phagocytosis. Similar observations were made in the brains of Tg2576 mice (expressing human mutated APPswe), in which activated microglia were concentrated in close proximity to amyloid plaques.19 Similarly, the aggregation of activated microglia around Aβ plaques was shown in vivo in 3–4-month-old double-transgenic mice expressing a mutant APP and PS1; chronic imaging experiments demonstrated that the number of plaque-associated microglia increased at the rate of about three cells per plaque per month.31
We found an increase in activated microglia in the CA1 area at 12 and 18 months, which correlates with the age of the appearance and development of Aβ plaques in the hippocampus of the 3 × Tg-AD animal model, even though in cortical areas they appear as early as at 6 months of age.23, 25, 26
We therefore suggest that the activation of microglia occurs in response to Aβ plaque formation and stabilisation. The activated microglia surrounded not only the fully formed plaques but also smaller deposits of Aβ (Figure 2h). This observation may be interpreted in either of two ways: (i) a result of plaque destruction and clearance, or (ii) the formation of small soluble aggregates before plaque consolidation. Activated microglia associated with Aβ plaques have been implicated in the phagocytosis of cerebral Aβ,15, 18 with reports indicating that these cells may be involved in the stabilisation or clearance of Aβ plaques in the brain.32 Our results corroborate these suggestions, as we found the accumulation of Aβ by activated microglia, as shown by MAC-1/Aβ double staining (Figure 2f and g). Incidentally, we did not observe a substantial activation of microglia at a pre- and/or early plaque stage at 9 months of age, indicating that microglial reactivity does not develop until the plaques are formed and/or consolidated. Similarly, a recent longitudinal study using multiphoton microscopy to image the brains of 5–6-month-old B6C3-YFP transgenic mice (bearing APPswe and PS1d9x-YFP genes) has also alluded to this, reporting that microglia are recruited to Aβ plaques only after they have been formed.33
Apart from confirming the AD-associated activation of microglia, we found the disease-specific increase in the density of resting microglial cells. To the best of our knowledge, this is the first attempt to characterise resting microglia in the AD. The immunoreactivity against tomato lectin, which is used as a specific probe for resting microglia, increased at all ages in 3 × Tg-AD animals, whereas there were no age-dependent changes in the control brains. An increase in the density of resting microglia was quite substantial – the number of cells had roughly doubled. This increase preceded both the plaque formation and the activation of microglia by extracellular Aβ accumulation. The mechanism of microglial proliferation and the origin of the additional resting microglial cells remain obscure and require further study. In particular, we cannot rule out the role for blood-borne macrophages, which have been shown to migrate across the blood–brain barrier in response to perivascular amyloid deposition, as well as the secretion of cytokines and chemoattractant peptides by microglia and astrocytes in these areas.21, 22 Upon entering the CNS, these cells function as reactive microglia, localising as phagocytes to amyloid plaques, secreting pro-inflammatory peptides and effectively accumulating Aβ.21, 34 We cannot exclude, however, that a similar infiltration can start before the active inflammatory Aβ-associated phase; at that stage, the intruding cells may transform to the resting microglia. All in all, we may suggest that AD pathology triggers a complex microglial reaction: at the initial stages of the disease the number of resting microglia increases, as if in preparation for the ensuing activation in an attempt to fight the extracellular Aβ load that is characteristic of the terminal stages of the disease.
An increase in the density of resting microglia associated with atrophic changes in astrocytes26 may also lead to a remodelling of the glial-related domain structure of the grey matter (Figure 3). This in turn can affect synaptic connectivity, because of both reduced astroglial coverage and the possible synaptolitic effects of multiplied microglia. Thus, early changes of neuroglia can represent a pathologically relevant step in the cognitive decline observed at the early stages of AD.

Glial reactions at the different stages of Alzheimer's disease. At the early, A plaque-free stages of AD, an increase in the density of resting microglia coincides with the initial atrophy of astrocytes.26 Consolidation of plaques at the later stages of AD triggers rapid and massive activation of microglia, reactive astrogliosis and neurodegeneration
Several therapeutic strategies have attempted to target microglia activity as a means of treating AD, each with limited success. Of these, immunotherapy protocols aimed to up-regulate the microglial clearance of cerebral Aβ,18, 35 whereas an alternative strategy used anti-inflammatory agents to reduce microglial neurotoxicity.36 These attempts reflect the well-known dichotomy of microglial function, which may be both neuroprotective and neurotoxic.2 Nonetheless, without a comprehensive understanding of microglial changes and roles in AD, and especially the balance between different stages of microglial activation, these attempts at therapeutic intervention may continue to be futile. Furthermore, an early increase in resting microglia, which precedes the Aβ plaque formation, may also have certain diagnostic significance.
Materials and Methods
All animal procedures were carried out in accordance with the United Kingdom Animals (for Scientific Procedures) Act of 1986 under the license from the Home Office. All efforts were made to reduce the number of animals by following the 3R's.
Mice
The procedure for generating 3 × Tg-AD mice has been described previously.23, 24 Briefly, human APP cDNA harbouring the Swedish mutation (KM670/671NL) and human P301L mutated four-repeat Tau were co-microinjected into single-cell embryos of homozygous PS1M146V knock-in mice. The background of the PS1 knock-in mice is a hybrid 129/C57BL6. The control mice used were also from the same strain and genetic background as the PS1 knock-in mice, but they express the endogenous wild-type mouse PS1 gene. All 3 × Tg-AD and control mice were obtained by crossing homozygous breeders. Mice were independently group housed and kept on a daily 12-h light–dark cycle dark schedule. All mice were given ad libitum access to food and water.
Fixation and tissue processing
Male 3 × Tg-AD and their respective non-transgenic controls (non-Tg) of different ages (9, 12 and 18 months of age; n=3–5) were anaesthetised with an intraperitoneal injection of sodium pentobarbital. Brains were fixed by perfusion through the aortic arch with 3.8% acrolein (TAAB, UK) in a solution of 2% paraformaldehyde and 0.1 M phosphate buffer (PB), pH 7.4, followed by 2% paraformaldehyde. The brains were removed from the cranium and cut into 4–5 mm coronal slabs of tissue containing the entire rostrocaudal extent of the SVZ. These slabs were subsequently post-fixed for 30 min in 2% paraformaldehyde before sectioning at 40–50 μm on a vibrating microtome (VT1000, Leica, Milton Keynes, UK). To remove excess reactive aldehyde groups, sections were treated with 1% sodium borohydride in PB for 30 min. Coronal vibratome sections at levels −1.58 mm/−2.46 mm (hippocampus) posterior to the bregma were selected for immunohistochemistry according to the mouse brain atlas of Paxinos and Franklin.37
Antibody and reagents
A polyclonal affinity-purified rat antiserum raised against CD11b (Mac-1; Serotec, Kidlington, UK) was used for the determination of activated microglial cells. A Mac-1 antibody was used to selectively label activated microglia in our brain sections. Mac-1 has a specific affinity to the microglial surface membrane receptor CD11b, which is highly expressed by reactive microglia,28 and is therefore routinely used as a specific marker of reactive microglia. Although we identified a small amount of low-intensity labelling for resting microglia in our tissue sections, the staining was observed to be largely specific for reactive microglia. For the identification of β-amyloid (Aβ) plaques, we used a monoclonal mouse antiserum that reacts with abnormally processed isoforms, as well as precursor forms of Aβ, recognizing an epitope within amino acids 3–8 (EFRHDS; anti-Aβ 6E10 [SIG-39320]; Signet Laboratories, Dedham, MA, USA). To assess for non-specific background labelling or cross-reactivity between antibodies derived from different host species, a series of control experiments were performed. Omission of primary and/or secondary antibodies from the incubation solutions resulted in a total absence of target labelling (data not shown). Resting microglia were identified by using biotinylated tomato lectin (Sigma-Aldrich, Poole, UK). Tomato lectin is a sugar-binding glycoprotein that has a specific affinity for poly-N-acetyl lactosamine residues occurring on the surface membranes of microglia and endothelial cells.27
Immunohistochemistry
To optimise the detection of Mac-1 and tomato lectin (resting microglia), sections through the hippocampus of all control and transgenic animals were processed by using the highly sensitive avidin–biotin peroxidise complex method.38 For this procedure, vibratome sections were first incubated for 30 min in 0.5% bovine serum albumin in 0.1 M Tris-buffered saline (TBS), pH 7.6, containing 0.25% Triton X-100 to minimise non-specific labelling. The tissue sections were then incubated for 48 h at room temperature in 0.1% bovine serum albumin in TBS containing 0.25% Triton X-100 that in turn contained either rat anti-Mac-1 (1 mg/ml) or biotinylated tomato lectin (2 mg/ml). Briefly, following repeated washing in TBS, sections for Mac-1 were incubated in 1:200 dilutions of biotinylated donkey anti-rat IgG followed by avidin-biotin complex (Elite kit; Vector Laboratories, Peterborough, UK). For tomato lectin identification the sections were directly incubated in the avidin-biotin complex. The peroxidase reaction product was visualised by incubation in a solution containing 0.022% 3,3′-diaminobenzidine (Aldrich, Gillingham, UK) in TBS containing 0.003% H2O2 for 6 min.
For the detection and determination of Mac-1-positive cells and their relationship with Aβ senile plaques, we used dual indirect immunofluorescence labelling. The sections were incubated for 48 h at room temperature in a primary antibody cocktail containing (1) mouse anti-β amyloid monoclonal antibody (Aβ; 1:2000; Covance, Emeryville, CA, USA) and (2) rat anti-Mac-1 (1:30.000; Sigma, Saint Louise, MO, USA) simultaneously. Subsequently, Aβ and Mac-1 were detected in a sequential manner on the same sections by incubation with rhodamine (TRITC)-conjugated goat anti-mouse and FITC-conjugated goat anti-rat (Invitrogen, Paisley, UK) IgG, respectively. Finally, sections were rinsed with 0.1 M TS for 30 min and permanently mounted in an aqueous medium (Vectashield; Vector Laboratories).
Cell counting
The Sv (number/mm2) of Mac-1 (Mac-1-IR) and TL-IR cells was determined from four non-consecutive coronal sections, separated by at least 80 mm, which were taken from representative sections of the dorsal hippocampus. The ImageJ 1.41 image processing software was used to outline and determine the area of CA1, by performing the quantification; then the number of either Mac-1-IR or TL-IR was determined. Cells were counted and images obtained using a Nikon E-80i widefield microscope and a confocal microscope (Leica SP5 upright). To ensure consistency and reproducibility, samples were counted blindly.
Statistical analysis
Data were expressed as mean±S.E. Analysis of variance (one-way) was used to examine differences in the mean number of Mac-1-IR and TL-IR cells in the 3 × Tg-AD and non-Tg animals during ageing, followed by a Bonferroni's post-hoc test where appropriate. Whenever two groups were compared, an unpaired t-test was applied.
Conflict of interest
The authors declare no conflict of interest.
Abbreviations
- 3xTg-AD:
-
triple-transgenic mice model of AD
- AD:
-
Alzheimer's disease
- Aβ:
-
amyloid β peptide
- APP:
-
Amyloid precursor protein
- CNS:
-
central nervous system
- CA1:
-
CA1 (Cornu Ammonis 1) region of hippocampus
- TL:
-
tomato-lectin
- Tg2576:
-
transgenic mice, expressing (expressing human mutated APPswe)
References
Del Rio-Hortega P . El tercer elemento de los centros nerviosos. I. La microglia en estado normal. II. Intervencíon de la microglia en los procesos patológicos. III. Naturaleza probable de la microglia. Bol de la Soc Esp de Biol 1919; 9: 69–120.
Ransohoff RM, Perry VH . Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 2009; 27: 119–145.
Kreutzberg GW . Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996; 19: 312–318.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8: 752–758.
Nimmerjahn A, Kirchhoff F, Helmchen F . Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308: 1314–1318.
Hanisch UK, Kettenmann H . Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007; 10: 1387–1394.
Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A . Glia: the fulcrum of brain diseases. Cell Death Differ 2007; 14: 1324–1335.
Alzheimer A . Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiat Psych-Gericht Med 1907; 64: 146–148.
Walsh DM, Selkoe DJ . Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004; 44: 181–193.
Selkoe DJ . Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 2001; 81: 741–766.
Selkoe DJ . Alzheimer's disease is a synaptic failure. Science 2002; 298: 789–791.
McGeer PL, Itagaki S, Tago H, McGeer EG . Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987; 79: 195–200.
Heneka MT, O’Banion MK . Inflammatory processes in Alzheimer's disease. J Neuroimmunol 2007; 184: 69–91.
Streit WJ . Microglia and Alzheimer's disease pathogenesis. J Neurosci Res 2004; 77: 1–8.
Hickman SE, Allison EK, El Khoury J . Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008; 28: 8354–8360.
Zilka N, Ferencik M, Hulin I . Neuroinflammation in Alzheimer's disease: protector or promoter? Bratisl Lek Listy 2006; 107: 374–383.
Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M . Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 1999; 154: 1673–1684.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000; 6: 916–919.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998; 152: 307–317.
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 2008; 28: 11650–11661.
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S . Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 2006; 49: 489–502.
Wegiel J, Imaki H, Wang KC, Wronska A, Osuchowski M, Rubenstein R . Origin and turnover of microglial cells in fibrillar plaques of APPsw transgenic mice. Acta Neuropathol 2003; 105: 393–402.
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM . Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 2003; 24: 1063–1070.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular A and synaptic dysfunction. Neuron 2003; 39: 409–421.
Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One 2008; 3: e2935.
Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A . Astroglia in dementia and Alzheimer's disease. Cell Death Differ 2009; 16: 378–385.
Acarin L, Vela JM, Gonzalez B, Castellano B . Demonstration of poly-N-acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J Histochem Cytochem 1994; 42: 1033–1041.
Solovjov DA, Pluskota E, Plow EF . Distinct roles for the α and subunits in the functions of integrin αMβ2 . J Biol Chem 2005; 280: 1336–1345.
McGeer PL, McGeer EG . The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 1995; 21: 195–218.
Amaral DG, Witter MP . The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience 1989; 31: 571–591.
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 2008; 28: 4283–4292.
Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW et al. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci 2001; 21: 858–864.
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 2008; 451: 720–724.
Boche D, Nicoll JA . The role of the immune system in clearance of A from the brain. Brain Pathol 2008; 18: 267–278.
Schenk D . Amyloid-beta immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci 2002; 3: 824–828.
Gasparini L, Ongini E, Wenk G . Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer's disease: old and new mechanisms of action. J Neurochem 2004; 91: 521–536.
Franklin KBJ, Praxinos G . The Mouse Brain in Stereotaxic Coordinates. Elsevier: Amsterdam, 2007.
Hsu SM, Raine L, Fanger H . Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 1981; 29: 577–580.
Acknowledgements
This research was supported by the Alzheimer's Research Trust (UK) Programme Grant ART/PG2004A/1 to AV and JJR; and the Grant Agency of the Czech Republic GACR 309/09/1696 to JJR and GACR 305/08/1384 to AV. We thank Chia-Yu Yeh for her technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Melino
Rights and permissions
This article is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rodríguez, J., Witton, J., Olabarria, M. et al. Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer's disease. Cell Death Dis 1, e1 (2010). https://doi.org/10.1038/cddis.2009.2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2009.2
Keywords
This article is cited by
-
An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease
Nature Communications (2023)
-
Different phenotypes of microglia in animal models of Alzheimer disease
Immunity & Ageing (2022)
-
Supercritical fluid chromatography-mass spectrometry enables simultaneous measurement of all phosphoinositide regioisomers
Communications Chemistry (2022)
-
An altered glial phenotype in the NL3R451C mouse model of autism
Scientific Reports (2020)
-
Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling
Journal of Neuroinflammation (2019)
