
Abstract
Background:
This phase I study assessed the maximum tolerated dose, dose-limiting toxicity (DLT) and pharmacokinetics of belinostat with carboplatin and paclitaxel and the anti-tumour activity of the combination in solid tumours.
Methods:
Cohorts of three to six patients were treated with escalating doses of belinostat administered intravenously once daily, days 1–5 q21 days; on day 3, carboplatin (area under the curve (AUC) 5) and/or paclitaxel (175 mg m−2) were administered 2–3 h after the end of the belinostat infusion.
Results:
In all 23 patients received 600–1000 mg m−2 per day of belinostat with carboplatin and/or paclitaxel. No DLT was observed. The maximal administered dose of belinostat was 1000 mg m−2 per day for days 1–5, with paclitaxel (175 mg m−2) and carboplatin AUC 5 administered on day 3. Grade III/IV adverse events were (n; %): leucopenia (5; 22%), neutropenia (7; 30%), thrombocytopenia (3; 13%) anaemia (1; 4%), peripheral sensory neuropathy (2; 9%), fatigue (1; 4%), vomiting (1; 4%) and myalgia (1; 4%). The pharmacokinetics of belinostat, paclitaxel and carboplatin were unaltered by the concurrent administration. There were two partial responses (one rectal cancer and one pancreatic cancer). A third patient (mixed mullerian tumour of ovarian origin) showed a complete CA-125 response. In addition, six patients showed a stable disease lasting ⩾6 months.
Conclusion:
The combination was well tolerated, with no evidence of pharmacokinetic interaction. Further evaluation of anti-tumour activity is warranted.
Similar content being viewed by others


Main
Aberrant gene transcription is one of the hallmarks of cancer. Transcriptional regulation is controlled by an interaction of many processes including histone tail modification (Grunstein, 1997). The acetylation and deacetylation of histone tails is achieved by competing the activity of histone acetyl transferases and histone deacetylases (HDACs) (Grunstein, 1997). HDAC enzymes catalyse the removal of acetyl groups from lysine residues of histone proteins, compact chromatin and thus repress the transcription of associated genes. Deregulation in the expression of HDAC enzymes has been implicated in the development of cancers (Marks et al, 2001), and as such, HDAC enzymes represent a potential therapeutic target. HDAC inhibitors can induce apoptosis, cell cycle arrest and cellular differentiation, as well as inhibit angiogenesis (Marks and Dokmanovic, 2005; Bolden et al, 2006b). In addition, HDAC inhibitors have been shown to affect multiple pathways involved in oncogenesis through acetylation of non-histone proteins including heat shock protein-90, tubulin and hypoxia-inducible factor-1α (Bolden et al, 2006b; Schemies et al, 2009). A variety of HDAC inhibitors have already shown some anti-tumour activity in both pre-clinical and clinical settings (Bolden et al, 2006b).
Belinostat (PXD101) is a low molecular weight HDAC inhibitor of the hydroxamate class, having nanomolar activity against both class I and II recombinant HDAC isoforms with the advantage of both inducing histone acetylation and tubulin acetylation (Khan et al, 2008). It shows growth-inhibitory effects in a variety of human tumour cell lines in vitro, and human tumour xenografts in vivo (Plumb et al, 2003). This in vitro and in vivo growth inhibition was associated with a marked increase in the levels of acetylated histone proteins (Plumb et al, 2003). In addition, belinostat exhibited additive to synergistic when combined with paclitaxel, docetaxel and carboplatin both in vitro and in vivo (Qian et al, 2006), the postulated mechanism being through belinostat enhancing the acetylation of tubulin induced by docetaxel and phosphorylation of H2AX (variant of the histone H2A family) induced by carboplatin.
In two phase I studies of belinostat in solid (Steele et al, 2008) and haematological tumours (Gimsing et al, 2008), the treatment was well tolerated at the maximum tolerated dose (MTD) of 1000 mg m−2 per day administered intravenously (i.v.), q21 days. The dose-limiting toxicities (DLT) in the solid tumour study included fatigue, diarrhoea and atrial fibrillation, whereas none were seen in the study of haematological tumours. Importantly, no significant myelosuppression was observed.
On the basis of (i) the hypothesis that belinostat shows additive-to-synergistic activity when combined with commonly used cytototoxic agents including platinums (Pts) and taxanes and (ii) the lack of overlapping toxicity with these agents, this phase I study was designed to determine the MTD, DLT and the pharmacokinetic (PK) profile of belinostat, carboplatin and paclitaxel (BelCaP). Furthermore, it planned to explore the anti-tumour activity of the combination in solid tumours and in an MTD expansion of patients with relapsed advanced ovarian cancer.
Materials and methods
Eligibility
Patients with histological or cytological confirmation of advanced solid malignancy refractory to standard therapy were eligible, provided that they met the following criteria: age ⩾18 years; Eastern Co-operative Oncology Group performance status ⩽2; adequate bone marrow, hepatic and renal function (neutrophils >1.0 × 109 l−1, platelets >100 × 109 l−1, total bilirubin ⩽1.5 × upper limit of normal, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase ⩽3 × upper limit of normal (or ⩽5 × upper limit of normal if liver metastases) and calculated creatinine clearance ⩾45 ml min−1); female patients of reproductive potential were required to have a negative pregnancy test.
Patients were excluded if they had co-existing significant medical conditions including prolonged QTc >500 ms; use of concomitant medication that may cause torsade de Pointes; neuropathy ⩾grade II because of previous treatment; or had received >3 previous lines of chemotherapy for metastatic disease.
The study protocol was approved by the institutional ethics committee of the three sites participating in the study and all patients gave written informed consent before any study procedures were performed. The study was conducted in line with Good Clinical Practice in accordance with the Declaration of Helsinki and its amendments.
Study design, objectives and treatment
This was an open-label phase I study with the primary objectives of determining the safety, DLT and MTD of BelCaP. Secondary objectives were to define the PK interaction between the agents as well as define preliminary anti-tumour activity.
Belinostat was administered in escalating doses of 600, 800 and 1000 mg m−2 per day as a 30-min i.v. infusion on days 1–5 q21. Carboplatin was administered as a 30-min i.v. infusion at area under the curve (AUC) 5 on day 3 q21 days, whereas paclitaxel was administered at 175 mg m−2 i.v. over 3 h. Cytotoxics were given 2–3 h after the administration of belinostat–paclitaxel followed by carboplatin. This schedule of belinostat followed by cytotoxics was chosen on the basis of pre-clinical in vitro and in vivo data supporting a higher degree of synergy, when HDAC inhibitors were administered before topoisomerase II therapy (Marchion et al, 2004, 2005). In addition, 48 h pre-exposure with belinostat was shown to be better than 24 h, which was better than 12 h. Carboplatin was dosed in accordance with the Calvert formula using ethylenediaminetetraacetic acid clearance to estimate glomerular filtration rate (GFR) (Calvert et al, 1989) at the European Union sites and the Jelliffe formula at sites in the United States (Jelliffe, 1973). Prophylactic anti-emetic and hypersensivity prophylaxis for paclitaxel were administered as per local standards.
A DLT was defined as an absolute neutrophil count <0.5 × 109 l−1 lasting for ⩾7 days, or absolute neutrophil count <0.5 × 109 l−1 with sepsis; or platelet count <25 × 109 l−1. In addition, any other drug-related non-haematological grade III or IV toxicity with the exceptions of alopecia, nausea and vomiting, diarrhoea, rash, arthralgias and myalgias was categorized as DLT. Treatment interventions were attempted to palliate toxicity symptoms before concluding a DLT had occurred. The patients who experienced DLT with documented clinical benefit (stable disease (s.d.) or an objective response) were allowed to continue treatment with BelCaP at the before dose level, if toxicity had resolved to less than grade II (except for alopecia).
Dose escalation was planned in a classical 3+3 design in up to five cohorts. In cohort 1A, belinostat was to be administered at 600 mg m−2 together with carboplatin AUC 5; in cohort 1B, paclitaxel was to be evaluated with paclitaxel 175 mg m−2. In cohort 2, belinostat at 600 mg m−2 was to be administered with both carboplatin AUC 5 and paclitaxel at 175 mg m−2. In cohorts 3 and 4, belinostat was to be administered at 800 and 1000 mg m−2, respectively, together with the same doses of BelCaP. Treatment cycles were to be repeated until disease progression, evidence of significant treatment-related toxicities, or withdrawal of consent. Up to two dose reductions were to be allowed of all three drugs. The patients who did not complete the first cycle were replaced.
Study assessments
Pre-treatment evaluation included history, physical examination, laboratory assessments, chest x-ray, electrocardiogram (ECG), urinalysis and tumour evaluation with computed tomography (CT) or magnetic resonance imaging (MRI). Laboratory assessments (complete blood count, clotting screen, urea, creatinine, electrolytes, liver function tests, uric acid, glucose and cholesterol) were performed pre-treatment and weekly thereafter as well as on day 4 of cycles 1 and 2 only. Electrocardiograms were performed before and within 1 h after treatment with belinostat in cycle 1 and on day 1 only of all subsequent cycles. Central ECG reading was carried out using a digital analysis method.
Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0. Efficacy was assessed by CT or MRI performed every two cycles using Response Evaluation Criteria in Solid Tumors (RECIST) (Therasse et al, 2000).
Drug preparation
Belinostat was supplied by Topotarget as 10-ml vials, each containing 50 mg ml−1 belinostat and the solubilizer L-arginine (100 mg ml−1). Belinostat was stored below 5°C. The assigned dose of belinostat was added to a 250-ml bag of sterile sodium chloride (0.9% solution or 5% dextrose solution) and immediately used. Carboplatin and paclitaxel were prepared as indicated in the summary of product characteristics for each agent.
Pharmacokinetic sampling and analysis
Blood samples for belinostat PK, when administered alone were taken pre-treatment, at the end of infusion and 5 min, 15, 30 min, 1 h, 2, 3 h, 3 h 15 min, 3 h 30 min, 4, 5, 6 h after infusion on days 1 and 3 of cycle 1. Samples for carboplatin or paclitaxel when administered after belinostat, were taken relative to the start of belinostat infusion at 3 h (pre-carboplatin or-paclitaxel), 3 h 15 min, 3 h 30 min, 4, 5 and 6 h in cycle 1 on day 3. In addition, samples were taken for BelCaP when administered together at 6 h 15 min, 6 h 30 min, 7, 8, 9 and 24 h after belinostat on day 3. Belinostat and paclitaxel levels were determined in lithium-heparinised plasma using validated solid-phase extraction high performance liquid chromatograpgy (HPLC)-tandem mass spectrometric detection (MS/MS) methods (Covance, Indianapolis, IN, USA). The lower limit of quantitation was 5 and 10 ng ml−1 for belinostat and paclitaxel, respectively. Belinostat plasma concentrations were determined at Covance using a validated method involving solid-phase extraction, separation with acetic acid acetonitril gradient on reversed phase C18 silica and MS/MS using penta-deuterated belinostat as the internal standard. Belinostat and the internal standard were detected using negative ion electron spray with the transition from 317 to 143 and 322 to 143 m z−1, respectively.
Paclitaxel and the internal standard, cephalomannine, are extracted from human plasma by solid-phase extraction on a cyanopropyl solid-phase extraction cartridge. After eluting with the mobile phase, the eluent is directly analysed using liquid chromatography with MS/MS. The standard curve range for this assay is from 10.0 to 2000 ng ml−1 for paclitaxel, using a plasma sample volume of 0.250 ml.
Human plasma samples were analysed for Pt from carboplatin by inductively coupled plasma-mass spectrometry after dilution with 0.01% Triton X-100 in 0.1% nitric acid. The samples were analysed without pre-digestion or extraction directly against matrix-matched standard solutions. Intensity, as counts per second from Pt, mass 195, was measured. Terbium, mass 159, was used as an internal standard. The results were calculated from the net intensity ratio of Pt to internal standard. The standard curve range was 2.00–1000 ng Pt ml−1 in plasma. Samples above the calibration range were diluted using blank human plasma.
Plasma concentration–time curves of all three analytes were analysed by non-compartmental methods using WinNonlin (version 4.0.1 Pharsight Corp., Cary, NC, USA). For all analytes the AUC was estimated from the start of infusion by the logarithmic-linear trapezoidal rule and extrapolated to infinity using the estimated value of the slope of the terminal logarithmic-linear disposition phase. Analysis was carried out on the non-dose-normalised plasma concentration of carboplatin, as dosing had already been normalised per GFR using Calvert's formula (Calvert et al, 1989).
Results
Patient characteristics
In all 23 patients with a median age of 56 years (range 39–66) were enroled between August 2005 and August 2006; the demographics are described on Table 1. A total of 139 complete cycles were administered (median 4; range 0–32; Table 2). A total of 10 patients completed ⩾6 cycles of treatment.
Safety
All patients were evaluable for safety. Dose escalation proceeded through all cohorts with no observed DLT attributable to the combination of BelCaP. There were no grade IV non-haematological adverse events (AE). Grade III AEs observed were peripheral sensory neuropathy (n=2), fatigue (n=1), vomiting (n=1) and myalgia (n=1) (Table 3). There was no cardiac toxicity; two events of investigator-reported QTc prolongation in two patients were later refuted by central ECG analysis. Grade III haematological AEs comprised (n) leucopenia (5), neutropenia (3), thrombocytopenia (3) and anaemia (1e) (Table 3). Three patients experienced grade IV neutropenia. There was no evidence that the incidence of haematological AEs increased with the addition of belinostat to BelCaP. As per the protocol no further dose escalation beyond 1000 mg m−2 per day was attempted, and the recommended dose was defined as belinostat (1000 mg m−2 per day) by 30-min i.v. infusion once daily for 5 days every 21 days, with paclitaxel (175 mg m−2) and carboplatin AUC 5 administered on day 3.
Pharmacokinetics
The belinostat pharmacokinetics was dose proportional at the dose range evaluated (Table 4). In five of seven patients, the pre-infusion values on day 3 were below the limit of quantitation, indicating that there was no accumulation of belinostat at the 1000 mg m−2 per day level. Administration of BelCaP on day 3 did not alter the PK of belinostat. Similarly, there was no effect of increasing the belinostat dose on the PK parameters of either paclitaxel or carboplatin and these were comparable with that reported in the literature (Tables 5 and 6) (Oguri et al, 1988; Bhalla et al, 1999; Goffin et al, 2005).
Efficacy
In all 14 patients completed at least two cycles and were evaluable for tumour response. Two patients showed a confirmed partial response (PR), including one patient with metastatic pancreatic cancer treated with belinostat and carboplatin and the other patient with metastatic rectal cancer treated with belinostat with BelCaP. The duration of response was 7 and 9 months, respectively. Figure 1 shows the reduction in the size of adrenal metastases in the patient with metastatic rectal carcinoma. These patients had not previously received carboplatin or paclitaxel, but the latter patient had received oxaliplatin. A third patient with a mixed mullerian ovarian tumour, with radiologically s.d. by RECIST, showed a complete CA-125 response. This patient had previously received both BelCaP. A fourth patient with metastatic transitional cell cancer of the bladder bones only, treated with belinostat and paclitaxel, showed a resolution of a bony metastatic deposit, but was otherwise rated s.d. by investigator. This patient was previously treated with carboplatin and gemcitabine with a best response of progressive disease.

Efficacy of BelCaP in a patient with rectal cancer. Response in a patient with metastatic rectal carcinoma treated with BelCaP showing an adrenal metastasis at baseline, (A) (encircled in red) measuring 7.7 cm, and after six cycles, (B) measuring 3.6 cm. The patient also had retroperitoneal lymph node metastases and with this, showed a confirmed PR. (See online version for color information).
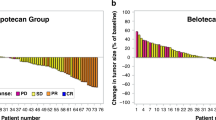
Overall, eight patients showed a best response of s.d. (median 6.6 months; range 1–29); in seven of these eight patients s.d. lasted ⩾ 6 months (range 6–29 months). These included patients with metastatic transitional cell carcinoma (TCC) bladder, metastatic cholangiocarcinoma, carcinoma of unknown primary (CUP), ovarian cancer, rectal cancer, mesothelioma and Ewing sarcoma. Five of these six patients had received Pt-based treatments previously. Figure 2 shows a waterfall plot of the 14 patients evaluable for response.

Waterfall plot showing best response in individual patients evaluable for responses assessment by RECIST 1.0 criteria. In all14 patients completed the first two cycles and were evaluable for response and featured in this plot. Dotted lines at +20% and −30% indicate the % level at which disease progression and disease response has occurred, respectively.
Discussion
The safety and MTD of belinostat have been previously described (Steele et al, 2008). We now present data from a phase I trial of a first-in-man combination of the HDAC inhibitor with carboplatin and/or paclitaxel, BelCaP. Carboplatin and paclitaxel were well tolerated with no evidence of DLT up to the maximum administered dose of 1000 mg m−2 per day belinostat with BelCaP at standard doses. The most common toxicities were gastrointestinal and constitutional, often associated with BelCaP alone and seen with single agent belinostat. However, there was no evidence that the incidence or severity of these, or of haematological toxicity, increased with the combination. It should be noted that there was no evidence of significant cardiac toxicity, which has been associated with HDAC inhibitors.
The toxicity profile of BelCaP was favourable and grade III/IV neutropenia was reported in 26% of the patients and this was probably attributed to BelCaP. Ramalingan et al reported that the combination of the HDAC inhibitor vorinostat with BelCaP, resulted in grade III/IV neutropenia in 50% of the patients, but in their study the dose of BelCaP was higher than in the present study (Ramalingam et al, 2007a). Furthermore, when valproic acid was combined with epirubicin, neurological DLTs were attributed to valproate with no exacerbation of epirubicin-related myelosuppression (Munster et al, 2007). However, grade II–III QTc prolongation was seen in approximately 25% of patients in that study and as previously reported, this was, in nearly all cases, associated with electrolyte abnormalities (Munster et al, 2007; Piekarz et al, 2006).
PK results for belinostat showed that there was a dose-proportional increase in belinostat exposure with increasing dose and that there was no accumulation with repeat dosing. There seemed to be no significant change in the PK parameters of belinostat with the addition of BelCaP when compared with the PK parameters seen in the single-agent phase I study (Steele et al, 2008). The similarity between the PK parameters of BelCaP in this study with historical controls, suggests that this combination is feasible, although such a comparison with historical data has its limitations.
Encouragingly, BelCaP showed evidence of clinical activity in heavily pre-treated patients, including those previously exposed to Pt therapy. As a result of the clinical benefit in a patient with metastatic TC of the bladder to bone, an expansion cohort of BelCaP was initiated in patients with this tumour type.
Clinical efficacy has been observed with other combinations of an HDAC inhibitor and cytotoxic chemotherapy. A total of 9 of 41 patients, with a variety of solid tumours, including prostate, pancreatic, melanoma and cervical cancer, responded to valproic acid with epirubicin; in a phase II portion of this study, 64% of patients with breast cancer responded to treatment (Munster et al, 2009). In the study of Ramalingam et al of 25 patients treated with vorinostat and BelCaP, 10 patients with non-small-cell lung and 1 patient with head and neck cancer showed a PR (Ramalingam et al, 2007a). However, the majority of patients (82%) was chemotherapy naive. The efficacy of this belinostat combination fares favourably and merits further evaluation.
In summary, the combination of belinostat (100 mg m2 per day) with carboplatin AUC5 and paclitaxel (175 mg m−2) is well tolerated, with no evidence of any PK interaction. Recruitment of patients to two expansion cohorts of ovarian cancer and TCC of bladder has recently been completed and an evaluation in patients with CUP is ongoing.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bhalla KN, Kumar GN, Walle UK, Ibrado AM, Javed T, Stuart RK, Reed C, Arbuck SG, Walle T (1999) Phase I and pharmacologic study of a 3-h infusion of paclitaxel followed by cisplatinum and 5-fluorouracil in patients with advanced solid tumors. Clin Cancer Res 5: 1723–1730
Bolden JE, Peart MJ, Johnstone RW (2006b) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5: 769–784
Calvert AH, Newell DR, Gumbrell LA, O'Reilly S, Burnell M, Boxall FE, Siddik ZH, Judson IR, Gore ME, Wiltshaw E (1989) Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 7: 1748–1756
Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P (2008) A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol 81: 170–176
Goffin JR, Anderson IC, Supko JG, Eder Jr JP, Shapiro GI, Lynch TJ, Shipp M, Johnson BE, Skarin AT (2005) Phase I trial of the matrix metalloproteinase inhibitor marimastat combined with carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin Cancer Res 11: 3417–3424
Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389: 349–352
Jelliffe RW (1973) Letter: creatinine clearance: bedside estimate. Ann Intern Med 79: 604–605
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409: 581–589
Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN (2004) Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem 92: 223–237
Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN (2005) In vivo synergy between topoisomerase II and histone deacetylase inhibitors: predictive correlates. Mol Cancer Ther 4: 1993–2000
Marks P, Rifkind RA, Richon VM (2001) Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1: 194–202
Marks PA, Dokmanovic M (2005) Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs 14: 1497–1511
Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, Simon G, Fishman M, Minton S, Garrett C, Chiappori A, Lush R, Sullivan D, Daud A (2007) Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol 25: 1979–1985
Munster PN, Marchion D, Thomas S, Egorin M, Minton S, Springett G, Lee JH, Simon G, Chiappori A, Sullivan D, Daud A (2009) Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer 101: 1044–1050
Oguri S, Sakakibara T, Mase H, Shimizu T, Ishikawa K, Kimura K, Smyth RD (1988) Clinical pharmacokinetics of carboplatin. J Clin Pharmacol 28: 208–215
Piekarz RL, Frye AR, Wright JJ, Steinberg SM, Liewehr DJ, Rosing DR, Sachdev V, Fojo T, Bates SE (2006) Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin Cancer Res 12: 3762–3773
Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R (2003) Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 2: 721–728
Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS, Jeffers M (2006) Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 5: 2086–2095
Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP (2007a) Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res 13: 3605–3610
Schemies J, Sippl W, Jung M (2009) Histone deacetylase inhibitors that target tubulin. Cancer Lett 280: 222–232
Steele NL, Plumb JA, Vidal L, Tjornelund J, Knoblauch P, Rasmussen A, Ooi CE, Buhl-Jensen P, Brown R, Evans TR, DeBono JS (2008) A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res 14: 804–810
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92: 205–216
Acknowledgements
We thank Lise-Lotte Fuglsang, Anni Norman, Britt Tryde and Holly Coulter for valuable assistance during study conduct; Thomas Hawthorne for pharmacokinetics support; and Nis Nissen for invaluable support throughout the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lassen, U., Molife, L., Sorensen, M. et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer 103, 12–17 (2010). https://doi.org/10.1038/sj.bjc.6605726
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605726
Keywords
This article is cited by
-
Complexation of histone deacetylase inhibitor belinostat to Cu(II) prevents premature metabolic inactivation in vitro and demonstrates potent anti-cancer activity in vitro and ex vivo in colon cancer
Cellular Oncology (2024)
-
Phase 1 study of belinostat and adavosertib in patients with relapsed or refractory myeloid malignancies
Cancer Chemotherapy and Pharmacology (2023)
-
Epigenetic regulation of pancreatic adenocarcinoma in the era of cancer immunotherapy
Journal of Gastroenterology (2022)
-
Phase I trial of belinostat in combination with 13-cis-retinoic acid in advanced solid tumor malignancies: a California Cancer Consortium NCI/CTEP sponsored trial
Cancer Chemotherapy and Pharmacology (2019)
-
Gemigliptin, a novel dipeptidyl peptidase-IV inhibitor, exerts a synergistic cytotoxicity with the histone deacetylase inhibitor PXD101 in thyroid carcinoma cells
Journal of Endocrinological Investigation (2018)
