
Abstract
Various types of stem cell lines have been derived from preimplantation or postimplantation mouse embryos: embryonic stem cell lines, epiblast stem cell lines, and trophoblast stem cell lines. It is not known if extraembryonic endoderm stem (XEN) cell lines can be derived from postimplantation mouse embryos. Here, we report the derivation of 77 XEN cell lines from 85 postimplantation embryos at embryonic day E5.5 or E6.5, in parallel to the derivation of 41 XEN lines from 69 preimplantation embryos at the blastocyst stage. We attain a success rate of 100% of XEN cell line derivation with our E5.5 whole-embryo and E6.5 disaggregated-embryo methods. Immunofluorescence and NanoString gene expression analyses indicate that the XEN cell lines that we derived from postimplantation embryos (post-XEN) are very similar to the XEN cell lines that we derived from preimplantation embryos (pre-XEN) using a conventional method. After injection into blastocysts, post-XEN cells contribute to extraembryonic endoderm in chimeras at E6.5 and E7.5.
Similar content being viewed by others


Introduction
Mouse preimplantation embryonic development culminates in the blastocyst stage. A blastocyst consists of three cell lineages: epiblast, trophectoderm, and primitive endoderm (PrE). The epiblast develops into most of the embryo proper, the amnion, and the extraembryonic mesoderm of the yolk sac; the trophectoderm gives rise ultimately to the fetal portion of the placenta; and the primitive endoderm forms the two extraembryonic endoderm lineages – the visceral endoderm (VE) and the parietal endoderm (PE) of the yolk sac1,2. The extraembryonic endoderm provides nutritive support to the embryo, and is required for several inductive events such as anterior patterning and formation of endothelial cells and blood islands3,4,5.
Stem cell lines have been derived from these three cell lineages6. Embryonic stem (ES) cell lines from epiblast were first reported in the 1980 s (refs 7 and 8), trophoblast stem (TS) cell lines from trophectoderm in the 1990 s (ref. 9), and extraembryonic endoderm stem (XEN) cell lines from PrE in the 2000 s (ref. 10). The conventional source of these cell lines is the blastocyst stage embryo. TS cell lines can also be derived from postimplantation embryos9,11,12. Moreover, mouse epiblast stem cell (EpiSC) lines, which resemble ES cell lines of human, can be derived from preimplantation embryos13 and postimplantation embryos14,15, and can be reverted to ES cells16. XEN cell lines are useful for the investigation of signaling pathways of cells of the extraembryonic endoderm lineages, and represent an in vitro model to identify patterning activities of the extraembryonic endoderm such as factors involved in cardiac induction17,18. Mouse fibroblasts pass via a XEN-like state on their way to induced pluripotent stem (iPS) cells by chemical reprogramming19.
There are three methods to derive mouse XEN cell lines20. The first method entails the direct derivation of XEN cell lines from blastocysts10. The second method involves the conversion of an existing ES cell line to a XEN or XEN-like cell line, either by forced expression of a transcription factor gene encoding Gata4 or Gata6 (refs 21, 22, 23) or Sox17 (refs 24 and 25), or by chemical modification of the culture medium such as by addition of retinoic acid and activin A26. A third, more recently reported method, derives induced XEN cells (iXEN) by reprogramming fibroblasts with the classical iPS reprogramming factors Oct4, Sox2, Klf4, and Myc; colonies from which iXEN cells can be derived, arise in parallel to iPS cells27.
Here, we show that XEN cell lines can be derived with very high efficiency from postimplantation embryos at E5.5 or E6.5, either from whole or disaggregated embryos. These so-called post-XEN cell lines are very similar to the pre-XEN cell lines that we derived directly from preimplantation embryos.
Results
Derivation of pre-XEN cell lines from blastocysts
To distinguish unambiguously the XEN cell lines that were derived from preimplantation embryos from the XEN cell lines that were derived from postimplantation embyros, we refer to these cell lines operationally as pre-XEN and post-XEN cell lines, respectively.
We first derived a set of conventional pre-XEN cell lines from blastocysts, in order to set up conditions in our laboratory and to provide a comparison for post-XEN cell lines. We collected 63 E1.5 embryos from three types of natural matings: from three heterozygous PDGFRa-GFP females28 mated with homozygous CAG::mRFP1 males29, two B6D2F1 females mated with hemizygous D4/XEGFP males30, and two homozygous D4/XEGFP females mated with DBA/2 N males (Table 1). PDGFRa is a XEN-cell marker26,31,32; CAG::mRFP1 is a transgene that expresses ubiquitously the red fluorescent protein; D4/XEGFP is a transgene integrated on the X-chromosome, and is not expressed from the inactive X-chromosome, enabling the future study of X-chromosome inactivation in XEN cell lines. We cultured the embryos in KSOM medium until the blastocyst stage, and then removed the zona pellucida using acid Tyrode solution. We transferred each blastocyst separately into a well of a 4 well-dish coated with 0.1% gelatin and covered with mouse embryonic fibroblasts (MEF), and switched to ES cell medium supplemented with leukemia inhibitory factor (LIF)20. All blastocyst cells including PrE cells express RFP from the transgene (Fig. 1a). After 3 days in culture, blastocysts started to form outgrowths (Fig. 1a), which we disaggregated on day 5. Cell line X47 was still in the process of becoming established (and was still displaying intrinsic red fluorescence) after 19 days in culture (Fig. 1a). We obtained a stable pre-XEN cell line X47 after 38 days of culture (Fig. 1a). The CAG promoter does not appear to generate sufficient mRFP1 to enable us to detect the intrinsic red fluorescence in XEN cell lines, similar to what has been reported for CAG::H2B-GFP and CAG::YFP strains26. We thus derived, using the conventional method20 with ES cell medium and LIF, a total of 36 pre-XEN cell lines from 63 blastocysts, at a 57% success rate (Table 1).

Derivation of pre-XEN cell lines from blastocysts.
(a) PDGFRa-GFP × CAG::mRFP1 blastocysts and pre-XEN cell line X47. From left to right: blastocysts, outgrowth of blastocyst after 3 days in culture, cells after disaggregation of the outgrowth and culture for 19 days, and established pre-XEN cell line on day 38. From top to bottom, first three columns: bright-field image, intrinsic green fluorescence of GFP, intrinsic red fluorescence of RFP, and combined green and red fluorescence/bright-field image. From top to bottom, right-most column: bright-field image, intrinsic green fluorescence of GFP, immunofluorescence for RFP, and combined green fluorescence and immunofluorescence/bright-field image. (b) Fluorescence analysis of pre-XEN cell line X42. Shown are eight pairwise combinations of intrinsic (indicated with an asterisk after GFP) green fluorescence from the gene-targeted PDGFRa locus and immunofluorescence (magenta), together with DAPI (blue). Cells are immunoreactive for XEN markers GATA4, GATA6, SOX7, SOX17, and DAB2. But cells are negative for ES cell markers OCT4 and NANOG, and for TS cell marker CDX2.
Next, we isolated by immunosurgery33 the inner cell mass (ICM) of blastocysts from a natural mating between an R26-tauGFP41 female and a Sox17-Cre male34. With Sox17 a XEN-cell marker, and R26-tauGFP41 a Cre reporter strain, this cross marks permanently cells that have been derived from Sox17-expressing cells. We obtained 6 ICMs from 6 blastocysts, which we had collected at E2.5 as morulae and cultured overnight in KSOM medium. We transferred each ICM separately into a well of a 4-well dish coated with gelatin and covered with MEF, and cultured the ICM outgrowths in ES medium with LIF. Seven days later we replaced the ES medium by TS medium with 25 ng/ml FGF4 and 1 μg/ml heparin (referred to as F4H) without passaging the cells. We changed the medium every two days, and on day 14 we passaged the cells to 12-well dishes. We thus established five XEN cell lines from six ICMs, at a success rate of 83% (Table 1).
Figure 1b shows immunofluorescence staining of pre-XEN cell line X42, from the cross PDGFRa-GFP × CAG::mRFP1. Cells also display intrinsic green fluorescence of GFP produced from the gene-targeted PDGFRa-GFP locus (indicated with the asterisk PDGFRa-GFP*). We find that this and other pre-XEN cell lines are immunoreactive for XEN cell markers GATA4, GATA6, SOX7, SOX17, and DAB2, but negative for ES cell markers OCT4 and NANOG, and negative for TS cell marker CDX2.
Derivation of post-XEN cell lines from whole E6.5 embryos
Next we collected E6.5 postimplantation embryos from three types of natural matings: two heterozygous Xist1loxGFP females35 mated with a wild-type DBA/2 N male, two heterozygous ROSA26-STOP-taulacZ females mated with a heterozygous Sox17-Cre male34, and one hemizygous Gata6-mTomato female36 mated with a homozygous Cdx2-GFP male37 (Table 1). Xist1loxGFP is a GFP-containing targeted mutation in the Xist locus on the X-chromosome; Sox17 and Gata6 are XEN-cell markers; and Cdx2 is a marker for trophoblast stem cells. We removed the ectoplacental cone of the embryos as much as possible, and transferred each embryo separately into a well of 4-well dish coated with 0.1% gelatin and covered with MEF in TS cell medium including 25 ng/ml FGF4 and 1 μg/ml heparin (referred to as F4H). One day later, the embryos had attached to the surface and started to form an outgrowth. The embryos had formed a large outgrowth after 5 days. We used TrypLE Express to disaggregate the outgrowths and passaged cells into a well of a 4-well dish. After cells reached 70–80% confluency, they were passaged into a well of a 12-well dish. After they reached 70–80% confluency again, cells were passaged into a well of a 6-well dish, and we then obtained stable post-XEN cell lines. The intrinsic red fluorescence of mTomato produced from the Gata6 promoter in the transgene was sufficiently high to detect it in the whole embryo and outgrowth, but not in the established post-XEN cell line at day 60 (Fig. 2a). We thus derived, using the whole-embryo method, a total of 30 post-XEN cell lines from 38 E6.5 embryos, at a 79% success rate (Table 1).

Derivation of post-XEN cell lines from whole E6.5 embryos.
(a) Gata6-mTomato × Cdx2-GFP embryo and post-XEN cell line X-E6.5-82278-4. From left to right: whole E6.5 embryo, large outgrowth of embryo after 5 days in culture, and established post-XEN cell line on day 60. The embryo and the outgrowth display intrinsic red fluorescence of mTomato, but the expression of mTomato in the post-XEN cell line is below the detection level of intrinsic red fluorescence from mTomato. (b) Immunofluorescence analysis of post-XEN cell line X-E6.5-82278-4. Expression of mTomato from the Gata6 promoter in the transgene is detectable with an antibody for RFP (first row, second column). Cells are immunoreactive (magenta) for XEN markers GATA4, GATA6, SOX7, SOX17, and DAB2. But cells are negative for ES cell markers OCT4 and NANOG, and for TS cell marker CDX2. Right-most column shows DAPI (blue).
We found that mTomato expressed from the Gata6 transgenic promoter in these cells can be detected by immunofluorescence with antibodies against RFP, together with GATA6 expressed from the endogenous Gata6 locus with antibodies against GATA6 (Fig. 2b). As was the case for our pre-XEN cell lines, our post-XEN cell lines are positive for XEN cell markers GATA4, GATA6, SOX7, SOX17, and DAB2 but negative for ES cell markers OCT4 and NANOG, and negative for TS cell marker CDX2.
Derivation of post-XEN cell lines from disaggregated E6.5 embryos
During the process of post-XEN cell line derivation from whole E6.5 embryos, we observed that some embryos had difficulty forming large outgrowths. We reasoned that some unidentified cell types in these embryos may inhibit XEN cell growth. We therefore proceeded to derive post-XEN cell lines from disaggregates of these embryos. We isolated 30 E6.5 embryos from three types of natural matings: a homozygous R26-tauGFP41 female with a heterozygous Sox17-Cre male36, a heterozygous ROSA26-STOP-taulacZ female with a heterozygous Sox17-Cre male, and a wild-type CD1 female with a heterozygous PDGFRa-GFP male28 (Table 1). We prepared disaggregates (Fig. 3a) by exposing the embryos to collagenase and deoxyribonuclease followed by TrypLE Express, and then gently pipetting the embryos in a glass pipette with a diameter of 50–60 μm. We plated the cell suspension, which consisted of a mixture of single cells and clumps of cells, from one disaggregated embryo separately into a well of 4-well dish coated with 0.1% gelatin and covered with MEF in standard TS medium including F4H. On day 3, XEN-like cells colonies appeared. We picked these colonies, disaggregated them by a glass pipette or by TrypLE Express for 5 min at 37 °C, and passaged them into a well of a 4-well dish. (We did not succeed in deriving post-XEN cell lines from single GFP+ cells that we placed in wells of a 96-well dish.) We thus derived, using the disaggregation method, a total of 30 post-XEN cell lines from 30 E6.5 embryos, at a 100% success rate (Table 1).

Derivation of post-XEN cell lines from disaggregates of E6.5 embryos.
(a) R26-tauGFP41 × Sox17-Cre E6.5 embryo and post-XEN cell line X-E6.5-Z0617-2. From left to right: whole E6.5 embryo, disaggregated embryo, XEN-like colony expressing GFP on day 3 of culture, and established post-XEN cell line on day 30. Intrinsic green fluorescence of GFP. (b) Fluorescence analysis of post-XEN cell line X-E6.5-Z0617-5. First column: intrinsic (indicated with an asterisk after GFP) green fluorescence of GFP expressed from the ROSA26 locus after activation by Cre recombinase that is expressed from the gene-targeted Sox17 locus. Second column: cells are immunoreactive (magenta) for XEN markers GATA4, GATA6, SOX7, and SOX17. Third column: cells are immunoreactive for XEN marker DAB2, but negative for ES cell markers OCT4 and NANOG, and negative for TS cell marker CDX2. Fourth column: DAPI (blue).
As was the case with our pre-XEN cell lines and post-XEN cell lines derived from E6.5 whole embryos, cells are immunoreactive for XEN markers GATA4, GATA6, SOX7, SOX17, and DAB2, but negative for ES cell markers OCT4 and NANOG, and negative for TS cell marker CDX2 (Fig. 3b). Thus, we increased the success rate of post-XEN cell derivation from 79% for whole E6.5 embryos to 100% for disaggregated E6.5 embryos.
Derivation of post-XEN cell lines from E5.5 whole embryos
In our final set of experiments, we derived post-XEN cell lines from 5.5 day-old embryos. We isolated 17 embryos from 33 E5.5 implantation sites from a mating of three homozygous R26-tauGFP41 females with three heterozygous Sox17-Cre males, and a mating of a homozygous ROSA-STOP-taulacZ female with a heterozygous Sox17-Cre male (Table 1). We removed as much of the ectoplacental cone as possible, and transferred one whole embryo separately into a well of a 4-well dish coated with 0.1% gelatin and covered with MEF, in TS medium including F4H (Fig. 4a). In our experience, the less ectoplacental cone and extraembryonic ectoderm remains in a dissected embryo, the easier it is to derive a post-XEN cell line. We surmise that ectoplacental cone cells and extraembryonic ectoderm cells (trophoblast-derived cells) compete with or inhibit XEN cells in culture. For 14/17 embryos, large outgrowths had appeared by day 5. We disaggregated these outgrowths with TrypLE Express on day 7, and passaged the cells into a well of a 4-well dish. On day 11, XEN-like colonies had accumulated. We picked these colonies, combined them, disaggregated them with a glass pipette or with TrpLE Express for 5 min at 37 °C, and passaged them into a well of a 4-well dish. We thus established 14 post-XEN cell lines after ~21 days in culture. For the remaining 3/17 embryos, trophoblast-derived cells surrounded the outgrowth. We performed a disaggregation by pipette on day 3, and switched to ES medium with LIF. When XEN cells became abundant, we passaged the cells onto gelatin-coated dishes without MEF, and switched back to TS medium including F4H. Three post-XEN cell lines were established after ~50 days culture. We thus derived a total of 17 post-XEN cell lines from 17 E5.5 embryos, at a 100% success rate (Table 1).

Derivation of post-XEN cell lines from whole E5.5 embryos.
(a) Sox17-Cre × R26-tauGFP41 E5.5 embryo and post-XEN cell line. From left to right: whole E5.5 embryo, large outgrowth of embryo after 5 days in culture, cells after outgrowth disaggregation and culture for 11 days, and established post-XEN cell line X-E5.5-8 on day 22. Combined intrinsic green fluorescence of GFP/bright-field image. (b) Fluorescence analysis of post-XEN cell line X-E5.5-10. First column: intrinsic (indicated with an asterisk after GFP) green fluorescence of GFP from the ROSA26 locus after activation by Cre recombinase that is expressed from the gene-targeted Sox17 locus. Second column: cells are immunoreactive (magenta) for XEN markers GATA4, GATA6, SOX7, and SOX17. Third column: cells are stained for XEN marker DAB2, but not for ES cell markers OCT4 and NANOG, and not for TS cell marker CDX2. Fourth column: DAPI (blue).
These post-XEN cell lines are immunoreactive for XEN cell markers GATA4, GATA6, SOX7, SOX17, and DAB2, but negative for ES cell markers OCT4 and NANOG, and negative for TS cell marker CDX2 (Fig. 4b).
Post-XEN cell morphology, population characteristics, and in vitro differentiation
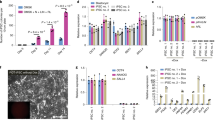
Like pre-XEN cell lines10, our cultures of post-XEN cell lines contain at least two cell morphologies: a rounded, highly refractile cell type and a more stellate epithelial-like cell type (Fig. 5a). At higher densities, post-XEN cells can form epithelial sheets (Fig. 5b) and often a lattice-type structure (Fig. 5c). To determine if these two cell morphologies represent two cell types in the cultures, we FACS-sorted single GFP+ post-XEN cells (X-E6.5-Z0617-5) directly into wells of a 96-well dish, and derived two subclones. Cells of these subclones continued to exhibit either round or epithelial-like cell morphologies (Fig. 5d).

Post-XEN cell morphology in culture, population characteristics, and in vitro differentiation with BMP4.
(a) Post-XEN cells X-E6.5-Z0617-5 cultured at low density contain a refractile, round cell type, and an epithelioid cell type. (b) Post-XEN cells X-E6.5-Z0617-5 cultured to near-confluency show the presence of individual cells and an epithelial sheet of cells. (c) Post-XEN cells X-E6.5-Z0617-5 participate in a lattice-type structure. (d) A subcloned cell line of X-E6.5-Z0617-5 continues to contain a refractile, round cell type, and an epithelioid cell type. (e) Analysis of population characteristics of the post-XEN cell lines X-E5.5-10, X-E6.5-Z0617-5, and X-E6.5-Z0617-2. Representative images of fields of view used to analyze Z0617-5 are shown. The GFP signal reflects intrinsic fluorescence, the GATA4 signal reflects immunoreactivity, and DAPI was used to counterstain the nuclei. The bottom right image shows the segmented nuclei of cells selected for marker analysis after the DAPI signal had undergone thresholding, declumping, and filtering for size and nuclear roundness. The individual cells are shown in multiple colors to clearly show the distinct cells that were used for analysis. Three sets of images were evaluated per cell line, and 500–1000 cells were evaluated per cell line. (f) Marker characteristics of the three cell lines are shown in the form of a doughnut chart, as determined by intrinsic fluorescence of GFP and immunoreactivity for GATA4. Cells are divided into four populations. (g,h) Immunofluorescence analysis of pre-XEN cell line X47 (g) and post-XEN cell line X-E6.5-Z0617-5 (h). Expression of the VE marker E-cadherin is higher after treatment with 10 ng/ml BMP4, in gelatin-coated plates. Left columns: E-cadherin (magenta). Right columns: DAPI signal (blue) merged with E-cadherin (magenta).
To evaluate the population characteristics of post-XEN cell lines, we analyzed three GFP-expressing cell lines (X-E6.5-Z0617-5, X-E6.5-Z0617-2 and X-E5.5-10) by immunofluorescence for GATA4 and counterstaining with DAPI. Sets of fluorescence images were captured for each line visualizing the intrinsic fluorescence of GFP, DAPI, and GATA4 immunoreactivity (Fig. 5e). The images were evaluated with a custom CellProfiler pipeline that segmented cells using the DAPI fluorescent signal. A cutoff for size and nuclear roundness was used to exclude the MEF population as much as possible. We then evaluated each cell for levels of GFP and GATA4 signal. In these three post-XEN cell lines, >94% of cells evaluated exhibited GFP fluorescence and are GATA4+ (Fig. 5f). The populations of cells that expressed either GFP or GATA4 but not both represented 0–3% of the cells evaluated. The remaining population of cells, which expressed neither GFP nor GATA4, may reflect another cell type, or MEFs that escaped exclusion during the cell identification step in the custom CellProfiler pipeline.
We asked if post-XEN cells can differentiate into a VE identity by incubation with BMP438,39. We cultured four post-XEN cell lines (X-E5.5-9, X-E6.5-Z0617-2, X-E6.5-Z0617-5 and X-E6.5-78097-7) and three pre-XEN cell lines (X42, X47, X-ICM-4) in gelatin-coated dishes in TS medium with F4H, plus 10 ng/ml BMP4. In parallel, we cultured the same cell lines in TS medium with F4H without BMP4. Four days later, we performed immunofluorescence for E-cadherin, a VE marker. We found that culture with BMP4 induces expression of E-cadherin in post-XEN cells, as well as in pre-XEN cells10 (Fig. 5g-h).
NanoString gene expression analyses of XEN and ES cell lines
We applied the NanoString multiplex platform for gene expression40,41,42 and agglomerative clustering, in order to compare the patterns of expression of selected genes in four pre-XEN cell lines, three post-XEN cell lines, and three ES cell lines that we had derived in other experiments (Fig. 6). We find that all our XEN cell lines have high levels of expression of XEN-specific genes, such as Dab2, Gata4, Gata6, Pdgfra, Sox7, and Sox17, versus low or no expression of ES cell-specific genes such as Nanog, Pou5f1/Oct4, Sox2, and Nr0b1. There is no expression of EpiSC-specific genes such as Cer1 and Fgf5 (data not shown). Thus, the NanoString gene expression analysis confirms and extends the immunofluorescence profiles.

NanoString gene expression analyses of XEN and ES cell lines.
Heatmap analysis of four pre-XEN cell lines, three post-XEN cell lines, and three ES cell lines. Red means high expression level, green means lowly expression level. Agglomerative clustering was performed with nSolver software. Among the classical XEN cell markers are Dab2, Pdgfra2, Gata6, Sox7, Sox17, and Gata4. Among the classical ES cell markers are Oct4/Pou5f1, Sox2, Ffg4, and Nanog. Pre-XEN and post-XEN cell lines share a gene expression profile that is typical for XEN cell lines.
Post-XEN cells contribute to the extraembryonic endoderm of chimeras after injection into blastocyts
A final and stringent test of the potency of stem cells is to assess their contribution to embryonic and extraembryonic tissues in vivo. We injected into blastocysts (B6D2F2, C57BL/6 J, or CD1) cells of two R26-tauGFP41+ pre-XEN cell lines (X-ICM-4 and X-ICM-5) and one PDGFRa-GFP+ pre-XEN cell line (X47) that we had derived from isolated ICMs or blastocysts, and cells from four R26-tauGFP41+ post-XEN cell lines (X-E6.5-Z0617-2, X-E6.5-Z0617-5, X-E5.5-6 and X-E5.5-9) and one PDGFRa-GFP+ post-XEN cell line (X-E6.5-Z0663-9) that we had derived from E5.5 embryos or disaggregated E6.5 embryos (Table 2). For pre-XEN cells, we identified 3 chimeras among 8 E6.5 embryos (38%), and 15 chimeras among 43 E7.5 embryos (35%), from a total of 82 injected blastocysts (18/82 = 22%). For post-XEN cells, we identified 19 chimeras among 49 E6.5 embryos (39%) and 30 chimeras among 83 E7.5 embryos (36%), from a total of 196 injected blastocysts (49/196 = 25%). The percentages of identifiable chimeras among embryos and with regard to injected blastocysts are thus comparable for pre-XEN and post-XEN cell lines.
We found that pre-XEN and post-XEN cells contribute to extraembryonic endoderm at E6.5 or E7.5 (Fig. 7), as is typical for XEN cells injected into blastocysts10,20. We sectioned the decidua of E6.5 or E7.5 embryos obtained by injecting pre-XEN or post-XEN cells into blastocysts, and performed immunofluorescence with antibodies against PDGFRa and E-cadherin. PDGFRa is expressed in the PE and VE32, and E-cadherin is expressed in the VE and epiblast16,43,44. Therefore, extraembryonic endoderm cells that are only immunoreactive for PDGFRa are PE, and cells that are immunoreactive for both PDGFRa and E-cadherin are VE. We analyzed 103 GFP+ cells in E6.5 chimeras obtained by injection of X-ICM-4 (pre-XEN), and 123 GFP+ cells in E7.5 chimeras with X-ICM-5 (pre-XEN), and found that all 226 GFP+ cells reside within the PE region and are immunoreactive for PDGFRa but not for E-cadherin; thus, no GFP+ cell contributed to the VE (Fig. 7a–c). We analyzed 75 GFP+ cells in E6.5 chimeras and 142 GFP+ cells in E7.5 chimeras obtained by injection of X-E6.5-Z0617-5 (post-XEN), and found that all 217 GFP+ cells reside within the PE region and are immunoreactive for PDGFRa but not for E-cadherin; thus, no GFP+ cell contributed to the VE (Fig. 7d–k).

XEN cells contribute to the parietal endoderm of chimeric embryos.
(a–c) Pre-XEN cell line X-ICM-4 contributes to the PE of E6.5 and E7.5 embryos. GFP+ cells are derived from the injected pre-XEN cells. (a) Wholemount of E7.5 embryo. (b,c) Sections of the decidua of E6.5 embryos, showing the merged image of immunofluorescence for PDGFRa (red) and E-cadherin (blue), and fluorescence of DAPI (white) and GFP (green). (d–k) Post-XEN cell lines (X-E5.5-9, X-E6.5-Z0617-5, and X-E6.5-Z0617-2) contribute to the PE of E6.5 or E7.5 embryos. (d–f) Wholemount. GFP+ cells derived from X-E5.5-9 contribute to an E7.5 embryo. (g) Wholemount. GFP+ cells derived from X-E6.5-Z0617-5 contribute to an E7.5 embryo. (h,i) Wholemount. GFP+ cells derived from X-E6.5-Z0617-2 contribute to an E6.5 embryo. (j,k) Sections of the decidua of E7.5 embryos containing GFP+ cells derived from X-E6.5-Z0617-5, with immunofluorescence for PDGFRa (red) and E-cadherin (blue), fluorescence of DAPI (white) and GFP (green), and merge. The single GFP+ cell in (k) apposes but does not belong to the VE, as it is not immunoreactive for E-cadherin. (a–g) and (j,k) were imaged using a Zeiss LSM 710 confocal microscope, and (h–i) were imaged using a Nikon SMZ25 stereofluorescence microscope. In (a) and (e–g) the red counterstain is propidium iodide.
Discussion
We report here that XEN cell lines can be derived efficiently from postimplantation embryos, and from a wide variety of strains and crosses. We believe that it is prudent to continue to refer to these two types of cell lines with regard to the embryonic stage from which they were derived. Additional work will be necessary to identify and characterize possible differences, subtle or substantial, between pre-XEN and post-XEN cell lines.
With our E5.5 whole-embryo method and E6.5 disaggregated-embryo method, we attained a success rate of 100%: we derived 17 post-XEN cell lines from 17 embryos and 30 post-XEN cell lines from 30 embryos, respectively. There are only ~11 PrE cells at the late blastocyst stage45. As E5.5 and E6.5 embryos contain ~95 and ~250 extraembryonic endoderm cells respectively46, these higher numbers of potential source cells may explain the 100% efficiency of XEN cell line derivation from postimplantation embryos. Other, experimental, reasons for the highly efficient derivation may be the reduction of the negative effects of trophoblast-derived cells on XEN cells in the disaggregated E.6 embryo method, and the promotion of XEN cells over trophoblast-derived cells by culturing in ES medium with LIF in the whole-embryo E5.5 embryo method. Regardless of these numerical and experimental explanations, a more exciting, and biological, explanation is that extraembryonic tissues preserve developmental plasticity through implantation.
Since the first report of XEN cell derivation from blastocysts in 2005 (ref. 10), there are three types of methods in place to derive XEN cell lines, either directly from blastocysts10,20 or by conversion from ES cells20,21,22,23,24,25,26 or, recently, by reprogramming from fibroblasts27. The success rate for derivation of XEN cell lines from blastocysts was 21% in TS medium including F4H and 56% in ES medium supplemented with LIF20, which is identical to our success rate of 57% in ES medium supplemented with LIF. Recently, LIF has been reported to support PrE expansion during pre-implantation embryo development47, and LIF could also be supporting XEN cell expansion. The conventional protocol to derive ES cell lines from blastocysts involves also ES medium supplemented with LIF48,49; this medium can be used to derive XEN cell lines from blastocysts, by picking XEN cell colonies or by removing ES cell colonies, but there is always the risk of deriving ES cell lines50. The indirect generation of XEN cell lines from ES cell lines requires, obviously, the prior generation or availability of such lines.
TS cell lines can be derived from the extraembryonic ectoderm of postimplantation embryos in TS medium including FGF4 and heparin9,11. Interestingly, we did not derive TS cell lines from either whole or disaggregated embryos using this medium. We did observe transiently cells and colonies with a morphology that is consistent with TS cells or EpiSCs, but they disappeared from the culture with time. In our immunofluorescence analyses, our XEN cell lines do not contain cells that express the trophectoderm marker Cdx2, and our Nanostring analyses do not reveal expression of TS-specific genes either. The cell lines that we derived from postimplantation embryos in TS medium are thus not TS cell lines, not ES cell lines, and not EpiSC lines, but represent XEN cell lines. Why then are XEN cell lines obtained rather than TS cell lines with our method, which cultures the cells in the same TS medium? The protocol for deriving TS cell lines from postimplantation embryos prescribes that the extraembryonic ectoderm be dissected out and that the embryo and the VE and PE be discarded12. By contrast, we remove the ectoplacental cone from the embryo. We speculate that the VE and PE surrounding the extraembryonic ectoderm inhibit TS cell growth, and/or that VE- and PE-derived cells divide at a faster rate than TS cells and dominate with time the cell culture.
Chemical reprogramming of mouse fibroblasts to the iPS cells involves a XEN-like intermediary stage as a bridge between somatic and pluripotent cells19. In another study, iXEN clones arise in parallel to iPS clones during OSKM-mediated reprogramming of mouse fibroblasts27. More knowledge about the derivation, biology, culture, and conversion of XEN cell lines may be beneficial to develop robust and efficient protocols for the derivation of iPS cell lines by chemical reprogramming with small molecules.
In future experiments, it will be interesting to determine the cell type(s) that are at the origin of the post-XEN cell lines.
Methods
Mouse strains
The PDGFRa-GFP strain28 was obtained from The Jackson Laboratory (JAX), strain 7669, strain name B6.129S4-Pdgfra<tm11(EGFP)>Sor/J. The CAG::mRFP1 strain29 was obtained from JAX, strain 5645, strain name Tg(CAG-mRFP1)1F1Hadj/J. The D4/XEGFP strain30 was obtained from JAX, strain 3116, strain name Tg(CAG-EGFP)D4Nagy/J. The Sox17-Cre strain34 was obtained from MMRRC, strain 036463-UNC, strain name Sox17<tm2(EGFP/cre)Mgn>/Mmnc. (Although this strain is reported to contain and express GFP, and we confirm the presence of GFP in this targeted insertion in the Sox17 locus, we cannot detect GFP expression in embryos and cell lines derived from them.) The Xist1loxGFP strain35 was provided by the RIKEN BioResource Center through the National BioResource Project of the MEXT (Japan), strain RBRC01260, strain name B6;129(Cg)-Xist<tm2Sado>. The GATA6-mTomato strain36 was provided by the RIKEN BioResource Center, strain RBRC04900, strain name B6;B6C3(129)-Tg(GATA6-mTomato)3Hmd. The Cdx2-GFP strain37 was obtained from JAX, strain 18983, strain name Cdx2<tm1Yxz>/J. The ROSA26-STOP-taulacZ reporter strain51 was generated by Dr. Ivan Rodriguez in the laboratory of P.M. at The Rockefeller University, and is publicly available from JAX, strain 6744, STOCK Gt(ROSA)26Sor<tm1.1Mom>/MomJ. The R26-tauGFP41 reporter strain52 was a gift from Dr. Uli Boehm, Universität des Saarlandes, Homburg, Germany. For the preparation of mouse embryonic fibroblasts, strain 3208, strain name Tg(DR4)1Jae was obtained from JAX.
TS cell medium
Advanced RPMI-1640 (Gibco, #12633-012) was supplemented with 20% (vol/vol) FBS (HyClone, #SH30071.03), 2 mM GlutaMAX Supplement (Gibco #35050), 1% penicillin/streptomycin (Specialty Media #TMS-AB2-C), 0.1 mM β-mercaptoethanol (Gibco #21985-023), 1 mM sodium pyruvate (Gibco #11360-039), supplemented with 25 ng/ml FGF4 (Peprotech #100-31) and 1 μg/ml heparin (Sigma #H3149).
ES cell medium
ES cell lines were maintained on MEF-coated, pregelatinized tissue culture dishes (Falcon) in DMEM (Specialty Media #SLM-220) supplemented with 15% FBS (HyClone #SH30071.03), 2 mM GlutaMAX Supplement, 1% penicillin/streptomycin, 1% β-mercaptoethanol (Specialty Media #ES-007-E), 0.1 mM nonessential amino acids (Gibco #11140-035), 1 mM sodium pyruvate, and 1000 IU/ml leukemia inhibitory factor (LIF) (Millipore #ESG1107).
Derivation of ES cell lines
Embryos were collected at the 2–8 cell stage by flushing oviducts using M2 medium (Sigma #M7167), and cultured in KSOM medium (Millipore #MR-106-D) to the blastocyst stage. The zona pellucida of blastocysts was then removed using acid Tyrode solution (Sigma #T1788). Blastocysts were transferred separately into a well of a 96-well dish (Falcon) coated with 0.1% gelatin (Specialty Media #ES-006-B) and covered with MEF in ES medium supplemented with LIF and 1 μM PD0325901 (Axon #1408) and 3 μM CHIR99021 (Axon #1386), a combination of chemicals that is typically referred to as “2i”. After 4 days TrypLE Express (Gibco #12604-013) was used to disaggregate the embryonic outgrowths, and cells were passaged to a well of 24-well dish to derive ES cell lines.
Derivation of pre-XEN cell lines from blastocysts
Embryos were collected at the 2–8 cell stage embryos and cultured in in KSOM medium to the blastocyst stage. The zona pellucida of blastocysts was removed using acid Tyrode solution. Blastocysts were transferred separately into a well of a 4 well-dish (Nunc #176740) coated with 0.1% gelatin and covered with MEF in ES medium supplemented with LIF. The XEN lines were derived as described20.
Derivation of post-XEN cell lines from whole E5.5 or E6.5 embryos
The ectoplacental cone was removed with forceps or needles. A whole embryo was placed in a well of a 4-well dish (Nunc #176740) coated with 0.1% gelatin and covered with MEF in TS medium including F4H. After the embryos formed a large outgrowth, TrypLE Express was used to disaggregate the outgrowths and passage cells into a 4-well dish. When cells reached 70–80% confluency, they were passaged into a well of a 12-well dish until XEN cell lines were obtained, which were then passaged into a well of a 6-well dish. If the outgrowth of an E5.5 embryo grew well and XEN cells thrived, we continued to culture cells in TS medium including F4H. But if the outgrowth grew slowly and XEN cells were surrounded by trophoblast-derived cells, we cultured cells in ES medium supplemented with LIF, culture conditions that would inhibit trophoblast-derived cells; when XEN cells started to become abundant, we switched to TS medium including F4H. To prepare XEN cells for RNA extraction, cells were cultured in dishes coated with 0.1% gelatin but without MEF in TS medium including F4H.
Derivation of post-XEN cell lines at E6.5 from disaggregated embryos
The ectoplacental cone was removed with forceps or needles. A whole embryo was treated with 0.1 mg/ml collagenase (Gibco #17104-019) and 0.01 mg/ml deoxyribonuclease (Gibco #D5025) for 20–30 min at room temperature, followed by 0.2 mg/ml TrypLE for 5 min at room temperature. The embryo was disaggregated into single cells using a glass pipette with a diameter of 50–60 μm. Cells were transferred into a well of a 4-well dish (Nunc #176740) coated with 0.1% gelatin and covered with MEF in TS medium including F4H. Three days later XEN colonies appeared. We picked these colonies, disaggregated them with a glass pipette or by TrypLE Express for 5 min at 37 °C, and passaged them into a well of a 4-well dish. When cells reached 70–80% confluency, they were passaged into a well of a 12-well dish until XEN cell lines were obtained, which were then passaged into a well of a 6-well dish.
Population characteristics
Images representing Fields of View of post-XEN cell lines were imaged for intrinsic fluorescence of GFP, fluorescence of DAPI, and GATA4 immunoreactivity. Automated cell population characteristics were determined with CellProfiler http://cellprofiler.org/ (Broad Institute, Cambridge, MA, USA). The DAPI signal was used to segment individual cells by thresholding, de-clumping, and applying size and roundness filter in an effort to evaluated single cells that are not MEFs. After filtering, the GFP and GATA4 signals were evaluated in each cell, and the population characteristics for these two markers were quantified.
In vitro differentiation
Gelatin-coated plates were prepared by coating with 0.1% gelatin overnight at room temperature. XEN cells were cultured in TS medium with F4H and with or without 10 ng/mL BMP4 (Peprotech, 120–05) on gelatin-coated plates for four days.
Immunofluorescence and imaging
Cell lines X42, X44, X47 (PDGFRa-GFP × CAG::mRFP1); X35, X36 (B2D6F1 × D4/EGFP); X97, X107 (D4/EGFP × DBA2/N); X-E6.5-81346-8 (ROSA-STOP-taulacZ x Sox17-Cre); X-E6.5-Z0617-2, X-E6.5-Z0617-5, X-E6.5-Z0617-8 (R26-tauGFP41 x Sox17-Cre); X-E6.5-78097-4 (Xist1loxGFP × DBA/2 N); X-E6.5-82278-4 (Gata6-mTomato × Cdx2-GFP); X-E6.5-Z0663-12 (CD1 × PDGFRa-GFP); and X-E5.5-6, X-E5.5-8, X-E5.5-9, X-E5.5-10, X-E5.5-13 (R26-tauGFP41 × Sox17-Cre) were cultured in 4-well or 24-well dishes. Cells were fixed in 4% paraformaldehyde at 4 °C overnight or room temperature for 30 min, permeabilized with 0.1% Triton X-100 in 1× PBS (1× PBST) for 30 min and blocked with 5% normal donkey serum (Jackson ImmunoResearch Laboratories, #017-000-121) diluted in 1× PBST (blocking solution) for 1 hr. Primary antibodies were diluted at 1:50–1:200 in blocking solution and samples incubated at 4 °C rotating overnight. After three 10-min washes in 1× PBST 10 min, samples were incubated for 1–1.5 hr at room temperature in a 1:500 dilution of secondary antibody in blocking solution, then washed and covered with 1× PBST containing DAPI. Images were taken with an AMG EVOS (Life Technologies), Zeiss LSM 710 confocal microscope, and Nikon SMZ25 stereofluorescence microscope.
Primary antibodies from Santa Cruz Biotechnology were against GATA4 (#SC-1237), DAB2 (#SC-13982), OCT3-4 (#SC-5279), NANOG (#SC-376915), and CDX2 (#SC-166830). Primary antibodies from R&D Systems were against GATA6 (#AF1700), SOX7 (#AF2766), SOX17 (#AF1924), and PDGFRa (#AF1062). Primary antibodies against E-cadherin (ECCD2) were from Invitrogen (#13-1900). Secondary antibodies from Jackson ImmunoResearch Laboratories were Cy5™ AffiniPure Donkey anti-goat IgG (H+L) (#705-175-147), and Cy5™ AffiniPure Donkey anti-rabbit IgG (H+L) (#711-175-152). Secondary antibodies from Invitrogen were Donkey anti-rabbit IgG (H+L) with Alexa Fluor 546 (#A10040), Goat anti-rat IgG (H+L) with Alexa Fluor 546 (#A11081), Donkey anti-mouse IgG with Alexa Fluor 546 (#A10036), Donkey anti-mouse IgG with Alexa Fluor 488 (#A21202), and Goat anti-chicken IgY (H+L) with Alexa Fluor 488 (#A11039).
NanoString multiplex gene expression analysis
Cells were cultured in 12-well plates with TS medium including F4H or ES medium supplemented with LIF. Dissociated cells were collected by trypsinization and centrifugation. Cell pellets were dispensed directly in RNAlater Stabilization Solution (Qiagen) and stored in −80 °C for later use. Cell pellets were lysed in RLT Lysis Plus Buffer using a TissueLyser LT (Qiagen) at 40 Hz for 2 min. Total RNA extraction was performed using RNeasy Plus Micro kit (Qiagen) according to manufacturer’s protocol. The custom NanoString CodeSet “Extra” was used. 100 ng of total RNA samples were hybridized at 65 °C for 18 hr and processed with the nCounter Analysis System GEN1 (NanoString Technologies). The reporter counts were processed using nSolver Analysis Software v2.5 (NanoString). Two normalizations were performed to the counts, the first normalization to the generic positive controls, followed by normalization to the reference genes, Actb and Gapdh. Normalized counts are displayed in a heatmap generated by the nSolver Analysis Software v2.5, using agglomerative clustering.
NanoString probe sequences
Below are the nucleotide sequences of the NanoString probes; first line is the capture probe, second line the reporter probe.
Actb
ACGATGGAGGGGCCGGACTCATCGTACTCCTGCTTGC
GGGTGTAAAACGCAGCTCAGTAACAGTCCGCCTAGAAGCACTTGCGGTGC
Dab2
GTCTCCTCGAGCATCAGGCACATCATCAATACCGATTAGCTTGGC
CCAGCTGCTGCCATTCCCTTGAGTTTCATCATAGAATCCTGACTCATTTT
Dnmt3l
GAGGCAGCGCATACTGCAGGATCCGGTGGAACTGGAACATG
CCATGAATATCCAGAAGAAGGGCCGCTGACTCTCCTGGC
Dppa4
CAAGTCTTTACAGTTGACTGCTGAACTGGTTATGACGCCCGTTGTGCTGG
CACTACAACCCAGGGAAGAGGACATGCATGCGGAGGCTACAGGTATAAGC
Dppa5a
CGCACGGCCCACAGCTCCAGGTTCAGGAAGTTTTAGTAC
CCTGCCAAGGAACCAGACTTCAGGGAAGACGAGATCAAGCTTATCCACCA
Esrrb
TTTCCAGAATGAACCGCTTCATCTTTAGGACATCCTGTCAACCCAAACCC
GAGCAGGTAAAGCCGGAGGACTTGTCATGAAAGTGGCGTGTCCAT
Fgf4
ATTCTGGTAACAAAATTCCAAAGATACAGTCTTGTCCCTGGGCGCAGGAA
ACAGACCGACTCGGTAACAGTGGCAGATACAGAGCAGAAACATCAAACCC
Foxa2
CACAGACAGGTGAGACTGCTCCCTTGAGGCCTGAAG
TCCCTTCCCTATTTAGAATGACAGATCACTGTGGCCCATCTATTTAGGGA
Foxq1
GCTGTCCTTACTCCGAGGTTTAGAGACTTTGAGCGGAAGACAAGCG
TTTTGATTGTTGGGTGAAGTGAGGAGTGGAGTGATAGAAGTTGGTGCAGT
Fst
AGGACTTTGTGATACACTTTCCCTCATAGGCTAATCCAATGGATCTGCCC
CTTGGAATCCCATAGGCATTTTTTCCCGCCGCCACACTGGATATCTTCAC
Fxyd3
TGAGCCCGCCGACTCGGAGGCTGTACCAATCATAGTAGAAAGGATCATTT
GCCACTCATAAGGACTATAATGCCCAGGGCACAGAGAATCCCTGCACAAA
Gapdh
ATACTTGGCAGGTTTCTCCAGGCGGCACGTCAGATCCACGACG
CCTCAGATGCCTGCTTCACCACCTTCTTGATGTCATC
Gata4
AGAGCCAGGTAACTGTCTGACTTAAGAGGGCTTGGCTTGG
GCAAACAGTCTGTATTTTCTAGACAAAGGATCTGTGCTGGAGAAAGTCCC
Gata6
ATCTGGACTGCTGGACAATATCAGACACAAGTGGTATGAGGCCTTCAGAG
GGCACAGAAATCACGCATCGAAGGAATGTTATGTCTGCATTTTTGCTGCC
Krt8
TTCCCATCTCGGGTTTCAATCTTCTTCACAACCACAGCCTTG
CAGTGGCCATTCACTTGGACACGACATCAGAAGACTCGGACACCAGC
Lama1
CATTGGCTAAATCGGCATGGCGGTCATCCTTGATACAGACAGAACTCAGA
CATAACCTTTCCTACATGGACACTGACCTGGCCACTTTC
Lamb1
CCAGGAAGGAATGCGGTCCTGAATGTACTGCCTTTCCACCACAATGAC
AAAAACTCCAAATAAGCCCCTTCAGGCACCCGGACGAACCCAGGTCCTGT
Nanog
CATATTTCACCTGGTGGAGTCACAGAGTAGTTCAGGAATAATTCCAAGGC
GAAGGAACCTGGCTTTGCCCTGACTTTAAGCCCAGATGTTGCGTAAGTCT
Nr0b1
CACTTGAAAAAGAAACTCTTGATGGCCTGGACCGCAGCAGCTGGGAGCAA
CCTTTCAGATAGGCATACTCTTTGGTGTCAATGTTCAGACTCCAG
Pdgfra
TATGGAGTAAGTCGCTCTCACACACTTACCACACCACCATGTTGGGAACA
AAACATGAACAGGGGTATCTGGAAGCCATCTTGTATTGGAAGACCCTTCC
Pou5f1
ACATGGTCTCCAGACTCCACCTCACACGGTTCTCAATG
AGTGATCTGCTGTAGGGAGGGCTTCGGGCACTTCAGAA
Pth1r
CCTGGGCACGGTGCAGCAGGAAAATCTGTTCCTCTTTGGTAAAGACATCG
TGCTGTGTGCAGAACTTCCTTGAGCAGCTTGTCACATTGCG
Sox2
CCCCGCCGCCCTCAGGTTTTCTCTGTACAAAAATAGTCCCCCAAAAAGAA
TGCGTAGTTTTTTTCCTCCAGATCTATACATGGTCCGATTCCCCCGCCCT
Sox7
AGAAATCAGCACACCCCAACACTTTTGTGGACAGACGTTTTAGGTTTCTA
TCATCTTTTGCATCTGTAGATAAGAGTATGCTACAGCTCTGCTCT
Sox17
GGCAGATACTGTTCGAATTCCGTGCGGTCCACCTC
TGTCCCTGGTAGGGAAGACCCATCTCGGGCTTATACACAAAG
Tet3
TTTTCAAAGAGCTGAATGAATGCACCAGGATTTTAGGATGGGCGTGTTTC
GTCAGGCCCAACCTCTCAATGTCACAGAAATTAAAGCACCAACGTTCTCA
Ethics statement
All animal studies were carried out in accordance with the German Animal Welfare Act, European Communities Council Directive 2010/63/EU, and institutional ethical and animal welfare guidelines of the Max Planck Institute of Biophysics and the Max Planck Research Unit for Neurogenetics. All experimental protocols were approved by the Regierungspräsidium Darmstadt and the Veterinäramt of the City of Frankfurt.
Additional Information
How to cite this article: Lin, J. et al. Efficient derivation of extraembryonic endoderm stem cell lines from mouse postimplantation embryos. Sci. Rep. 6, 39457; doi: 10.1038/srep39457 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Enders, A. C., Given, R. L. & Schlafke, S. Differentiation and migration of endoderm in the rat and mouse at implantation. Anat. Rec. 190, 65–77 (1978).
Gardner, R. L. & Rossant, J. Investigation of the fate of 4.5 day post-coitum mouse inner cell mass cells by blastocyst injection. J. Embryol. exp. Morph. 52, 141–152 (1979).
Byrd, N. et al. Hedgehog is required for murine yolk sac angiogenesis. Development 129, 361–372 (2002).
Damert, A., Miquerol, L., Gertsenstein, M., Risau, W. & Nagy, A. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development 129, 1881–1892 (2002).
Rossant, J. & Tam, P. P. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 136, 701–713 (2009).
Artus, J. & Hadjantonakis, A. K. Troika of the mouse blastocyst: lineage segregation and stem cells. Curr. Stem Cell Res. Ther. 7, 78–91 (2012).
Evans, M. J. & Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981).
Martin, G. R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 78, 7634–7638 (1981).
Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A. & Rossant, J. Promotion of trophoblast stem cell proliferation by FGF4. Science 282, 2072–2075 (1998).
Kunath T. et al. Imprinted X-inactivation in extra-embryonic endoderm cell lines from mouse blastocysts. Development 132, 1649–1661 (2005).
Uy, G. D., Downs, K. M. & Gardner, R. L. Inhibitation of trophoblast stem cell potential in chorionic ectoderm coincides with occlusion of the ectoplacental cavity in the mouse. Development 129, 3913–3924 (2002).
Hayakawa, K., Himeno, E., Tanaka, S. & Kunath, T. Isolation and manipulation of mouse trophoblast stem cells. Curr. Protoc. Stem Cell Biol. doi: 10.1002/9780470151808.sc01e04s32 (2015).
Najm, F. et al. Isolation of epiblast stem cells from preimplantation mouse embryos. Cell Stem Cell 8, 318–325 (2011).
Brons, I. G. et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195 (2007).
Tesar, P. J. et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199 (2007).
Bao, S. et al. Epigenetic reversion of postimplantation epiblast to pluripotent embryonic stem cells. Nature 461, 1292–1295 (2009).
Brown, K. et al. eXtraembryonic ENdoderm (XEN) stem cells produce factors that activate heart formation. PLoS One 5, e13446 (2010).
Liu, W., Brown, K., Legros, S. & Foley, A. C. Nodal mutant eXtraembryonic ENdoderm (XEN) stem cells upregulate markers for the anterior visceral endoderm and impact the timing of cardiac differentiation in mouse embryoid bodies. Biol. Open 1, 208–219 (2012).
Zhao, Y. et al. A XEN-like state bridges somatic cells to pluripotency during chemical reprogramming. Cell 163, 1678–1691 (2015).
Niakan, K. K., Schrode, N., Cho, L. T. & Hadjantonakis, A. K. Derivation of extraembryonic endoderm stem (XEN) cells from mouse embryos and embryonic stem cells. Nat. Protoc. 8, 1028–1041 (2013).
Fujikura, J. et al. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev. 16, 784–789 (2002).
Shimosato, D., Shiki, M. & Niwa, H. Extra-embryonic endoderm cells derived from ES cells induced by GATA factors acquire the character of XEN cells. BMC Dev. Biol. 7, 80 (2007).
Wamaitha, S. E. et al. Gata6 potently initiates reprogramming of pluripotent and differentiated cells to extraembryonic endoderm stem cells. Genes Dev. 29, 1239–1255 (2015).
Shimoda, M. et al. Sox17 plays a substantial role in late-stage differentation of the extraembryonic endoderm in vitro. J. Cell. Sci. 120, 3859–3869 (2007).
Niakan, K. K. et al. Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes Dev. 24, 312–236 (2010).
Cho, L. T. et al. Conversion from mouse embryonic to extra-embryonic endoderm stem cells reveals distinct differentiation capacities of pluripotent stem cell states. Development 139, 2866–2877 (2012).
Parenti, A., Halbisen M. A., Wang, K., Latham, K. & Ralston, A. OSKM induce extraembryonic endoderm stem cells in parallel to induced pluripotent stem cells. Stem Cell Reports 6, 447–455 (2016).
Hamilton, T. G., Klinghoffer, R. A., Corrin, P. D. & Soriano, P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell. Biol. 23, 4013–4025 (2003).
Long, J. Z., Lackan, C. S. & Hadjantonakis, A. K. Genetic and spectrally distinct in vivo imaging: embryonic stem cells and mice with widespread expression of a monomeric red fluorescent protein. BMC Biotechnol. 5, 20 (2005).
Hadjantonakis, A. K., Gertsenstein, M., Ikawa, M., Okabe, M. & Nagy, A. Non-invasive sexing of preimplantation stage mammalian embryos. Nat. Genet. 19, 220–222 (1998).
Plusa, B., Piliszek, A., Frankenberg, S., Artus, J. & Hadjantonakis, A. K. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 135, 3081–3091 (2008).
Artus, J., Panthier, J. J. & Hadjantonakis, A. K. A role for PDGF signaling in expansion of the extra-embryonic endoderm lineage of the mouse blastocyst. Development 137, 3361–3372 (2010).
Lin, J. et al. Defects in trophoblast cell lineage account for the impaired in vivo development of cloned embryos generated by somatic nuclear transfer. Cell Stem Cell 8, 371–375 (2011).
Choi, E. et al. Dual lineage-specific expression of Sox17 during mouse embryogenesis. Stem Cells 30, 2297–2308 (2012).
Sado, T., Hoki, Y. & Sasaki, H. Tsix silences Xist through modification of chromatin structure. Dev. Cell. 9, 159–165 (2005).
Takaoka, K., Yamamoto, M. & Hamada, H. Origin and role of distal visceral endoderm, a group of cells that determines anterior-posterior polarity of the mouse embryo. Nat. Cell. Biol. 13, 743–752 (2011).
McDole, K. & Zheng, Y. Generation and live imaging of an endogenous Cdx2 reporter mouse line. Genesis 50, 775–782 (2012).
Paca, A. et al. BMP signaling induces visceral endoderm differentiation of XEN cells and parietal endoderm. Dev. Biol. 361, 90–102 (2012).
Artus, J. et al. BMP4 signaling directs primitive endoderm-derived XEN cells to an extraembryonic visceral endoderm identity. Dev. Biol. 361, 245–262 (2012).
Geiss, G. K. et al. Direct multiplex measuement of gene expression with color-coded probe pairs. Nat. Biotechnol. 26, 317–325 (2008).
Khan, M., Vaes, E. & Mombaerts, P. Regulation of the probability of mouse odorant receptor gene choice. Cell 147, 907–921 (2011).
Khan, M., Vaes, E. & Mombaerts, P. Temporal patterns of odorant receptor gene expression in adult and aged mice. Mol. Cell. Neurosci. 57, 120–129 (2013).
Kadokawa, Y., Fuketa, I., Nose, A., Takeichi, M. & Nakatsuji, N. Expression pattern of E- and P-cadherin in mouse embryos and uteri during the periimplantation period. Dev. Growth Differ. 31, 23–30 (1989).
Viotti, M., Nowotschin, S. & Hadjantonakis, A. K. SOX17 links gut endoderm morphogenesis and germ layer segregation. Nat. Cell Biol. 16, 1146–1156 (2014).
Morris, S. A. et al. Origin and formation of the first two distinct cell types of the inner cell mass in the mouse embryo. Proc. Natl. Acad. Sci. USA 107, 6364–6369 (2010).
Snow, M. H. L. Gastrulation in the mouse: Growth and regionalization of the epiblast. J. Embryol. exp. Morph. 42, 293–203 (1977).
Morgani, S. & Brickman, J. LIF supports primitive endoderm expansion during pre-implantation development. Development 142, 3488–3499 (2015).
Pettitt, S. J. et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495 (2009).
Behringer, R., Gertsenstein, M., Vintersten, K. & Nagy, A. Manipulating the Mouse Embryo: A Laboratory Manual, 4th ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (2014).
Kang, M., Piliszek, A., Artus, J. & Hadjantonakis, A. K. FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development 140, 267–279 (2013).
Mombaerts, P. et al. Visualizing an olfactory sensory map. Cell 87, 675–686 (1996).
Wen, S. et al. Genetic identification of GnRH receptor neurons: a new model for studying neural circuits underlying reproductive physiology in the mouse brain. Endocrinology 152, 1616–1526 (2011).
Acknowledgements
P.M. thanks the Max Planck Society for generous support.
Author information
Authors and Affiliations
Contributions
J.L. designed research, performed experiments, and analyzed data. M.K. and B.Z. performed experiments and analyzed data. P.M. supervised research and wrote the manuscript. All authors contributed to editing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lin, J., Khan, M., Zapiec, B. et al. Efficient derivation of extraembryonic endoderm stem cell lines from mouse postimplantation embryos. Sci Rep 6, 39457 (2016). https://doi.org/10.1038/srep39457
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39457
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
