
Abstract
Time-resolved two-photon photoemission (TR-2PPE) spectroscopy is employed to probe the electronic states of a C60 fullerene film formed on highly oriented pyrolytic graphite (HOPG), acting as a model two-dimensional (2D) material for multi-layered graphene. Owing to the in-plane sp2-hybridized nature of the HOPG, the TR-2PPE spectra reveal the energetics and dynamics of photocarriers in the C60 film: after hot excitons are nascently formed in C60 via intramolecular excitation by a pump photon, they dissociate into photocarriers of free electrons and the corresponding holes, and the electrons are subsequently detected by a probe photon as photoelectrons. The decay rate of photocarriers from the C60 film into the HOPG is evaluated to be 1.31 × 1012 s−1, suggesting a weak van der Waals interaction at the interface, where the photocarriers tentatively occupy the lowest unoccupied molecular orbital (LUMO) of C60. The photocarrier electron dynamics following the hot exciton dissociation in the organic thin films has not been realized for any metallic substrates exhibiting strong interactions with the overlayer. Furthermore, the thickness dependence of the electron lifetime in the LUMO reveals that the electron hopping rate in C60 layers is 3.3 ± 1.2 × 1013 s−1.
Similar content being viewed by others


Introduction
Research on the two-dimensional (2D) graphene material, which consists of a sp2 carbon network, has recently received a great deal of attention owing to its remarkable physical properties1,2, such as high electron conductivity and chemical stability, as well as optical transparency. While many practical possible usages of graphene have been proposed, a combination of graphene and an organic molecular film is of particular interest3,4,5,6,7,8,9,10,11,12,13,14, because they nicely suit the requirements for the next generation of low-cost, flexible, large area optical and electronic devices. From these viewpoints, an investigation of the electronic interaction and charge carrier dynamics at the graphene-organic interface is one of the key issues to resolve in the dawning era of graphene-based functional devices. Although the characteristics of organic molecules or functional nanomaterials deposited on a graphene surface have been investigated in this decade with scanning probe microscopy4,6,8,9,12, there have been few spectroscopic investigations relevant to the electronic structures and carrier dynamics at the hetero surface and interface.
Fullerene C60, consisting of a sp2 carbon network, is one of the materials most studied as a functional organic semiconductor since its discovery15 and it is also known to be a good electron acceptor for organic photovoltaic (OPV) devices, including solar cells, owing to its high electron affinity16,17,18,19,20,21. Indeed, graphene-C60 based OPV devices have been recently fabricated by several groups3,7,11,14. Since the transport gap (Et) of crystalline C60 is believed to be 2.3–2.4 eV22, it is likely that free photocarriers are generated with a photon energy (hν) considerably higher than 2.3–2.4 eV by the dissociation of highly-excited excitons (hot exciton). In fact, efficient photoconductance appears in a C60 film at hν > 2.3 eV23. So far, the ultrafast dynamics of photoexcited electronic states of C60 has been studied by several research groups24,25,26,27,28,29,30, but there are still insufficient spectroscopic investigation tracking precisely the photocarrier electron generated in C60 thin films.
In this article, we present a spectroscopic study of photocarrier electron dynamics of C60 deposited on a highly-oriented pyrolytic graphite (HOPG) as a model for multi-layered graphene by means of time-resolved two-photon photoemission (TR-2PPE) spectroscopy. The TR-2PPE unveils the ultrafast dynamics of electronically excited states on a surface system with femtosecond (fs = 10−15 s) time resolution31,32, and it has lately begun to be applied to graphene-related surfaces33,34.
The experimental results reveal that the energy levels of C60, which are independent of the C60 coverage, are little influenced by the 2D electronic nature of the in-plane sp2 carbon network; the overlayer molecular C60 film is electronically decoupled from the HOPG substrate. The photoexcited electron behavior in the lowest unoccupied molecular orbital (LUMO) of C60 suggests that free photocarrier electrons are generated by the dissociation of hot excitons without forming lower-lying excitons. The decay rate of photocarrier electrons into the HOPG substrate is very low (1.31 × 1012 s−1) compared with that on metallic substrates, which is also a characteristic of the 2D substrate. Moreover, thanks to the 2D nature of the material, it is revealed that the photocarrier electron hopping rate in C60 layers is 3.3 ± 1.2 × 1013 s−1, from the C60 thickness dependence of the TR-2PPE measurements.
Results and Discussion
Energy levels of C60 monolayer on HOPG
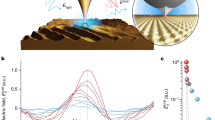
Figure 1a shows 2PPE spectra for a C60 monolayer (1 ML, see Supplementary Figs S1 and S2 for the coverage calibration) formed on HOPG by changing the incident hν at ~0.05 eV intervals. The photoelectrons emitted with surface normal (k// = 0) were recorded with a limited acceptance angle of ± 0.6°. The horizontal axis shows the final state energy (Efinal) relative to Fermi level (EF). Six spectral features labeled as L0, L2, F0, F1, F2, and H appear by the formation of C60 film. The vacuum level (Evac) is located at 4.60 eV above EF obtained from the energy of the low energy cut-off of the spectra with lower photon energy, while the intensity around the cut-off becomes stronger at hν ≥ Evac because of signals from direct (one-photon) photoemission. When the peak positions in the 2PPE spectra, including a threshold of H (red open triangle), are plotted in Fig. 1b versus hν, they can be assigned to an initial, intermediate, or final state35; an occupied state below EF (initial state: E = Efinal−2 hν), an unoccupied state between EF and Evac (intermediate state: E = Efinal−1 hν), or an unoccupied state above Evac (final state: E = Efinal) are distinguished, whose shifts are proportional to 2 hν, 1 hν, and 0 hν, respectively. Note that the intensities of final states are enhanced at specific hν marked by larger symbols in Fig. 1b owing to a resonance with an intermediate unoccupied state at EF + 1.8 eV, labeled by L1. In fact, the larger symbols are consistently fitted by a slope of 1 hν as a dotted line superimposed in Fig. 1b. Note that, the L1 structure appears more clearly in the 2PPE spectra with a wider acceptance angle (Supplementary Fig. S1) or in angle-resolved 2PPE (Supplementary Fig. S3), because it has an electronic resonance with dispersive F0–F2, as discussed below. Including the state at EF + 1.8 eV, the 2PPE spectra unveil seven electronic states; one occupied state located at EF − 2.2 eV (H), three unoccupied states between EF and Evac at EF +0.7 (L0), +1.8 (L1), and +2.7 eV (L2), and three unoccupied states above Evac at EF +5.8 (F0), +6.0 (F1), and +6.5 eV (F2). Although the peak H (red solid triangle) is broadened below hν = 4.6 eV, which is assignable to an occupied state, judging from the plot of the slope aligning with 2 hν, including that of higher energy thresholds (red open triangle).

2PPE of 1 ML C60 on HOPG.
(a) hν dependences of 2PPE spectra for 1 ML C60 film on HOPG aligned with Efinal, where the hν range was 4.13‒4.77 eV. (b) Energy positions of 2PPE structures including the onset of H against hν, which are assignable to the slope of “0 hν”, “1 hν”, or “2 hν”. Factors obtained by liner fits of the plots are indicated nearby the data. The dotted line shows the “1 hν” slope by an unoccupied level at EF + 1.8 eV (see text). (c) Total 2PPE yield integrated whole energy region versus hν in which the maximum is corresponding to that optical absorption (1T1u) of C60 film. Note that recovering intensity at the highest hν is due to a contribution of one photon photoemission.
The series of F0–F2 states show large band dispersions parallel to the surface from an angle-resolved 2PPE measurement, while the others have little dispersion (see Supplementary Fig. S3). The result suggests that F0–F2 are assignable to a series of higher-lying superatomic molecular orbitals (SAMOs)36,37,38,39,40,41; the wavefunctions of SAMOs generally diffuse outside the molecular framework, which would form highly dispersive band structures in a molecular film with a periodic surface structure. As shown in Fig. 1a, F0–F2 are enhanced only around non-dispersive L1 (Supplementary Fig. S3), suggesting that the observation of SAMOs in 2PPE requires an initial or intermediate electronic state to allow the resonance. It is known that the lowest (1s-) SAMO is experimentally observed at around EF + 3.3–3.8 eV in a C60 monolayer on noble metal substrates with a strongly delocalized nature36,38,39,40. The absence of the 1s-SAMO in our study is attributable to the 2D electronic structure of the HOPG substrate; since the electronic structures of graphene below and above EF are governed by sp2 hybridized orbitals oriented parallel to the graphene plane, the chemical interaction with the overlayer material is very weak compared to metallic materials8. In fact, the energetics of a C60 film are almost independent (±0.1 eV) of the C60 coverage from 1 to 30 ML (see Supplementary Fig. S4), while metallic substrates considerably perturb the electronic states of C60, especially for the first layer, by strong metal-molecule coupling at the interface. Owing to the weak chemical interaction, an electron excitation through the conduction band of the substrate into the unoccupied states of C60 film would be unlikely. For C60 on HOPG, electron excitation into the 1s-SAMO is allowed only from an occupied level (e.g. the highest occupied molecular orbital (HOMO)) of C60 with intra- or inter-molecular excitation. Since the onset of the HOMO is EF–1.8 eV from the ultraviolet photoelectron spectroscopy (UPS) (Supplementary Fig. S5), the HOMO electron cannot access energetically to the 1s-SAMO with the current hνs (4 – 5 eV). Indeed, the 1s-SAMO observed in a 1 ML C60 film on Ag(111) with a similar hν quickly disappeared on additional C60 deposition38, indicating that injection of electrons from a metal substrate is essential to resolve the 1s-SAMO with the hν range used here.
More importantly, we now focus on the observed energy levels of H and L0–L2, which are essential to understand the electronic and optical properties of the C60-HOPG system, rather than higher-lying C60 SAMOs. Comparing with UPS result (see Supplementary Fig. S5), the peak H is assignable to HOMO of C6022,42.
For the assignments of L0, Dutton et al. had initially assigned it to LUMO of C6028, where they observed very similar 2PPE spectra with the present result for >2 ML C60 coverages of Cu(111) substrate. However, they subsequently reassigned the corresponding peak (L0) to the next LUMO (LUMO + 1) involved in Frenkel excitions29,43. The main reason of their change in the assignment relied on the energy matching with a theoretical result for the optical transition in solid C6044. They have also argued that the LUMO (exciton) is expected to be located at slightly above EF, which cannot be resolved using their experimental conditions. Another 2PPE group using a pump photon of 3 eV has insisted on other assignments of 2PPE structures; peaks observed at HOMO + 2.2 eV, and +3 eV are attributed to free photocarrier electron in LUMO, and LUMO + 1, respectively26,27. In spite of their previous (re)assignments, judging from our experimental findings on HOPG substrate, it should be more reasonable to assign L0 located at HOMO + 2.9 eV as a photocarrier electron in LUMO for the following reasons.
C60 has a HOMO (hu) and LUMO (t1u), with an optical gap (Eopt) of 1.7–1.9 eV45,46,47. The UPS and inverse photoemission spectroscopy (IPES) for a C60 film have shown that the LUMO is observed at EF + 0.6 eV, which provides the HOMO-LUMO gap (ΔE) of 2.35 eV48. The energy difference (ΔE−Eopt) is attributed to on-site Coulomb energy (exciton binding energy), U.
In the 2PPE spectroscopy, occupied states in a cationic final state are probed similar to UPS, whereas unoccupied states reflect a neutral or cationic final state depending on the excitation processes. There are two photoemission pathways via an unoccupied state: (1) When electrons are supplied from a donor state with a pump photon, the molecule can be regarded as an anion, and then the electron occupying originally unoccupied states is emitted by a probe photon. A probe photon of 2−6 eV can emit the photoelectron mainly from molecular anions in the top layer owing to the limited escape depth of electrons. In fact, the 2PPE spectra for 1 ML C60/Au(111), which show a strong molecule-substrate interaction are markedly changed at 2 ML29, suggesting that the photoelectron escape depth is around or smaller than the thickness of one monolayer.
The other pathway of (2) is via an intra-molecular excitation, in which a photo-excited e-h pair is localized at a molecule, resulting in a larger excitonic interaction between the electron and hole. After the intra-molecular excitation, the electron is emitted with the probe photon, where there are two channels of photoelectrons (2a) from the exciton and (2b) from photocarrier electrons. The latter is generated by the hot exciton dissociation into a free electron and hole.
For the C60 in the outermost layer, furthermore, the number of surrounding C60 molecules is smaller than that inside the C60, and then the polarization energy to stabilize a charged molecule becomes smaller for the surface molecules. In other words, the less crowded surroundings result in the increase of U, and the ΔE of the outermost layer is larger than that in the bulk. This effect should be paid attention to when the values are compared among the results of UPS, IPES, and 2PPE spectroscopy.
In bulk C60, ΔE has been estimated to be 2.35 eV from the thresholds of UPS and IPES49, while ΔE is 2.9 eV in the 2PPE spectroscopy obtained from H and L0 peaks. Based on the assignment of L0, indeed, the successive L1 and L2 peaks are assignable to LUMO + 1 and LUMO + 2 with reasonable energy positions observed by IPES22,42. These consistent results show that L0 is assignable to the LUMO of a neutral C60, where the photoemission from L0 occurs via process (2b).
It is known that ΔE for a C60 monolayer on a metal substrate is small compared to that in the bulk C60, because the on-site charge is more effectively screened by the substrate than by a C60 film, resulting in a reduction of U49,50,51. As mentioned above, the energy positions are almost identical regardless of the C60 coverage on HOPG (Supplementary Figs S1 and S4), which shows that the screening effect from HOPG is much smaller than that from a metal substrate, due to its low density of states near EF. Furthermore, as discussed below, electrons in L0 have a long lifetime of several picoseconds, also indicating that L0 does not originate from the LUMO + 1 involved in Frenkel excitions, as pervious reassignments29,43, otherwise it would decay rapidly into lower-lying states within a few hundred fs52,53,54. Another 2PPE group has observed a peak at HOMO + 2.2 eV using a 3 eV pump photon, and they have assigned it to the free photocarrier electron in the LUMO26,27. However, we believe the assignment is not appropriate, because the peak to peak energy separation of 2.2 eV is too small to assign to a free photocarrier in the LUMO, considering that the ΔE of 2.3 eV defined by the respective onset edges in the UPS and IPES spectra.
Despite the above, it seems reasonable that hot excitons are generated by pump photon in 2PPE spectroscopy. Figure 1c shows the hν dependence of the total 2PPE yield, which reaches a maximum at hν = 4.3–4.4 eV, implying a resonant photo-excitation in the C60 film. This energy indeed corresponds to the optical absorption of a C60 film (280 nm in wavelength)23,55, assignable to the third optically allowed resonance of 31T1u56 marked in Fig. 1c. As seen in Fig. 1a, the intensity of LUMO + 2 at EF + 2.7 eV is much weaker than that of the LUMO and LUMO + 1, also implying that electrons are dominantly excited from the HOMO at EF−2.2 eV with current hν of 4.8 eV at most; while the contribution of occupied states in HOPG is rather small. Therefore, it can be considered that hot excitons are nascently generated in the C60 film by a pump photon and subsequently dissociate into a free electron and hole without relaxing into lower-lying excitons. The excited electron temporally occupies LUMOs of surrounding non-excited C60, and is finally detected as a photoelectron excited by a probe photon. One may wonder why the hot exciton (31T1u) is not resolved as an individual peak in the 2PPE spectra. The absence of the initially-excited hot exciton in 2PPE spectra would imply a very fast (<sub 10 fs) process of hot exciton dissociation, which broadens the spectral feature of the excitonic state (lifetime broadening)32. The observed energy levels and the excitation and detection scheme in 2PPE process are schematically illustrated in Fig. 2a,b. Here, it is not exclusive for a charge transfer (CT) state of C60+C60− to be involved, and the CT state is actually predicted to be located at HOMO + 2.3 eV in theoretical calculations57. Formation of the lowest-lying excitons such as S1 and T1 would be another pathway of hot electron relaxation that may induce the photoemission by process 2a described above. The central energy of S1 (0-0 transition) is known to be 2.0 eV, whereas the absorption band of S1 distributes from 430 to 670 nm in wavelength58. Taking into account the energy of the HOMO (EF−2.2 eV), the higher energy component of S1 is expected to appear around the low energy cut-off (EF + 0−0.7 eV in the intermediate energy (Efinal−hν)) and also to exhibit relaxation dynamics on the ps-ns time scale. However, such a spectroscopic feature in the low energy region and the dynamics could not be observed (See Figs 1a and 3a shown below), showing that the formation of lower-lying excitonic states is a minor process in the current hν range employed of 4–5 eV. The formation of S1 and T1 has been actually reported in 2PPE with different experimental conditions26,27,30; a 3 eV pump photon results in a lower efficiency of photocarrier generation than that with hν ~ 4.5 eV. Furthermore, they employed a higher laser intensity (~sub-μJ/pulse)26 with low repetition rates (250 kHz) and measured for thicker C60 film (20 nm ≈ 25 ML), which may favorably induce excitons by paring the free photocarriers in C60 film due to a high photoexcitation density. It should be emphasized again that the enormous intensity around the cut-off with hν > Evac in Fig. 1a must be due to direct one photon photoemission from just below EF.

Energy diagram of observed energy levels.
(a) Energy diagram of occupied and unoccupied levels near EF as well as a hot exciton state of 1T1u. F0–F2 located above Evac are not addressed in (a,b) Drawing of the hot exciton dissociation and hopping processes during the 2PPE.

TR-2PPE spectra.
(a) TR-2PPE spectra (hν = 4.33 eV) at various Δts. Horizontal axis stands for the intermediate energy defined as Efinal−1 hν. (b) Intensity trace of LUMO + 1 against Δt, where the intensity is obtained from an integration of 0.1 eV width around EF + 1.8 eV marked in a. The trace is fitted by a convolution of Gaussian autocorrelation (116 fs in FWHM) and single exponential decay functions. The autocorrelation function was determined in advance from the intensity trace of TR-2PPE for bare HOPG at around EF + 1.5 eV (grey circle). (c) Intensity trace for LUMO. Fitted components of the first (Autocorrelation) and second terms (rise and decay) in eq. (1) are also overwritten as solid lines. Inset shows a wider Δt region (±1 ps).
TR-2PPE results
Figure 3a shows TR-2PPE spectra taken with various pump-probe delays (Δt), where a spectrum at Δt = 9 ps is subtracted as a background. The LUMO peak survives even at Δt = 3 ps, while LUMO + 1 decays more quickly within a few hundred fs. The energy positions of these peaks remain almost the same, which again suggests that free electron photocarriers are generated in the early regime. Otherwise, they would subsequently form lower-lying excitons, (e.g. S1 or T1), which recombine. Figure 3b shows the intensity of LUMO + 1 against Δt. The electron lifetime in the level is 48 ± 6 fs, evaluated by a convolution of a Gaussian autocorrelation (AC) (116 fs in full width at half maximum (FWHM)) and a single exponential decay, where the AC is determined from the intensity trace of the TR-2PPE for bare HOPG at the energy of EF + 1.2–1.7 eV; the lifetime of the electron in the intermediate energy is very short (<20 fs)59 as compared to the true AC width. As shown in Fig. 3c, interestingly, the signal intensity for LUMO is once again recovered around Δt = 100–250 fs after the excitation and gradually decreases. Assuming exponential rise and decay processes with time constants of τr and τd, the time evolution of 2PPE intensity, I(Δt), can be described as

where a and b are weight factors. In addition to the rise and decay processes of LUMO electrons, the second term of the AC component is taken into account, because photoelectrons at the corresponding energy may include two components of (i) coherent 2PPE from occupied states of HOPG and HOMO−1 of C60 (see Supplementary Fig. S5) and (ii) short-lived hot electrons in the HOPG and C60 film, both of which are not regarded as the photocarrier electrons in the LUMO. The TR-2PPE trace for these components should be very similar to the shape of the true AC for the current pulse duration. Eq. (1) reproduces the evolution of the LUMO with τr,1ML = 60 ± 9.8 fs and τd,1ML = 760 ± 24 fs. Note that the rise time τr,1ML is close to the lifetime of LUMO + 1 electron (48 ± 6 fs), suggesting that the LUMO electrons are mainly supplied from highly excited states via LUMO + 1. The LUMO lifetime on HOPG is significantly longer than those for C60 on metal substrates29,43, e.g. Dutton and co-workers have reported that the lifetime increases with the C60 thickness on Au(111) from 79 fs at 2 ML to 340 fs at 30 ML, while the corresponding peak has been assigned to a LUMO + 1* exciton29,43. Since 2D material substrate of HOPG exhibits weak electronic coupling toward adsorbed species, the decay rate into HOPG is less compared to metal substrates. In fact, the energy positions in the 2PPE spectra for C60 films on HOPG show little changes with the C60 coverage (Supplementary Figs S1 and S4).
One should also be concerned about the effect of the excitation density on the TR-2PPE results; the recombination of photocarriers generated at different C60 molecules (bimolecular recombination) would contribute to the relaxation dynamics. However, the effect is negligible because of the sufficiently low excitation density used in the current study. In the TR-2PPE measurement, assuming a photon flux of ~3 × 1010 photons·cm−2 at most (76 MHz, 10 mW, and 0.1 mm spot diameter), three photons are irradiated in an area of 10 × 10 nm2 area per pulse (an area consisting of about a hundred of C60 molecules). Therefore, the excitation density would be too low to cause bimolecular recombination.
Thickness dependence of LUMO lifetime
Since the sp2 hybridized nature of the HOPG substrate reduces photocarrier relaxation, the photocarrier hopping rate in a C60 film can be evaluated by the C60 thickness dependence of the LUMO lifetime. Figure 4a shows the time evolution of LUMO intensities obtained for various coverages of C60 films. The recovery of LUMO intensity around Δt = 100–250 fs becomes unclear at ≥2 ML, while the lifetime of LUMO + 1 is independent of the coverage (see Supplementary Fig. S6). As discussed in the previous section for spectral assignments, 2PPE probes the photocarrier electrons remaining in the topmost C60 layer even for the multilayered films. On the other hand, the photocarrier electrons are generated by the pump photon penetrating into whole C60 layers. When the photocarrier electrons at the topmost layer initially originate not only from the topmost layer but also at the deeper layers inside, the rise time behavior should become vaguer in the multilayer films, as seen in Fig. 4a. The result therefore indicates that the created photocarriers move around in the C60 film with a finite hopping rate. To evaluate the photocarrier hopping rate in the C60 film, the thickness dependence of the decay phenomena was modelled for the LUMO electrons.

Thickness dependence of LUMO lifetime.
(a) Intensity of LUMO against Δt for 1, 2, 3, and 5 ML C60. Decay times are fitted using eq. (1) for 1–3 ML, while that for 5 ML is estimated by a slope at Δt > 1 ps (dotted lines). (b) Simulated results with the interlayer hopping model for 2 ML (blue line), 3 ML (pink line), and 5 ML (black line). τd obtained in a are also shown as exp (−t/τd) (dotted lines) to compare with the simulation (solid lines).
As shown in Fig. 4a, the LUMO lifetime lengthens with the thickness, and the fitting with eq. (1) shows that τd for 1, 2, 3, and 5 ML are 730 ± 22, 1520 ± 120, 2060 ± 270, and 2510 ± 210 fs, respectively. With increases in the C60 thickness, a uniform layer-by-layer structure grows without the presence of non-uniform islands (see Supplementary Fig. S7). Since the lifetime of a free electron carrier in solid C60 is much longer (ns or μs) than the observed τd60, the decay of photocarriers mainly occurs at the first C60 layer interfaced with the substrate. When the photocarrier electron hopping rate from an original C60 site to the nearest neighboring C60 is defined as kC60, the rate to the specific neighboring C60 should be 1/12 kC60, because of the fcc crystal structure of a C60 film exhibiting a coordination number of twelve. Considering that the fcc’s C60(111) surface faces to the vacuum, the interlayer hopping rate from original C60 site at the i th layer to the i + 1 or i − 1 th layer is 3/12 kC60 each (six neighboring C60 molecules exist at the i th layer). In addition, it can be assumed that the interlayer hopping from top and bottom layers are modified to be 3/9 kC60 owing to the absence of a layer to one side. Based on these concepts, here, the electron population of each layer, Nn,m, is evaluated, in which n is the total number of C60 layers, and m is the targeted C60 layer (1 ≤ m ≤ n). First, we simply evaluate the value of kC60 for the result for 2 ML: the number of LUMO electrons at the first interface layer (N2,1) and the top layer (N2,2) can be simply described by


where kd is the decay rate of the photocarrier electron density by relaxing into the substrate from the first C60 monolayer, i.e. kd = 1/τd,1ML = 1.37 × 1012 s−1. Since the irradiated photon penetrates through the C60 films, it is reasonable to assume that all of C60 layers are uniformly photoexcited as N2,1 = N2,2 = 1 at t = 0, whereas the time trace of the TR-2PPE reflects the photocarrier at the top layer (N2,2). Then, the above differential equations can be solved numerically for the variable kC60. The blue solid line in Fig. 4b is the simulated result for N2,2, where kC60 is evaluated to be 3.3 ± 1.2 × 1013 s−1. So far, the electron hopping rate in unoccupied energy levels has been evaluated by analysis of the resonant autoionization spectrum which has been established as core-hole-clock-spectroscopy61. In this method, however, the accessible frequency range of 1016 > k > 1014 s−1 is limited by the lifetime of the core hole (~a few fs). In fact, Brühwiler et al. have evaluated the hopping rate of a LUMO + 1 electron in solid C60 to be ~1.7 × 1014 s−1, while they could not evaluate it for the LUMO62. The difference in the hopping rates depending on the energy levels can be explained by the width of the density of states63; the hopping rate would become higher for a wider band width. From the resonance window of LUMO + 1 (~0.5 eV) with SAMOs (supplemental material, S3), the band width is larger than that of LUMO (~0.2 eV in FWHM).
The above simulation can also be applied to the result for 3 ML. By using the central value, 3.3 × 1013 s−1, for kC60 as obtained above, however, the simulation for 3 ML slightly overestimates the experimental data (not shown). This could be caused by the polarization effect; owing to the reduction of U at the topmost C60, it can be qualitatively deduced that the hopping rate of electrons from an interior C60 layer to the topmost layer should be smaller than that from the topmost to an interior layer. Indeed, the result for 3 ML could be well simulated when the former and latter rates are modified to be 2.1 × 1013 and 4.5 × 1013 s−1 (pink solid line in Fig. 4b), respectively, as upper and lower limits of the error for kC60 (±1.2 × 1013 s−1). The reasonable modification falling within its uncertainty suggests the validity of our interlayer hopping model where hopping within an in-plane intralayer does not contribute explicitly. Note that the simulation for 5 ML (N5,5) overestimates the experimental result (τd,5ML), possibly due to thickness inhomogeneity (see Supplementary Fig. S7 for scanning tunneling microscopy images) that could open a decay channel through an edge state of C60 layers.
We think that the hopping rate obtained with the above analysis is intrinsic for organic species, but independent of the substrate. The analysis will therefore be also applicable universally for the determination of kd at the interface between an organic film and a metallic substrate, where kd is typically too large to be resolved experimentally because of the strong substrate-molecule interaction.
Summary
We observed the hot exciton dissociation and decay dynamics of generated photocarrier electrons in a C60 film on a HOPG substrate through the use of TR-2PPE spectroscopy. Unlike metal substrates, the 2D material substrate of HOPG substrate suppresses the electronic interaction with overlayered C60 film, which enables us to detect clear time required for photocarrier generation within a hundred fs. The time evolutions of the 2PPE intensity and their C60 thickness dependence reveal that the generated photocarriers randomly and frequently hop in the film until decaying into the substrate, where the electron hopping rate in C60 is independently and directly evaluated to be 3.3 ± 1.2 × 1013 s−1. The results have clarified fundamental aspects of photocarrier generation and carrier transportation in organic materials with time-domain measurements, and will provide a key to improving the performance of graphene-based organic devices.
Methods
Sample preparation and 2PPE measurements
Prior to C60 deposition, an HOPG substrate was cleaved in air and cleaned by heating (670 K, 50 h) in an ultrahigh vacuum (UHV) chamber, where the base pressure was better than 1 × 10−8 Pa. C60 was then deposited in the UHV system with a rate of 0.07 ML/min, monitoring with a quartz microbalance. The C60 coverage was calibrated from 2PPE measurements (Supplementary Figs S1 and S2). TR-2PPE measurements were carried out using the third harmonics (hν = 4.13–4.77 eV, p-polarization) of a Ti:sapphire laser (~100 fs, 76 MHz), which was separated into pump and probe photons with a beam splitter. Both pump and probe photons are recombined at the sample in UHV with a concave mirror (f = 400 mm). In this optical configuration (“skew configuration”) the autocorrelation of incident photons can approximate a Gaussian peak function without interference oscilation31. Photoelectrons emitted in the normal direction were detected by a hemispherical electron energy analyzer (VGSCIENTA: R3000), where the energy of photoelectron was calibrated by UPS of an Au plate providing a clear Fermi edge as zero binding energy at Efinal = EF + hν (21.22 eV) for Efinal (Supplementary Fig. S5). A sample bias of −3 V was applied to collect low-energy photoelectrons around the cut-off energy (Evac). Energy and time resolutions of the TR-2PPE system were about 20 meV and 20 fs, respectively. The sample temperature was 293 K during the TR-2PPE measurement.
Additional Information
How to cite this article: Shibuta, M. et al. Direct observation of photocarrier electron dynamics in C60 films on graphite by time-resolved two-photon photoemission. Sci. Rep. 6, 35853; doi: 10.1038/srep35853 (2016).
References
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nature Mater. 6, 183–191 (2007).
Castro Neto, A. H., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. Mod. Phys. 81, 109–162 (2009).
Wu, J. et al. Organic solar cells with solution-processed graphene transparent electrodes. Appl. Phys. Lett. 92, 263302 (2008).
Wang, Q. H. & Hersam, M. C. Room-temperature molecular resolution characterization of self-assembled organic monolayers on epitaxial graphene. Nature Chem. 1, 206–211 (2009).
Mao, J. H. et al. Tunability of Supramolecular Kagome Lattices of Magnetic Phthalocyanines Using Graphene-Based Moiré Patterns as Templates. J. Am. Chem. Soc. 131, 14136–14137 (2009).
Dichtel, W. R. et al. J. Oriented 2D Covalent Organic Framework Thin Films on Single-Layer Graphene. Science 332, 228–231 (2011).
Yu, D., Park, K., Durstock, M. & Dai, L. J. Fullerene-Grafted Graphene for Efficient Bulk Heterojunction Polymer Photovoltaic Devices. J. Phys. Chem. Lett. 2, 1113–1118 (2011).
Cho, J. et al. Structural and Electronic Decoupling of C60 from Epitaxial Graphene on SiC. Nano Lett. 12, 3018–3024 (2012).
Lu, J. et al. Using the Graphene Moiré Pattern for the Trapping of C60 and Homoepitaxy of Graphene. ACS Nano 6, 944–950 (2012).
Salzmann, I. et al. Epitaxial Growth of π-Stacked Perfluoropentacene on Graphene-Coated Quartz. ACS Nano. 6, 10874–10883 (2012).
Schlierf, A., Samorì, P. & Palermo, V. Graphene-organic composites for electronics: Optical and electronic interactions in vacuum, liquids and thin solid films. J. Mater. Chem. C. 2, 3129–3143 (2014).
Yin, Z. et al. Graphene-Based Materials for Solar Cell Applications. Adv. Energy Mater. 1, 1–19 (2014).
Christodoulou, C. et al. Tuning the Electronic Structure of Graphene by Molecular Dopants: Impact of the Substrate. ACS Appl. Mater. Interfaces 7, 19134–19144 (2015).
Kim, K. et al. Structural and Electrical Investigation of C60 Graphene Vertical Heterostructures. ACS Nano 9, 5922–5928 (2015).
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F. & Smalley R. E. C60: Buckminsterfullerene. Nature 318, 162–163 (1985).
Yu, G., Gao, J., Hummelen, J. C., Wudl, F. & Heeger, A. J. Polymer photovoltaic cells: enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 270, 1789–1791 (1995).
Yoo, S., Domercq, B. & Kippelen, B. Efficient thin-film organic solar cells based on pentacene/C60 heterojunctions. Appl. Phys. Lett. 85, 5427–5429 (2004).
Kim, Y. et al. A strong regioregularity effect in self-organizing conjugated polymer films and high-efficiency polythiophene:fullerene solar cells. Nature Mater. 5, 197–203 (2006).
Kandada, A. R. S. et al. Ultrafast Energy Transfer in Ultrathin Organic Donor/Acceptor Blend. Sci. Rep. 3, 2073, 10.1038/srep02073 (2013).
Grancini, G. et al. Hot exciton dissociation in polymer solar cells. Nat. Mater. 12, 29–33 (2013).
Lane, P. A., Cunningham, P. D., Melinger, J. S., Esenturk, O. & Heilweil, E. J. Hot photocarrier dynamics in organic solar cells. Nature Commun. 6, 7558, 10.1038/ncomms8558 (2015).
Lof, R. W., van Veenendaal, M. A., Koopmans, B., Jonkman, H. T. & Sawatzky, G. A. Band gap, excitons, and Coulomb interaction in solid C60, Phys. Rev. Lett. 68, 3924–3927 (1992).
Kazaoui, S., Ross, R. & Minami, N. Intermolecular charge-transfer excitation in C60 films: Evidence from luminescence and photoconductivity. Phys. Rev. B 52, R11665–R11667 (1995).
Dick, D. et al. Transient spectroscopy of excitons and polarons in C60 films from femtoseconds to milliseconds. Phys. Rev. Lett. 73, 2760–2763 (1994).
Fleischer, S. B. et al. Phototransformation in visible and near-IR femtosecond pump-probe studies of C60 films. Appl. Phys. Lett. 69, 296–298 (1996).
Jacquemin, R., Kraus, S. & Eberhardt, W. Direct observation of the dynamics of excited electronic states in solids: F-Sec time resolved photoemission of C60 . Solid State Commun. 105, 449–453 (1998).
Link, S., Scholl, A., Jacquemin, R. & Eberhardt, W. Electron dynamics at a Ag/C60 metal–semiconductor interface. Solid State Commun. 113, 689–693 (2000).
Dutton, G. & Zhu, X.-Y. Unoccupied States in C60 Thin Films Probed by Two-Photon Photoemission. J. Phys. Chem. B 106, 5975–5981 (2002).
Dutton, G. Quinn, D. P., Lindstrom, C. D. & Zhu, X.-Y. Exciton dynamics at molecule-metal interface: C60/Au(111). Phys. Rev. B 72, 045441 (2005).
Rosenfeldt, A. C., Göhler, B. & Zacharias, H. Time-resolved photoelectron spectroscopy of low-energy excitations of 4 × 4 C60/Cu(111). J. Chem. Phys. 133, 234704 (2010).
Petek, H. & Ogawa, S. Femtosecond time-resolved two-photon photoemission studies of electron dynamics in metals. Prog. Surf. Sci. 56, 239–310 (1997).
Echenique, P. M. et al. Decay of electronic excitations at metal surfaces. Surf. Sci. Rep. 52, 219–317 (2004).
Armbrust, N., Güdde, J., Jakob, P. & Höfer, U. Time-Resolved Two-Photon Photoemission of Unoccupied Electronic States of Periodically Rippled Graphene on Ru(0001). Phys. Rev. Lett. 108, 056801 (2012).
Niesner, N. et al. Trapping Surface Electrons on Graphene Layers and Islands. Phys. Rev. B 85, 081402(R) (2012).
Giesen, K., Hage, F., Himpsel, F. J., Riess, H. J. & Steinmann, W. Two-photon photoemission via image-potential states. Phys. Rev. Lett. 55, 300–303 (1985).
Feng, M., Zhao, J. & Petek, H. Atomlike, Hollow-Core-Bound Molecular Orbitals of C60 . Science 320, 359–362 (2008).
Zhao, J., Feng, M. & Petek, H. The Superatom States of Fullerenes and Their Hybridization into the Nearly Free Electron Bands of Fullerites. ACS Nano 3, 853–864 (2009).
Dutton, G. J., Dougherty, D. B., Jin, W., Reutt-Robey, J. E. & Robey, S. W. Superatom orbitals of C60 on Ag(111): Two-photon photoemission and scanning tunneling spectroscopy. Phys. Rev. B 84, 195435 (2011).
Chan, W.-L. et al. Communication: Momentum-resolved quantum interference in optically excited surface states. J. Chem. Phys. 135, 031101 (2011).
Feng, M., Zhao, J., Huang, T., Zhu, X.-Y. & Petek, H. The Electronic Properties of Superatom States of Hollow Molecules. Acc. Chem. Res. 44, 360–368 (2011).
Mignolet, B., Johansson, J. O., Campbell, E. E. B. & Remacle, F. Probing rapidly-ionizing super-atom molecular orbitals in C60: A computational and femtosecond photoelectron spectroscopy study. ChemPhysChem 14, 3332–3340 (2013).
Rudolf, P., Golden, M. S. & Brühwiler, P. A. Studies of fullerenes by the excitation, emission, and scattering of electrons. J. Elect. Spectrosc. Relat. Phenom. 100, 409–433 (1999).
Dutton, G. & Zhu, X.-Y. Distance-Dependent Electronic Coupling at Molecule-Metal Interfaces: C60/Cu(111). J. Phys. Chem. B 108, 7788–7793 (2004).
Dresselhaus, M. S., Dresselhaus, G. & Eklund, P. C. Science of Fullerenes and Carbon Nanotubes. (Academic Press, 1996).
Kremer, R. K. et al. High-temperature conductivity study on single-crystal C60 . Appl. Phys. A 56, 211–214 (1993).
Beu, T. A., Onoe, J. & Hida, A. First-principles calculations of the electronic structure of one-dimensional C60 polymers. Phys. Rev. B 72, 155416 (2005).
Pudlak, M. & Pincak, R. Energy gap between highest occupied molecular orbital and lowest unoccupied molecular orbital in multiwalled fullerenes. Phys. Rev. A 79, 033202 (2009).
Zhao, W. & Kahn, A. Charge transfer at n-doped organic-organic heterojunctions. J. Appl. Phys. 105, 123711 (2009).
Brühwiler, P. A., Maxwell, A. J., Nilsson, A., Mårtensson, N. & Gunnarsson, O. Auger and photoelectron study of the Hubbard U in C60, K3C60, and K6C60 . Phys. Rev. B 48, 18296–18299 (1993).
Tzeng, C. T., Tsuei, K. D., Cheng, H. M. & Chu, R. Y. Covalent bonding and hole–electron Coulomb interaction U in C60 on Be(0001) surfaces. J. Phys.: Condens. Matter 19, 176009, 10.1088/0953-8984/19/17/176009 (2007).
Torrente, I. F., Franke, K. J. & Pascual, J. I. Spectroscopy of C60 single molecules: the role of screening on energy level alignment. J. Phys.: Condens. Matter 20, 184001, 10.1088/0953– 8984/20/18/184001 (2008).
Tahara, T. Advances in Multi-Photon Processes and Spectroscopy, Vol. 16, Chap. 1 (ed. Lin, S. H., Villaeys, A. A. & Fujimura, Y. ) (World Scientific Publishing Company, 2004).
Varene, E., Bogner, L., Bronner, C. & Tegeder, P. Ultrafast Exciton Population, Relaxation, and Decay Dynamics in Thin Oligothiophene Films. Phys. Rev. Lett. 109, 207601 (2012).
Shibuta, M., Hirata, N., Eguchi, T. & Nakajima, A. Probing of an Adsorbate-Specific Excited State on an Organic Insulating Surface by Two-Photon Photoemission Spectroscopy. J. Am. Chem. Soc. 136, 1825–1831 (2014).
Cheville, R. A. & Halas, N. J. Time-resolved carrier relaxation in solid C60 thin films. Phys. Rev. B 45, 4548–4550 (1992).
Bauernschmitt, R., Ahlrichs, R., Hennrich, F. H. & Kappes, M. M. Experiment Values Time Dependent Density Functional Theory Prediction of Fullerene Electronic Absorption. J. Am. Chem. Soc. 120, 5052–5059 (1998).
Shirley, E. L., Benedict, L. X. & Louie, S. G. Excitons in Solid C60 . Phys. Rev. B 54, 10970–10977 (1996).
Pavlovich, V. S. & Shpilevsky, E. M. Absorption and fluorescence spectra of C60 fullerene concentrated solutions in hexane and polystyrene at 77–300 K. J. Appl. Spec. 77, 362–369 (2010).
Moos, G., Gahl, C., Fasel, R., Wolf, M. & Hertel, T. Anisotropy of Quasiparticle Lifetimes and the Role of Disorder in Graphite from Ultrafast Time-Resolved Photoemission Spectroscopy. Phys. Rev. Lett. 87, 267402 (2001).
Lee, C. H., Yu, G., Kraabel, B., Moses, D. & Srdanov, V. I. Effects of oxygen on the photocarrier dynamics in a C60 film: Studies of transient and steady-state photoconductivity. Phys. Rev. B 49, 10572–10576 (1994).
Föhlisch, A. et al. Direct observation of electron dynamics in the attosecond domain. Nature 436, 373–376 (2005).
Brühwiler, P. A. et al. C 1s autoionization study of electron hopping rates in solid C60 . Phys. Rev. Lett. 71, 3721–3724 (1993).
Brédas, J. L., Calbert, J. P., da Silva Filho, D. A. & Cornil, J. Organic semiconductors: A theoretical characterization of the basic parameters governing charge transport. Proc. Natl. Acad. Sci. USA 99, 5804–5809 (2002).
Acknowledgements
This work is partly supported by the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan, and by JSPS KAKENHI of Grant-in-Aids for Young Scientists (B) Grant Number 25810010 and for Scientific Research (A) Grant Number 15H02002.
Author information
Authors and Affiliations
Contributions
M.S., K.Y. and T.O. performed the TR-2PPE experiments and data analysis. M.N. and T.E. took the STM images of C60 film on graphite. M.S., T.E. and A.N. wrote the manuscripts. A.N. supervised the research project.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shibuta, M., Yamamoto, K., Ohta, T. et al. Direct observation of photocarrier electron dynamics in C60 films on graphite by time-resolved two-photon photoemission. Sci Rep 6, 35853 (2016). https://doi.org/10.1038/srep35853
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35853
This article is cited by
-
Al13− and B@Al12− superatoms on a molecularly decorated substrate
Nature Communications (2022)
-
Strong modification of the transport level alignment in organic materials after optical excitation
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
