
Abstract
The aim of our study was to assess the utility of next generation sequencing (NGS) for predicting toxicity and clinical response to thiopurine drugs in paediatric patients with inflammatory bowel disease. Exome data for 100 patients were assessed against biochemically measured TPMT enzyme activity, clinical response and adverse effects. The TPMT gene and a panel of 15 other genes implicated in thiopurine toxicity were analysed using a gene based statistical test (SKAT-O test). Nine patients out of 100 (Crohn’s disease- 67, ulcerative colitis- 23 and IBDU-10) had known TPMT mutations associated with deficient enzyme activity. A novel and a highly pathogenic TPMT variant not detectable through standard genotyping, was identified through NGS in an individual intolerant to thiopurines. Of the 14 patients intolerant to thiopurines, NGS identified deleterious TPMT variants in 5 individuals whereas the biochemical test identified 8 individuals as intolerant (sensitivity 35.7% and 57.14%; specificity 93.75% and 50% respectively). SKAT-O test identified a significant association between MOCOS gene and TPMT activity (p = 0.0015), not previously reported. Although NGS has the ability to detect rare or novel variants not otherwise identified through standard genotyping, it demonstrates no clear advantage over the biochemical test in predicting toxicity in our modest cohort.
Similar content being viewed by others


Introduction
Thiopurine drugs, which include azathioprine and 6-mercaptopurine (6-MP) have been effectively used in inflammatory bowel disease for more than 30 years. They are also widely used in the treatment of patients with neoplastic conditions, post-organ transplantation and a wide range of autoimmune and inflammatory conditions. However concerns over toxicity and adverse reactions frequently result in discontinuation of treatment and a switch to alternative therapies1,2. Known adverse reactions include bone marrow suppression, severe gastric intolerance, pancreatitis, hepatotoxicity, skin reactions, susceptibility to infections, risk of malignancy and flu-like symptoms2,3.
Thiopurine S-methyl transferase (TPMT) is a key enzyme involved in the metabolism of thiopurine drugs and functions by catalysing the S-methylation of aromatic and heterocyclic sulfhydryl groups4. TPMT is encoded by a gene located on chromosome 6p22, consisting of 10 exons, encoding one protein domain5. Although the precise mode of action of thiopurines is still unclear, the most important mechanism is thought to be the incorporation of 6-TGNs (thioguanine nucleotides) into the cell DNA, resulting in an impaired DNA synthesis and cell death2,6 (Fig. 1). The TPMT gene is known to exhibit genetic heterogeneity resulting in wide inter-individual differences, both in terms of clinical efficacy and toxicity profiles based on the enzyme activity2,7. Approximately 4–11% of individuals of Caucasian origin are heterozygous for a mutant TPMT allele with intermediate enzyme activity, whereas approximately 1 in 300 individuals are homozygous or compound heterozygous with a consequent low or absent TPMT activity3,7. Current clinical guidelines recommend determining TPMT status in a given patient before commencement of thiopurine therapy; this is achieved by measuring TPMT enzyme activity in the circulating red blood cells or through genotyping known TPMT variants associated with enzyme deficiency2,8,9.

Schematic diagram showing thiopurine drug metabolism and genes implicated in thiopurine-induced toxicity.
There are three main catabolic pathways for thiopurine drugs, following conversion of AZA to 6-MP: (1) Phosphorylation to 6-thioguanines (6-TGN) which are active metabolites; 6-MP is first converted by hypoxanthine guanine phosphoribosyl-transferase (HGPRT) to 6-thioinosine monophosphate (6-TIMP), which is then phophorylated to 6-TGN with inosine 5-monophosphate dehydrogenase (IMPDH1 and IMPDH2) and guanosine monophosphate synthetase (GMPS) (2) Methylation by of 6-MP by TPMT to form 6-methyl-MP (6-MMP) which is an inactive metabolite and not a substrate for IMPDH) (3) Catabolism of 6-MP to 6-thiouracil (6-TU) via xanthine dehydrogenase (XDH, synonym- Xanthine oxidase) or aldehyde oxidase 1 (AOX1). TPMT competes with IMPDH for their common substrate 6-TIMP to form 6- methylmercaptopurine nucleotides (6-MMPN). 6-TIMP can be phosphorylated by kinases to 6-thioinosine triphosphate (6-TITP), which can get dephosphorylated by inosine triphosphatase (ITPase) to form 6-TIMP again. Although the precise mode of action of thiopurines is still unclear, the most important mechanism is thought to be the incorporation of 6-TGNs into the cell DNA, resulting in an impaired DNA synthesis and cell death2,6. Abbreviations: ABCC4- ATP-binding cassette, sub-family C (CFTR/MRP), member 4; AZA- azathioprine; FSLT5- Follistatin-Like 5; GST- glutathione s-transferase; HGPRT- hypoxanthine phosphoribosyltransferase;IL6ST- Interleukin 6 signal transducer; 6-MP- 6- mercaptopurine; 6-MMPN- 6- methyl mercaptopurine nucleotides; MOCOS- Molybdenum cofactor sulfurase; MTHFR- Methyl-enetetrahydrofolate reductase ;NUDT15- Nudix (nucleoside diphosphate linked moiety X)-type motif 15; PACSIN2- Protein kinase C and casein kinase substrate in neurons 2; 6-TXMP- 6-thioxanthosine monophosphate)
The rationale for assessing the TPMT status before commencement of thiopurine therapy is to minimise the risk of adverse effects whilst aiming for an optimal clinical response. Thiopurines are best avoided in individuals who are deficient or have extremely low TPMT activity and administered at a reduced dose if TPMT activity is intermediate or low normal2,9,10.
Although TPMT is the most crucial pharmaco-gene involved in the metabolism of thiopurines, previous studies have highlighted the role of other genes, whose products substantially alter drug metabolism and consequently impact clinical efficacy or toxicity11,12,13,14,15,16,17,18. It is plausible that some proportion of adverse effects observed whilst on treatment in the context of a normal TPMT status (genotype or phenotype), could be explained by variation in genes encoding the other enzymes involved in thiopurine metabolism.
In this study, we identify genes known to be involved in thiopurine metabolism and determine all coding mutations in these genes in a cohort of 100 children with IBD.
We assess the joint effect of rare and common variants within the TPMT gene and other genes implicated in thiopurine toxicity on TPMT enzyme activity through the application of a gene based statistical test (SKAT-O). The test was also conducted to investigate the association of these variants with thiopurine tolerance and clinical response.
Materials and Methods
Study Population
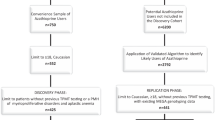
Patients were identified through the paediatric gastroenterology service database based at the University Hospital Southampton (UHS), recruited from outpatient clinics and followed through their treatment. All children were diagnosed using the Porto diagnostic criteria19 and treated according to British Society of Paediatric Gastroenterology, Hepatology and Nutrition (BSPGHAN) published guidelines9. Data for one hundred paediatric patients with TPMT phenotype defined as red blood cell enzyme activity and concurrent exome data were analysed.
Ethical Approval
The study was ethically approved by Southampton and South West Hampshire Research Ethics Committee (09/H0504/125). Informed consent was obtained from all participants before recruitment to the study. All methods were carried out in accordance with the approved and published guidelines.
TPMT Phenotype Determination
TPMT enzyme activity was measured using standard high performance liquid chromatographic technique20. TPMT enzyme activity level groups were defined as previously described (Fig. 2)20,21.

TPMT phenotype frequency distribution for the Wessex paediatric population and the research cohort.
Figure shows the TPMT phenotype frequency distribution for 524 paediatric patients, in the Wessex region (524 patients ≤ 18 years, between Dec 2010–April 2015, includes patients with/without IBD) and the TPMT phenotype distribution in our research cohort. Within our cohort we observe a statistically significant difference between the sub-category with TPMT values between 21–40 units (p = 0.001) compared to the Wessex paediatric population. This difference is not observed between the sub-categories with TPMT values between 41 to161 (p = 0.90)
Use of Thiopurines and Monitoring for Adverse Effects
British Society of Paediatric Gastroenterology, Hepatology and Nutrition (BSPGHAN) recommend initiation of treatment with thiopurines for maintenance of remission in individuals who relapse in less than 6 months, have 2 or more relapses per year following initial successful therapy and in all steroid-dependent patients. Practice with regards to initiation of treatment varies among clinicians, but is usually commenced and monitored as per the BSPGHAN guidelines9,19, with regular blood tests to monitor adverse effects such as bone marrow suppression, pancreatitis, hepatotoxicity and patients are clinically followed up to assess progress through treatment. For this study, bone marrow suppression was defined as leucopoenia (WBC < 3000 mm−3) and/or thrombocytopenia (platelets < 100,000 mm−3); liver toxicity was alanine transaminase (ALT), gamma-glutamyl transpeptidase (GGT) or alkaline phosphatase more than twice their normal levels; acute pancreatitis was defined as significant abdominal pain within 3 months of starting thiopurines, accompanied by a serum amylase or lipase level of greater than twice their normal levels as per our local laboratory values22.
Evaluation of response to therapy
Assessment of clinical poor response or non-response was based on one of the following: (1) Inability to achieve clinical improvement as assessed by global clinical assessment after at least 6 months of thiopurine therapy; (2) Corticosteroid dependence after at least 3 months of thiopurine therapy; (3) Relapse within 6 months of therapy; (4) Use of biologics within 6 months of therapy with thiopurines and; (5) Disease progression needing surgery within 6 months of thiopurine therapy commencement.
Intolerance to treatment was based on all of the following: (1) Occurrence of adverse side effects (including bone marrow suppression, severe gastric intolerance, pancreatitis, hepatotoxicity, skin reactions, flu-like symptoms); (2) Adverse effects unexplained by disease course or other concomitant co-morbidities; and (3) Partial or complete resolution of the observed adverse effects following discontinuation of therapy.
Genes Implicated in Thiopurine Toxicity
A systematic search was conducted through ovidsp using MEDLINE and EMBASE from inception to the end of March 2015. Only studies in humans describing TPMT genetic variants and other genes involved in thiopurine metabolism and toxicity were included. We applied the TPMT nomenclature committee website (http://www.imh.liu.se/tpmtalleles) that outlines all reported TPMT variants23, to cross-reference against variants identified in this study.
Whole Exome Sequencing and Data Analysis
DNA Extraction
Genomic DNA was extracted from peripheral venous blood samples collected in EDTA, using the salting out method as previously described24,25.
Exome Sequencing
Whole-exome capture was performed using Agilent SureSelect Human all Exon 51 Mb (versions 4 and 5) capture kit as previously described25. A bespoke script was used to assign individual variants as “novel” if they were not previously reported in the dbSNP137 databases26, 1000 Genomes Project (1 KG)27, the Exome Variant Server (EVS) of European Americans of the NHLI-ESP project with 6500 exomes [http://evs.gs.washington.edu/EVS/], in 46 unrelated human subjects sequenced by Complete Genomics28 or in the Southampton database of reference exomes.
Burden of Mutation Testing
The burden of genetic variations within the genes was conducted using a gene-based statistical test (the sequence kernel association optimal unified test- SKAT-O)29. To conduct the test, a group file of non-synonymous, synonymous, splicing, frameshifts and non-frameshift, stop gain and stop loss mutations was created for each of the genes analysed. SKAT-O was executed with the small sample adjustment, by applying MAF threshold of 0.05 to define rare variations within the whole cohort, and using default weights. The EPACTS software package30 was use to perform this test. The test was conducted to assess the impact of variations on TPMT enzyme activity, drug tolerance and clinical response.
Results
Clinical Data
Of the 100 patients, 78 initiated thiopurines as part of their clinical management while 22 maintained remission without recourse to this therapy. The median duration of follow up for the 78 patients commenced on thiopurines was 46 months (7–156) from diagnosis and 43 months (6–119) from starting therapy with thiopurines (Table 1).
Genes identified in TMPT metabolism and toxicity
A systematic search of genes implicated in TPMT metabolism and toxicity identified 15 genes with robust evidence from a review of over 3,000 articles (Fig. 1 and Supplementary Table S1) for which we could ascertain variation from exome data. Variation in TMPT and these genes was included in downstream analysis of: (1) biochemically assessed TMPT activity; (2) tolerance to thiopurines and; (3) response to treatment.
Biochemically measured TPMT activity in red blood cells
The TPMT enzyme activity was unexpectedly bimodal in distribution across the 100 patients examined in this study. We compared the distribution in our pIBD cohort against the same measure for an independent control group of individuals with various clinical diagnoses, aged ≤18 years from the Wessex region for whom TPMT activity was assessed by the same laboratory over the same period and observed a statistically significant distribution between the groups (P = 7.69 × 10−12) (Fig. 2). While approximately half of our cohort samples had biochemical activity levels within the normal range and half in the intermediate range, almost 80% of control samples had biochemical activity levels falling within the normal range. We believe this may reflect an ascertainment bias in our study cohort due to the fact that exome sequencing was preferentially conducted on children with most severe disease at earliest onset. This will have enriched for children with poor response to first-line treatments.
TPMT gene variants - their correlation with biochemically measured TPMT enzyme activity, thiopurine tolerance and response
We identified five TPMT variants across our cohort (Table 2). These included two non-synonymous variants (A154T and Y240C) previously known to impact TMPT function that were found to co-segregate in 9 individuals. The mean biochemical measurement of TMPT activity for this group was 36 mU/L (range 16–56), which is significantly different (p = 0.003) from that found in the 91 individuals without these mutations (mean = 68.1 mU/L). Although exome data are insufficient to resolve haplotypes, we postulate these two variants occur on a single haplotype in all nine individuals, consistent with the complex TPMT allele referred to as TPMT*3A, previously described as the most prevalent TPMT deficiency allele in Caucasians31. Of these 9 patients, 8 were commenced on thiopurine treatment and four were later identified as intolerant to this therapy. All four patients that tolerated thiopurine drugs showed therapeutic response.
One novel non-synonymous variant (p.A73V) was identified in a female patient with Crohn’s disease and a biochemical activity level of 55 mU/L. In silico tools indicate this variant is strongly conserved (Phylop = 0.99) and likely to have a deleterious impact on enzyme function (GERP = 4.98). The patient was initiated on thiopurine treatment at a reduced dose (1 mg/kg) but developed severe persistent nausea and treatment was discontinued. This variant has since been catalogued as a functionally significant allele by the TPMT nomenclature committee (http://www.imh.liu.se/tpmtalleles) with a unique allele number TPMT*39.
One UC patient harboured a rare intronic variant (in heterozygote form) proximal to the exon 7 splice junction (c.420-4G > A), this patient had a biochemical activity level of 32 mU/L, was administered thiopurine treatment which was tolerated and subsequently demonstrated therapeutic response. The MaxEnt score (0.95) for this splice junction variant predicts minimal impact on splicing within the gene. While this is positively consistent with drug tolerance, it does not explain the relatively low biochemical activity level observed in this patient.
Finally, we observed a common synonymous variant (I158I) at high frequency (37 heterozygotes and 57 homozygotes). Mean biochemical activity level is 67.1 mU/L (32–99) and 66 mU/L (152-145) across the subgroup of individuals carrying the variant in heterozygous and homozygous state respectively. The very common frequency of this variant and the fact it does not alter amino acid composition is consistent with silent variation having no impact on function.
Burden of mutation testing within TPMT and additional genes implicated in thiopurine toxicity
We interrogated exome data across fifteen additional genes implicated in thiopurine metabolism and toxicity. We observed mutations across 11 genes (no variation in HPRT1, GSTM1, FSLT5 and MTHFR).
SKAT-O test was applied to investigate the joint effect of rare, low frequency and common variations within these genes on TPMT enzyme activity, thiopurine tolerance and response.
Significant evidence for association was observed within MOCOS (p = 0.0015) and TPMT (p = 0.0017) gene with TPMT biochemical activity levels. The test also detected a nominal association between GMPS and drug tolerance (p = 0.0212) as well as variations within IL6ST (p = 0.0084) and ABBC4 (p = 0.0452) and drug response (Table 3, Supplementary Tables S2 and S3).
Individual tolerance and intolerance
Thiopurine drugs were discontinued in 14 individuals due to adverse effects. Eight of these patients had TPMT enzyme activity in the intermediate category and the rest had TPMT activity in the normal range.
Four of the eight individuals with TPMT enzyme activity in the intermediate range had known TPMT mutations associated with deficient enzyme activity and one individual had a novel TPMT mutation described in the previous section. In the rest of the nine individuals, there was enrichment for deleterious variants within the MOCOS gene and the AOX1 gene (Table 4).
Prediction of thiopurine toxicity showed a specificity of 93.75% through detection of TPMT risk variants compared to 50% for the biochemical test. Both tests had a low sensitivity of 37% and 57% respectively for predicting toxicity (Table 5). However, all the five patients with deleterious variants within the TPMT gene detected through NGS were also identified as potentially intolerant through the biochemical test. Although the biochemical test identified a higher number of individuals intolerant to thiopurines, the difference between the two approaches in predicting toxicity was not statistically significant (P value 0.45, Fisher’s exact test). The clinical data of individuals intolerant to thiopurines, is shown in Table 6 and of the entire cohort in Supplementary Table S4.
Discussion
It is well established that in a small percentage of patients, TPMT genotyping alone or in combination with TPMT phenotype is insufficient to predict tolerance to thiopurine drugs2. Several TPMT variants associated with deficient enzyme activity have been described23, however an appreciable subset of patients with intermediate to low activity do not harbour known risk alleles32. Our study identified nine individuals with the known TPMT mutations (9% compared to 11% reported in previous studies)31. All nine individuals had TPMT enzyme activity levels in the intermediate range in line with expectations; mean TPMT value across this group was significantly different compared to individuals without these mutations (36.6 mU/L compared to 68.1 mU/L, p = 0.003). However 43% of individuals with TPMT activity in the intermediate range did not have the known TPMT mutations. Our results indicate that although prediction of thiopurine toxicity through NGS has a higher specificity compared to the biochemical test, the sensitivity of both methods is clinically suboptimal (35.7% and 57.14% respectively). Among the 14 individuals intolerant to thiopurines, all the 5 patients who harboured deleterious TPMT variants would also have been predicted as potentially intolerant to thiopurines through the biochemical test. Furthermore, all the nine individuals with deleterious variants within the TPMT gene across the cohort of 100 patients would also have been identified as potentially intolerant as all these nine individuals had TPMT enzyme activity levels within the intermediate range. Hence based on first principles, NGS did not have a clear advantage over the biochemical test in predicting thiopurine toxicity.
We identified a highly pathogenic novel variant in TPMT in an individual who had TPMT enzyme activity level of 55 mU/L, but developed severe gastrointestinal toxicity despite a reduced dose. This suggests that thiopurine toxicity can develop in individuals harbouring rare or yet unknown variants, not detected through standard genotyping. As a widely used practical approach, TPMT genotyping of known variants is considered only when biochemical tests suggest a deficiency or if a patient has recently been transfused. However, a normal genotype for known variants cannot exclude the possibility of rare variation causing TPMT deficiency and development of adverse effects2. Exome sequencing therefore can be a powerful tool in identifying individuals at risk of toxicity who could have been missed through standard TPMT genotyping.
The SKAT-O test identified a significant association between variations within the MOCOS gene and TPMT enzyme activity (p = 0.0015). Molybdenum cofactor sulfurase (MOCOS) is a protein-coding gene located on 18q12, which sulfurates the molybdenum cofactor in XDH and AOX1, key enzymes involved in the degradation of thiopurines33. Previous studies have suggested a role for MOCOS gene in thiopurine metabolism with possible impact on clinical outcomes in patients with mutations, however an association between MOCOS and TPMT enzyme activity has not been explored. This is the first study to identify a significant role for this gene with variations causing alterations in biochemical enzyme activity. Further work is required to determine how MOCOS influences TPMT function.
We also detected a nominal association between GMPS and drug tolerance (p = 0.0212). GMPS is involved in the phosphorylation of 6-TIMP (6-thioinosine monophosphate) to thioguanine nucleotides, which is an important step for thiopurines to exert their cytotoxic effects. Further mechanistic studies will be required to elucidate the molecular mechanisms and clearly define the role of these genes involved in the thiopurine metabolic pathway.
A limitation of this study is that only children who had undergone exome analysis with concurrent TPMT values were selected for the study. While approximately half of our cohort patients had TPMT activity in the intermediate range, only 20% of control samples had TPMT activity in the intermediate category. Exome sequencing was preferentially conducted on children with the most severe disease phenotype, which would have enriched for patients with poor response to first-line treatments. Secondly, the selected population is predominantly Caucasian and a relatively small cohort, thereby limiting the general applicability of the results. Following a systematic search of literature to identify genes implicated in thiopurine toxicity, a panel of 15 genes was prioritised for assessment. It is it is possible that potentially pathogenic variants in other genes may have been missed through this approach. However extension of the list of genes examined would compromise power in the highest priority candidate genes.
Although there is no clear advantage of NGS over the biochemical test in predicting toxicity, our study demonstrates the strength of NGS as a powerful tool in identifying pathogenic variants in patients not detected through standard genotyping. As high throughput sequencing becomes more accessible and affordable, a more objective approach to assessing pharmaco-genomic variation in all genes involved in the drug metabolism pathway may be indicated in selected patients to guide treatment strategies. Replication in larger studies would be required for a comprehensive curation of candidate genes, possibly paving the way for a targeted gene panel as a reliable predictor of toxicity.
Additional Information
How to cite this article: Coelho, T. et al. Genes implicated in thiopurine-induced toxicity: Comparing TPMT enzyme activity with clinical phenotype and exome data in a paediatric IBD cohort. Sci. Rep. 6, 34658; doi: 10.1038/srep34658 (2016).
References
Present, D. H. et al. Treatment of Crohn’s disease with 6-mercaptopurine. A long-term, randomized, double-blind study. The New England journal of medicine 302, 981–987, doi: 10.1056/NEJM198005013021801 (1980).
Benkov, K. et al. Role of thiopurine metabolite testing and thiopurine methyltransferase determination in pediatric IBD. Journal of pediatric gastroenterology and nutrition 56, 333–340, doi: 10.1097/MPG.0b013e3182844705 (2013).
Liu, Y. P. et al. Association between Thiopurine S-methyltransferase Polymorphisms and Thiopurine-Induced Adverse Drug Reactions in Patients with Inflammatory Bowel Disease: A Meta-Analysis. PloS one 10, e0121745, doi: 10.1371/journal.pone.0121745 (2015).
Tai, H. L. et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. American journal of human genetics 58, 694–702 (1996).
Hamdan-Khalil, R. et al. Identification and functional analysis of two rare allelic variants of the thiopurine S-methyltransferase gene, TPMT*16 and TPMT*19. Biochemical pharmacology 69, 525–529, doi: 10.1016/j.bcp.2004.10.011 (2005).
Haglund, S. Interindividual differences in thiopurine metabolism . Linkoping University Medical Dissertations (2011).
Weinshilboum, R. M. & Sladek, S. L. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. American journal of human genetics 32, 651–662 (1980).
Relling, M. V. et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clinical pharmacology and therapeutics 89, 387–391, doi: 10.1038/clpt.2010.320 (2011).
Sandhu B. K., Fell, J., Beattie, R. M. & Mitton, S. G. Guidelines for the Management of Inflammatory Bowel Disease in Children in the United Kingdom. J Pediatr Gastroenterol Nutr. 2010 Feb;50 Suppl 1, S1–13 (2010).
Relling, M. V. et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clinical pharmacology and therapeutics 93, 324–325, doi: 10.1038/clpt.2013.4 (2013).
Palmieri, O. et al. Sequential evaluation of thiopurine methyltransferase, inosine triphosphate pyrophosphatase, and HPRT1 genes polymorphisms to explain thiopurines’ toxicity and efficacy. Alimentary pharmacology & therapeutics 26, 737–745, doi: 10.1111/j.1365-2036.2007.03421.x (2007).
Farfan, M. J. et al. Prevalence of TPMT and ITPA gene polymorphisms and effect on mercaptopurine dosage in Chilean children with acute lymphoblastic leukemia. BMC cancer 14, 299, doi: 10.1186/1471-2407-14-299 (2014).
Kudo, M. et al. Genetic variations in the HGPRT, ITPA, IMPDH1, IMPDH2, and GMPS genes in Japanese individuals. Drug metabolism and pharmacokinetics 24, 557–564 (2009).
Kurzawski, M., Dziewanowski, K., Safranow, K. & Drozdzik, M. Polymorphism of genes involved in purine metabolism (XDH, AOX1, MOCOS) in kidney transplant recipients receiving azathioprine. Therapeutic drug monitoring 34, 266–274, doi: 10.1097/FTD.0b013e31824aa681 (2012).
Stocco, G. et al. PACSIN2 polymorphism influences TPMT activity and mercaptopurine-related gastrointestinal toxicity. Human molecular genetics 21, 4793–4804, doi: 10.1093/hmg/dds302 (2012).
Ban, H. et al. The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. Journal of gastroenterology 45, 1014–1021, doi: 10.1007/s00535-010-0248-y (2010).
Yang, S. K. et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nature genetics 46, 1017–1020, doi: 10.1038/ng.3060 (2014).
Karas-Kuzelicki, N., Jazbec, J., Milek, M. & Mlinaric-Rascan, I. Heterozygosity at the TPMT gene locus, augmented by mutated MTHFR gene, predisposes to 6-MP related toxicities in childhood ALL patients. Leukemia 23, 971–974, doi: 10.1038/leu.2008.317 (2009).
Ibd Working Group of the European Society for Paediatric Gastroenterology, H. & Nutrition. Inflammatory bowel disease in children and adolescents: recommendations for diagnosis–the Porto criteria. Journal of pediatric gastroenterology and nutrition 41, 1–7 (2005).
Ford, L. T. & Berg, J. D. Determination of thiopurine S-methyltransferase activity in erythrocytes using 6-thioguanine as substrate and a non-extraction liquid chromatographic technique. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences 798, 111–115 (2003).
Ford, L., Graham, V. & Berg, J. Whole-blood thiopurine S-methyltransferase activity with genotype concordance: a new, simplified phenotyping assay. Annals of clinical biochemistry 43, 354–360 (2006).
Heap, G. A. et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nature genetics 46, 1131–1134, doi: 10.1038/ng.3093 (2014).
Appell, M. L. et al. Nomenclature for alleles of the thiopurine methyltransferase gene. Pharmacogenetics and genomics 23, 242–248, doi: 10.1097/FPC.0b013e32835f1cc0 (2013).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research 16, 1215 (1988).
Andreoletti, G. et al. Exome analysis of patients with concurrent pediatric inflammatory bowel disease and autoimmune disease. Inflammatory bowel diseases 21, 1229–1236, doi: 10.1097/MIB.0000000000000381 (2015).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic acids research 29, 308–311 (2001).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65, doi: 10.1038/nature11632 (2012).
Drmanac, R. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81, doi: 10.1126/science.1181498 (2010).
Ionita-Laza, I. e. a. “Sequence Kernel Association Tests for the Combined Effect of Rare and Common Variants.” American journal of human genetics 92.6 (2013): 841–53. Web. 13 Jan. 2014.
Kang, H. M., Z, X., Sim, X. & Ma, C. Biostatistics Dept, Univ Michigan, Ann Arbor, Ann Arbor, MI. “EPACTS (Efficient and Parallelizable Association Container Toolbox)”. N. p., n.d.
Schaeffeler, E. et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 14, 407–417 (2004).
Colombel, J. F. et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 118, 1025–1030 (2000).
Ichida, K., Matsumura, T., Sakuma, R., Hosoya, T. & Nishino, T. Mutation of human molybdenum cofactor sulfurase gene is responsible for classical xanthinuria type II. Biochemical and biophysical research communications 282, 1194–1200, doi: 10.1006/bbrc.2001.4719 (2001).
Acknowledgements
The authors are grateful to all patients and their families. We thank Rachel Haggarty, senior research nurse for recruitment of patients; Nikki J Graham and Sylvia J Diaper for technical assistance in the DNA laboratory; Eleanor G Seaby for assistance with diagrams and Olivia Kaye for help with biochemical data extraction. The work presented here, was financially supported by The Crohn’s in Childhood Research Association (CICRA) and The Gerald Kerkut Charitable Trust.
Author information
Authors and Affiliations
Contributions
T.C. and G.A. contributed equally to the paper; wrote the main manuscript text. J.J.A. prepared figures and helped with clinical data extraction. A.B. and N.A.A. helped with clinical data extraction. Y.G. and A.P.W. contributed to the outline of the paper, helped with the interpretation of findings. R.M.B. supervised the work, helped with clinical data extraction, contributed to the outline of the paper. S.E. supervised the work throughout, scrutinised every detail of the paper, designed the overall structure of the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Coelho, T., Andreoletti, G., Ashton, J. et al. Genes implicated in thiopurine-induced toxicity: Comparing TPMT enzyme activity with clinical phenotype and exome data in a paediatric IBD cohort. Sci Rep 6, 34658 (2016). https://doi.org/10.1038/srep34658
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34658
This article is cited by
-
A review article of inflammatory bowel disease treatment and pharmacogenomics
Beni-Suef University Journal of Basic and Applied Sciences (2023)
-
Genome-Wide Association Study for the Genetic Determinants of Thiopurine Methyltransferase Protein Expression in Human Livers and Racial Differences
Pharmaceutical Research (2023)
-
A bioinformatics approach to the identification of novel deleterious mutations of human TPMT through validated screening and molecular dynamics
Scientific Reports (2022)
-
Azathioprine/mercaptopurine
Reactions Weekly (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
