
Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most aggressive and lethal malignancies. Long non-coding RNAs (lncRNAs) are a novel class of non-protein-coding transcripts that have been implicated in cancer biogenesis and prognosis. By repurposing microarray probes, we herein analysed the lncRNA expression profiles in two public PDAC microarray datasets and identified 34 dysregulated lncRNAs in PDAC. In addition, the expression of 6 selected lncRNAs was confirmed in Ren Ji cohort and pancreatic cell lines, and their association with 80 PDAC patients’ clinicopathological features and prognosis was investigated. Results indicated that AFAP1-AS1, UCA1 and ENSG00000218510 might be involved in PDAC progression and significantly associated with overall survival of PDAC. UCA1 and ENSG00000218510 expression status may serve as independent prognostic biomarkers for overall survival of PDAC. Gene set enrichment analysis (GSEA) analysis suggested that high AFAP1-AS1, UCA1 and low ENSG00000218510 expression were correlated with several tumorigenesis related pathways. Functional experiments demonstrated that AFAP1-AS1 and UCA1 were required for efficient invasion and/or proliferation promotion in PDAC cell lines, while ENSG00000218510 acted the opposite. Our findings provide novel information on lncRNAs expression profiles which might be beneficial to the precise diagnosis, subcategorization and ultimately, the individualized therapy of PDAC.
Similar content being viewed by others


Introduction
Pancreatic cancer remains one of the most aggressive and lethal malignancies with an extremely poor prognosis worldwide1. It is the fourth and seventh leading cause of cancer-related deaths in USA and China, respectively2,3, and is projected to rank the second by 2030 in USA4. Despite 50 years of research and therapeutic development, it has a dismal overall median survival of 6 months5 and 5-year survival rate of approximately 7% currently2. Among all pancreatic cancer cases, pancreatic ductal adenocarcinoma (PDAC) accounts for approximately 85% whose most effective therapy is surgery plus appropriate chemotherapeutic strategy, but only 15 to 20% of patients are eligible for surgical resection due to late diagnosis and early metastasis6. Hence, novel and effective strategies against this devastating disease are urgently needed. Better understanding of the genetic and molecular disorders of the disease is the key to early diagnosis, appropriate treatment and improved prognosis of patients with PDAC.
In recent years, integrative genomic studies have revealed that the vast majority of the human transcriptome is noncoding RNAs (ncRNAs), which are divided into short noncoding RNAs (<200 nucleotides: containing rRNAs, miRNAs, snRNAs, snoRNAs, siRNAs, and piRNAs) and long noncoding RNAs (lncRNAs, >200 nucleotides)7,8,9. They all have limited or no protein-coding capacity. miRNAs, which have been the most extensively investigated, are found to be dysregulated and involved in most human carcinogenesis and other disease processes10,11. More recently, the roles of lncRNAs have attracted considerable attention and exploded interest9. Accumulating evidence indicates that lncRNAs are frequently aberrantly expressed in cancers8,9,13,14, and some of them play a significant role in oncogenic or tumor-suppressive pathways and have similar diagnostic and prognostic power to that of mRNA and miRNA signatures8,14,15,16. LncRNAs are believed to be implicated in diverse biological, developmental and pathological processes and act through mechanisms such as chromatin reprogramming, cis or trans regulation at neighboring genes and post-transcriptional regulation of mRNA processing12,17,18. LncRNAs achieve these regulatory specificity through modularity, assembling diverse combinations of proteins and possibly RNA (eg, mRNA, miRNA) and DNA interactions12,18. For example, the well-known lncRNA HOTAIR is overexpressed in breast cancer where it binds polycomb repressive complex 2 (PRC2). This leads to silence of a portion of the HOXD locus, inducing altered histone H3K27 methylation and gene expression, which further enhances cancer invasiveness and metastasis19. HOTAIR is also increased in expression in many other types of cancers, including pancreatic cancer20. It was found that HOTAIR was a negative prognostic factor for pancreatic cancer patients and exhibited pro-oncogenic activity in both vitro and vivo bioassays21. Another lncRNA, the lncRNA-activated by TGF-b (lncRNA-ATB) could promote hepatocellular carcinoma (HCC) cell invasion by competitively binding the miR-200 family, upregulating ZEB1 and ZEB2, and then inducing EMT. And enforced lncRNA-ATB in HCC metastases was associated with poor prognosis22. Although a set of human lncRNAs have been identified, but lncRNA expression patterns and their characteristics in PDAC remain largely unexplored. Previous study demonstrated that lncRNA profiling could be obtained by mining the existing gene expression microarray data because a large amount of lncRNA-specific probes were fortuitously represented on the commonly used microarray platforms23,24,25,26, allowing the identification of a wealth of lncRNA profiling data in PDAC.
In this study, we aimed at conducting lncRNA expression profiles and identifying a series of differentially expressed lncRNAs in PDAC compared with adjacent nontumor pancreatic tissues by analyzing a cohort of previously published PDAC microarray datasets from the Gene Expression Omnibus (GEO). The identified lncRNAs specific to different tissue types were then verified in another independent dataset. After that, we selected the top 8 differentially expressed lncRNA probes (corresponding to 6 lncRNAs) and validated them in Ren Ji cohort and pancreatic cell lines by quantitative real-time PCR (qRT-PCR). We further investigated whether the 6 lncRNAs expression levels were associated with PDAC patients’ clinicopathological features and prognosis and explored their possible biological pathways and processes involved and cell biological functions. Our findings provide novel information on lncRNAs expression profiles which might be beneficial to the precise diagnosis, subcategorization (such as clinical phenotypes and molecular subtypes) and ultimately, the individualized therapy of PDAC.
Results
Characteristics of microarray datasets and clinical samples
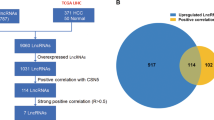
Our systematic study included two PDAC gene expression data from GEO: GSE16515 and GSE15471. GSE16515 consisted of 36 PDAC samples and 16 normal pancreatic tissue samples, a total of 52 samples. 16 pairs of samples consisted of both tumor and normal expression data, whereas the remaining 20 samples consisted of only tumor data. In GSE16515, there were 14 females and 22 males, with age ranging from 49 to 84 years old. GSE15471 contained 78 samples, including 39 PDAC tumors and 39 matching adjacent noncancerous tissue samples that were obtained from resected pancreas of 36 pancreatic cancer patients. 3 pairs were carried out replicate microarray hybridizations. Ren Ji cohort included 80 PDAC tumor samples paired with non-tumoral pancreatic tissue controls. Table 1 provides the clinical and histopathologic parameters for the 80 patients. While GSE16515 was used to derive a set of lncRNA expression signatures in PDAC, GSE15471 acted as the testing dataset to confirm the results. The selected distinctive lncRNA expressions were further to be verified in Ren Ji cohort and pancreatic cell lines. The work flow of the entire study designed is illustrated in Fig. 1.

Flowchart of the study overview.
LncRNA expression profiles were retrieved from Affymetrix HG-U133 Plus 2.0 microarray through a lncRNA-mining approach. LncRNA expression analyses were performed in the training dataset (GSE16515) first and then validated in the testing dataset (GSE15471). The 6 selected lncRNAs were then verified in Ren Ji cohort and pancreatic cell lines.
Distinctive lncRNA expressions between PDAC samples and normal pancreatic tissues
Using the GSE16515 dataset as a training set for discovery and LIMMA with an adjusted P-value of less than 0.01 with a minimum fold change cut-off of 1.60 as a threshold, we first compared the lncRNA expression profiles of 16 normal pancreatic tissues to 36 PDAC tumors and identified 39 probe sets (corresponding to 34 lncRNAs) that were differentially expressed in PDAC (Table 2). Of these 39 deregulated probe sets, 24 probe sets (20 lncRNAs) were found to be up-regulated and 15 probe sets (14 lncRNAs) to be down-regulated. The top five most up-regulated lncRNAs (7 probe sets) in PDAC tumors were CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1 and UCA1, while the top five most down-regulated lncRNAs (5 probe sets) were ENSG00000218510, ENSG00000244020, ENSG00000228536, ENSG00000251161 and ENSG00000236333. Nearly half of these lncRNAs don’t have an official Human Genome Nomenclature Committee (HGNC) symbol. The non-random partitioning of samples by unsupervised hierarchical clustering of these 39 probes (34 lncRNAs) clearly separated PDAC tissues from normal tissues (Fig. 2A). Only 6 samples (5 tumor samples and 1 normal samples) were misclassified by the clustering analysis. To independently confirm our results, we conducted the same analysis to identify the lncRNA signatures on the test dataset GSE15471 and found that 25 out of 34 differentially expressed lncRNAs identified by above analysis in GSE16515 also showed significant expression changes (adjusted P < 0.01, FC ≥ 1.60) with the same direction in GSE15471. And unsupervised hierarchical clustering of all samples in the GSE15471 dataset using the 39 lncRNA probe sets revealed similar distinctions between normal pancreatic tissues and PDAC samples: one group containing 28 of 39 non-tumoral samples and 5 PDAC samples, a second group containing 34 out of 39 PDAC samples and 11 normal samples (Fig. 2B). To further make this result convincing, we determined the correlation of the distribution of all 2448 probe sets (1970 lncRNAs) and the acquired 39 distinctive probe sets (34 lncRNAs) expression differentials between the experimental dataset GSE16515 and the validation dataset GSE15471. As shown in Fig. 2C,D, both of the two distribution of expression differentials was significantly concordant between the two datasets (r = 0.717, P < 0.0001; r = 0.767, P < 0.0001, respectively), suggesting a high consistence in expression patterns of these genes among different sample sets. Thus, the lncRNA signatures identified here were likely to be representative.

Distinctive lncRNA expressions between PDAC tissues and normal pancreas.
(A) One-way hierarchical clustering of the 39 lncRNA probe sets (corresponding to 34 lncRNAs) identified as significantly different between PDAC tissues and normal pancreas in the training dataset (GSE16515). (B) Validation of the 39 probe signatures in the testing dataset (GSE15471). Each column represents one sample and each row represents one lncRNA probe set. Gene expression levels are indicated as follows: red, high expression (+3.0); green, low expression (−3.0). The bar colors in the dendrogram represent the sample types as indicated: blue, tumor; yellow, normal. (C,D) The distribution of all 2448 lncRNA probe sets (corresponding to 1970 lncRNAs) and the acquired 39 distinctive probe sets expression differentials between the experimental dataset GSE16515 and the validation dataset GSE15471.
Validation of candidate lncRNAs in PDAC patients and cell lines
To further corroborate the validity of our prior microarray findings, we selected the top 8 differentially expressed lncRNA probes (6 lncRNAs: CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1, ENSG00000218510) from the 39 deregulated probe sets between PDAC versus normal samples and analysed their expression by qRT-PCR in Ren Ji cohort and pancreatic cell lines. Ren Ji cohort included 80 PDAC samples and 80 matching adjacent normal tissue samples while cell lines contained 8 human PDAC cell lines and 2 normal pancreatic ductal epithelial cell lines. Of the 6 selected lncRNAs, 5 lncRNAs (CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1) were up-regulated and 1 lncRNA (ENSG00000218510) was down-regulated, and the fold change in mean expression intensity was ≥2.50 in GSE16515. The qRT-PCR results showed that CRNDE, NR_036488, ENSG00000244649, AFAP1-AS and UCA1 expression was significantly increased (P = 0.0112, P = 0.0003, P = 0.0007, P < 0.0001, P < 0.0001, respectively, Fig. 3A–E) and ENSG00000218510 expression was remarkably decreased (P < 0.0001, Fig. 3F) in PDAC tissues compared with paired normal pancreatic tissues. Similar result was also observed in PDAC and normal pancreatic ductal epithelial cell lines as demonstrated by the data (Fig. 3G–L and Supplementary Fig. S1A–F). Consistent with the findings in PDAC tissues, CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1 and UCA1 expression was dramatically enhanced and ENSG00000218510 reduced in all or most of 8 PDAC cell lines in relative to the nonmalignant HPDE6-C7 cells or hTERT-HPNE cells at the RNA level. Our results thus highly coincided with our prior microarray findings in lncRNA expression profiles of PDAC.

Validation of candidate lncRNAs in PDAC patients and cell lines by qRT-PCR analysis.
(A–F) Analysis of CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1 and ENSG00000218510 relative expression in human PDAC and their matched normal pancreas samples, Ren Ji cohort, n = 80, paired sample t-test. (G–L) Relative expression level of CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1 and ENSG00000218510 in 8 PDAC cell lines compared to the nonmalignant HPDE6-C7 cell line. *P < 0.05 (Student’s t-test), ns: not significant. The qRT-PCR expression data were all shown as mean ± SD and normalised by GAPDH. GAPDH: glyceraldehyde-phosphate dehydrogenase; NP: normal pancreas.
Relationship of the 6 selected lncRNAs expression with clinicopathological parameters and prognosis in PDAC patients
The relative expression of the 6 selected lncRNAs in PDAC tissues were categorized into low expression group (n = 40) and high expression group (n = 40) using the median value as cutoff. To explore the clinical significance of the 6 lncRNAs expression in PDAC, we analyzed the relationship between their expression and corresponding patients’ clinicopathological characteristics including age, gender, tumor location, TNM stage, tumor size, T classification, lymph node metastasis, distant metastasis, perineural invasion and histological differentiation in Ren Ji cohort. The results showed that in PDAC tissues AFAP1-AS1 was significantly associated with tumor size (≤3 cm vs. >3 cm; P = 0.044), UCA1 expression was significantly correlated with T classification (T1-2 vs. T3-4; P = 0.034), and ENSG00000218510 was prominently correlated with tumor size (≤3 cm vs. >3 cm; P = 0.044), distant metastasis (absent vs. present, P = 0.005) and histological differentiation (well/moderate vs. poor; P = 0.044), whereas no significant relevance was found with the other clinicopathological features and no significant associations were discovered between CRNDE, NR_036488, ENSG00000244649 expression and all of the clinicopathological parameters (Table 3).
To further evaluate the prognostic significance of the 6 selected lncRNAs in PDAC patients, the correlations between their expression and corresponding clinical follow-up information were analyzed by Kaplan-Meier analysis and log-rank test. The results showed that AFAP1-AS1, UCA1 and ENSG00000218510 expression was remarkably associated with PDAC patients’ overall survival in Ren Ji cohort (P = 0.0125, P = 0.0011, P = 0.0197, respectively, Fig. 4A–C), which indicates that patients with higher AFAP1-AS1, UCA1 expression and lower ENSG00000218510 expression have significantly shorter survival time than those with lower AFAP1-AS1, UCA1 expression and higher ENSG00000218510 expression. The other 3 lncRNAs CRNDE, NR_036488 and ENSG00000244649 didn’t exhibit prognostic value in Ren Ji cohort (Figure not shown). Furthermore, univariable and multivariable analyses were performed to identify the risk factors correlated with patients’ prognosis. Univariable Cox regression analyses showed that tumor size (>3 cm vs. ≤3 cm), lymph node metastasis (present vs. absent), histological differentiation (poor vs. well/moderate), expression of AFAP1-AS1 (high vs. low), expression of UCA1 (high vs. low) and expression of ENSG00000218510 (high vs. low) were significant prognostic factors for overall survival prediction (Table 4). Meanwhile, multivariable Cox regression analysis displayed that lymph node metastasis (present vs. absent), expression of UCA1 (high vs. low) and expression of ENSG00000218510 (high vs. low) were independent predictors of PDAC patients’ overall survival after pancreatectomy (Table 4). Taken together, these data illustrated above suggested that part of the disregulated lncRNAs such as up-regulated AFAP1-AS1, UCA1 and down-regulated ENSG00000218510 could predict poor prognosis and might contribute to tumor progression in PDAC.

The potential value of candidate lncRNAs expression in predicting PDAC and patient prognosis.
(A–C) Kaplan-Meier analysis of overall survival in Ren Ji cohort. Patients were scored as low and high expression group using the median value as cutoff according to lncRNAs expression. Results showed that patients with higher AFAP1-AS1, UCA1 and lower ENSG00000218510 expression have a poorer overall survival after surgery than their corresponding counterparts in PDAC. P-values were calculated by log-rank test. (D) ROC curve analyses of CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1 and ENSG00000218510 for prediction of PDAC using qRT-PCR-based expression level in Ren Ji cohort. AUC: the area under curve.
Furthermore, receiver operating characteristic (ROC) curve analyses were conducted to investigate the prediction of diagnostic value according to the 6 lncRNAs expression. Results demonstrated that the area under curve (AUC) of CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1 and ENSG00000218510 in Ren Ji cohort was 0.589, 0.624, 0.622, 0.733, 0.715, 0.931, respectively (Fig. 4D). These data indicated that part of the disregulated lncRNAs such as AFAP1-AS1, UCA1 and ENSG00000218510 may also be practical predictors for diagnosis in PDAC patients.
Identification of lncRNAs associated biological pathways and processes
To gain further insight into the biological pathways and processes involved in PDAC pathogenesis through AFAP1-AS1, UCA1 and ENSG00000218510 pathway, Gene Set Enrichment Analysis (GSEA) was performed in tumor samples of GSE16515 datasets. NOM p-val < 0.05 and FDR q-val < 0.25 were considered as significant gene sets. Gene set differences in AFAP1-AS1 high vs low patients (median split) indicated that AFAP1-AS1 may regulate gene sets associated with I kappa B kinase NF-kappa B cascade, cell cycle phase and process, mitosis, mitotic cell cycle and so on (Supplementary Table S1A). High UCA1 expression was accompanied with up-regulation of gene sets associated with positive regulation of I kappa B kinase NF-kappa B cascade, regulation of growth, regulation of cell growth, cell migration, anti-apoptosis, growth and so on (Supplementary Table S1B). For ENSG00000218510, the gene signatures of cell cycle process and phase, mitosis, mitotic cell cycle, DNA replication, I kappa B kinase NF-kappa B cascade and so on were more correlated with patients with ENSG00000218510 lower expression versus patients with ENSG00000218510 higher expression (Supplementary Table S1C). These related pathways and processes were reported to be associated with tumorigenesis, and thus the lncRNAs signature might be involved with.
Three signature lncRNAs regulate the proliferation and/or invasion abililty of PDAC cell lines
The oncogenic function of AFAP1-AS1 has been reported in a recent study27. To evaluate the effects of UCA1 and ENSG00000218510 on cell biological behaviors, specific small interfering RNAs (siRNAs) were employed to knockdown UCA1 and ENSG00000218510 expression in human PDAC cell lines. According to their expression status in PDAC cell lines as described above, we selected the relative higher expression cell line MIA PaCa-2 for UCA1 experiment and Capan-2 for ENSG00000218510, respectively. The transfection efficiency was confirmed by qRT-PCR (Fig. 5A). Cell Counting Kit-8 (CCK-8) assays indicated that cell proliferation was decreased when UCA1 was knocked down, while ENSG00000218510 exhibited no difference (Fig. 5B). The results suggested that UCA1 played a physiological role in regulating cell proliferation. We further used Transwell assay to monitor the effect of manipulating UCA1 and ENSG00000218510 expression on cell invasiveness. Knockdown of UCA1 significantly reduced the number of PDAC cells that penetrated the Transwell filter, which demonstrated a substantial loss of cell invasion ability, while ENSG00000218510 the opposite (Fig. 5C).

LncRNA UCA1 and ENSG00000218510 regulate the proliferation and/or invasion abililty of PDAC cell lines.
(A) UCA1 and ENSG00000218510 knockdown efficiency was confirmed by qRT-PCR in MIA PaCa-2 and Capan-2 cell line, respectively. (B) The effect of UCA1 and ENSG00000218510 knockdown on PDAC cell line proliferation was determined by CCK-8 assays. *P < 0.05, Student’s t-test. Data are represented as the mean ± SD. (C) Representative images of transwell assay after UCA1 and ENSG00000218510 knockdown in MIA PaCa-2 and Capan-2, respectively.
Discussion
Since the beginning of genomic technologies, many groups have tried to identify molecular biomarkers that would help elucidate the mechanisms of PDAC progression and malignant transformation1. LncRNAs, as a novel class of regulatory functional molecules involved in cancer biology, may yield valuable information and provide a different perspective8,13. In recent years, an increasing number of lncRNAs have been identified and associations between lncRNAs and various types of cancer have been investigated8,9. However, only a handful of studies have been reported in current PDAC cancer literature regarding lncRNA expression changes. LncRNA expression patterns and their characteristics in PDAC remain to be further systematically explored. Here, We investigated the lncRNA expression signatures in PDAC and evaluated their potential clinical and prognostic significance of several lncRNAs among them, hoping to shed more light on progression of the illness and of course, possible targets to develop new therapeutic drugs.
In this study, we profiled lncRNAs expression in PDAC tissues by mining two cohort of existing microarray gene expression datasets using Affymetrix HG-U133 Plus 2.0 array platform, since a great number of lncRNAs were interrogated on many commonly used commercial arrays. The robustness of this mining approach has been applied by several researchers and it is proved to be feasible, reliable and of low cost24,25,26. Based on this method, we identified a set of 34 lncRNAs that differentiated (adjusted P < 0.01, FC ≥ 1.60) between PDAC and normal pancreatic tissues. Such differentiation implied their potential roles in pancreatic tumorigenesis. Among the 34 dysregulated lncRNAs, some have been demonstrated to be related to PDAC. For example, AFAP1-AS1, which was significantly up-regulated (with an average fold change of about 3) in our data, has been reported to be overexpressed in PDAC and its overexpression was associated with lymph node metastasis, perineural invasion and poor survival27. AFAP1-AS1 suppression resulted in attenuated PDAC cell proliferation, migration, and invasion in vitro functional experiments, while ectopic expression the opposite. In vivo inhibition of AFAP1-AS1 impaired pancreatic cancer cell tumorigenicity. Our results also confirmed its aberrant expression and prognostic significance in PDAC, but in our study, AFAP1-AS1 expression was only found to be correlated with tumor size, its relation with lymph node metastasis and perineural invasion wasn’t significant, which may have been a consequence of the small sample size in Ren Ji cohort. Another candidate, lncRNA PVT1, was found to be up-regulated (with an average fold change of 1.84) in our study. This finding was in concordance with recent reports that increased expression of lncRNA PVT1 in PDAC was correlated with clinical stage and N-classification, and PVT1 might act as a potential molecular biomarker for predicting the prognosis of patients with PDAC28. Besides, PVT1 was also identified as a regulator of Gemcitabine sensitivity and functional inactivation of it led to enhanced Gemcitabine sensitivity in human pancreatic cancer cells29. More interestingly, recent researches identified several new risk loci significantly associated with susceptibility to pancreatic cancer through conducting genome-wide association study (GWAS), among which one locus is at 8q24.21 located 455 kb telomeric of PVT130 and one at 17q25.1 (LINC00673)31. Coincidentally, PVT1 and NR_036488 (LINC00673) expression were both elevated in PDAC in our result. Whether there exist interactions between them remains to be investigated. In addition, we performed a comparsion between our results and the findings of a recent study32, which identified differential expression of 43 lncRNAs (FDR < 0.05, |log2FC| > 1.6) in six PDAC and five normal control tissues from 432 lncRNAs detected using massive analysis of cDNA ends (MACE). The contrast result showed that 6 elevated lncRNAs NR_036488 (LINC00673), AFAP1-AS1, NR_026812 (RUNX1-IT1), ENSG00000222041 (LINC00152), ENSG00000233429 (HOTAIRM1), PVT1 and 4 diminished lncRNAs ENSG00000218510 (LINC00339), ENSG00000236333 (TRHDE-AS1), NR_026887 (CEBPA-AS1), ENSG00000213373 (LINC00671) in our result were also in their list with the same trend. However, functional investigations of most differentially expressed lncRNAs identified in our study, so far, have not been implicated in PDAC. But a handsome amount of lncRNAs such as ENSG00000222041 (LINC00152)33, DLEU234, NCRNA00275 (ZFAS1)35, NR_024284 (ZEB1-AS1)36, ENSG00000241684 (ADAMTS9-AS2)37 have been reported to be involved in other types of cancers. Whether these lncRNAs play important roles in the development and progression of PDAC deserves further clarification.
Furthermore, our own qRT-PCR data confirmed the up-regulation of CRNDE, NR_036488, ENSG00000244649, AFAP1-AS1, UCA1 and down-regulation of ENSG00000218510 in PDAC and the subsequent analysis of their correlations with clinicopathological features and prognosis also indicated that part of them such as up-regulated AFAP1-AS1, UCA1 and down-regulated ENSG00000218510 may contribute to tumor progression and predict unfavourable prognosis in PDAC patients. This was further supported by examination of associated molecular pathways by GSEA analysis of GSE16515 tumors showing that up-regulated AFAP1-AS1, UCA1 and down-regulated ENSG00000218510 were more likely to involve with cell cycle, proliferation, I kappa B kinase NF-kappa B cascade and so on. They were reported to be associated with tumorigenesis, and thus the lncRNAs signature might be involved with. And a recent study27 and our following cell functional experiments further confirmed that the three lncRNAs regulate the proliferation and/or invasion abililty of PDAC cell lines. In addition, we attempted to seek some information and evidences regarding their sequence similarity, motifs and subcellular localization by searching the previous studies or several biomedical websites and softwares. For example, through RegRNA 2.038, an integrated web server for identifying functional RNA motifs and sites, we could predict their possible motifs and some motifs in the results (Supplementary Table S3) such as UCA1 transcriptional regulatory motifs c-Myb, C/EBP have also been reported and validated in human bladder cancer39,40. Past studies also indicated that UCA1 is located in the cytoplasm by in situ hybridization41. We are also interested in conducting more experimental investigations on these lncRNAs’ information in PDAC in the next work. Moreover, the ROC curve analyses suggested a possible diagnostic value of AFAP1-AS1, UCA1 and ENSG00000218510. However, this was based on a limited numbers of patients. Larger scale studies are needed to confirm our findings and the function as well as mechanism underlying the dysregulation of these cancer-associated lncRNAs deserves further studies. Among the 6 selected lncRNAs, the functional study of AFAP1-AS1 in PDAC has been reported as described above, whereas the other 5 lncRNAs’ function has not been implicated in PDAC currently. But there have been emerging evidence that some of them played an important role in many other human malignancies. For example, CRNDE has been characterized as a carcinogenesis promoter in colorectal cancer42, and glioma43. Earlier study with microarrays has also shown that depletion of CRNDE (lincIRX5) with shRNAs targeting exon-containing transcripts resulted in the alteration of a series of genes associated with tumorigenesis44. Another lncRNA UCA1 has also been demonstrated to participate and manifest a oncogenic function in multiple types of cancers such as bladder cancer45, colorectal cancer46, breast cancer47, esophageal squamous cell carcinoma48, non-small cell lung cancer49, hepatocellular carcinoma50, gastric cancer51 and so on. Also, lncRNA NR_036488 (LINC00673, LOC100499467) was found up-regulated in non-small cell lung cancer25,52 and glioma24 through qRT-PCR or microarray analysis. The relevant study on the other two lncRNAs ENSG00000244649 and ENSG00000218510 in cancer has not been found so far and the roles they play warrant further study.
As is known to us, identification of biomarkers closely correlated with disease progression is particularly meaningful. Cancer is initiated by a series of accumulative genetic and epigenetic alterations that affect normal cellular identity, growth and differentiation. Gene expression profiling offers a comprehensive molecular understanding of cancer that may grant insights into its pathophysiology and yield relevant information for subtype classification, staging, prognosis and therapeutic decision-making. Nowadays, increasing studies suggest that lncRNAs are promising biomarkers of cancer and have the potential to act such a role. One of the most prominent example is PCA3, a lncRNA highly expressed in prostate cancer53. The functions of lncRNAs are more likely to closely associated with their expression level as they do not encode proteins26. Thus lncRNAs may have higher specificity than protein-coding mRNAs and be more suitable to serve as prognostic and/or predictive markers for PDAC. Moreover, lncRNAs have the advantages of being detectable in the blood8 and urine9,12,53 of cancer patients by PCR methods. The distinctive expression patterns of lncRNAs in PDAC may potentially aid the development of biomarkers and, subsequently, the diagnosis of PDAC. Differential expression profiles of lncRNAs may also guide future development of novel therapies. Currently, therapies designed to target cancer-driving lncRNAs are also under intensive investigation54. To this end, rapid advances in oligonucleotide and nanoparticle technology create realistic optimism for delivering the RNA interference (RNAi)-mediated gene silencing technology to regulate lncRNA levels in vivo.
The limitations of this study should be acknowledged. First of all, HG-U133 Plus 2.0 arrays represent only portion of the possible lncRNAs present. Secondly, for many of the the signature lncRNAs, information regarding their possible functional roles and mechanisms is still limited. More experimental investigations on these lncRNAs are needed. Thirdly, the sample size of each dataset is relatively small. Our analysis may also ignore some lncRNAs that other groups have demonstrated to be involved in PDAC progression due to the different distributions of the patient populations in terms of age, gender and stage of PDAC. We are also interested in exploring which lncRNAs are differentially expressed in different stages of PDAC. Unfortunately, the supplementary files of both two PDAC datasets we adopted didn’t provide related information. Last but not least, PDAC is a heterogeneous malignancy notable for its profuse desmoplastic stroma comprised of activated fibroblasts, leukocytes, and extracellular matrix55, thus it is ambiguous which of the genomic alterations and expression changes are due to neoplastic ductal epithelial cells and which simply reflect the differences in cellular composition.
In summary, we have identified and validated multiple novel lncRNAs which differentially expressed in human PDAC. Our findings indicate the potential roles of lncRNAs in PDAC, and may provide useful information in PDAC diagnosis, classification, prognosis and therapeutic evaluation. Future studies will focus on the verifications of identified lncRNA signatures and the functional elucidation of these lncRNAs.
Materials and Methods
The methods were carried out in accordance with the approved guidelines.
The study was approved by the Research Ethics Committee of Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University and was performed in agreement with the approved ethical standards laid down in the 1964 declaration of Helsinki and all subsequent revisions.
PDAC datasets preparation
PDAC microarray datasets and corresponding clinical data in this study were directly downloaded from GEO database. These datasets corresponded to all available public datasets fulfilling the following criteria: (i) they used PDAC tissue and normal pancreatic tissue for comparison; (ii) they used the same chip platform (Affymetrix HG-U133 Plus 2.0 array); (iii) they contained more than three samples meeting the quality control standard in experimental and controlled group. Two panels of PDAC gene expression datasets were included in our study: GSE16515 and GSE15471. We adopted the training-validation method. While GSE16515 was first used to identify the gene expression signatures, GSE15471 served as a validation dataset to evaluate the proportion of misclassifications.
LncRNA profile mining
LncRNA expression profiles on Affymetrix GeneChip Human Genome HG-U133 Plus 2.0 array were pointed out in the light of the NetAffx annotation of the probe sets and the Refseq and Ensembl annotations of lncRNAs as described previously by Zhang24. To sum up, all transcripts represented on the microarray were first identified as protein-coding RNAs or ncRNAs, of which only the ncRNAs were retained and further screened by discarding pseudogenes, rRNAs, tRNAs, microRNAs and other short ncRNAs. Ultimately, total 2448 Affymetrix probe sets (corresponding to 1970 lncRNAs genes) were included in our following analysis. Among them, 725 probe sets (510 genes) mapping to lncRNAs were annotated by both the Refseq and the Ensembl databases; 512 probe sets (379 genes) were annotated only by the Refseq database, and 1211 probe sets (1081 genes) were annotated only by the Ensembl database. Those probe sets with controversial definitions in the two databases were excluded.
Microarray data processing and analysis
The expression data of raw CEL files were normalized, log2 transformed and background adjusted utilizing a Bioconductor package Robust MultiArray Average (RMA)56 through R 3.2.0 software. After that, a set of probe ID-centric gene expression values were retrieved for downstream analysis. The normalized data were then analyzed with linear models for microarray data (LIMMA), a modified t-test incorporating the Benjamini–Hochberg multiple hypotheses correction technique57. The probe sets of which the adjusted P-value was below 0.01 and the expression level differed by ≥1.60-fold between two comparsion groups were characterized as significantly different lncRNAs.
Hierarchical clustering analysis (HCA) of the lncRNA profiles was performed using MeV 4.9.0 software58. The normalized expression values of the lncRNAs were centred on the median before performing unsupervised hierarchical clustering. Clustering was done with complete linkage and Euclidean distance. HCA was used to visually inspect the result.
GSEA was performed by the JAVA program (http://software.broadinstitute.org/gsea/index.jsp) using the GO gene sets database (c5.all.v5.1.symbols.gmt [gene ontology]) from the Molecular Signatures Database–MsigDB. The patients were stratified by the median of AFAP1-AS1, UCA1 and ENSG00000218510 expression level and significantly enriched biological pathways between classes under comparison were identified, which produced a nominal P-value of 0.05 and FDR of 0.25.
Patients and samples
A total of 80 freshly-frozen primary pancreatic cancer and matched adjacent non-tumor tissues (Ren Ji cohort) were collected from patients who underwent pancreatic surgical resection at Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University between January 2012 and November 2014, and the pathological information was retrieved from the Pathology Department. None of the patients had received radiotherapy, chemotherapy, hormone therapy or other related anti-tumor therapies before surgery. For all samples from 80 patients, clinical information was available. The follow-up time was calculated from the date of surgery to pancreatic cancer-related death, or November 17, 2015, the ultimate deadline. All the patients were provided with written informed consent before enrolment, and the study was approved by the Research Ethics Committee of Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University.
Cell culture and treatment
Human PDAC cell lines AsPC-1, BxPC-3, Capan-2, CFPAC-1, HPAC, MIA PaCa-2, PANC-1 and SW1990 were purchased from the Cell Resource Center, Shanghai Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, China) and two immortalized normal human pancreatic ductal epithelial cell lines HPDE6-C7 and hTERT-HPNE were obtained from Professor ZG Zhang (Shanghai Cancer Institute, Shanghai, China). Cells were maintained in indicated medium according to ATCC protocols, supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% antibiotics (100 μg/ml streptomycin and 100 U/ml penicillin) at 37 °C in a humidified incubator with a 5% CO2 atmosphere.
The siRNAs targeting human UCA1 and ENSG00000218510 were transfected into the PDAC cells using the DharmaFECT 1 siRNA transfection reagent (Thermo Scientific Dharmacon lnc.), whereas nonspecific siRNA acted as negative controls. The siRNAs were purchased from Genepharm Technologies (Shanghai, China) and sequences are listed in Supplementary Table S2A.
RNA extraction and qRT-PCR
Total RNA was extracted from tissues and cells by using Trizol reagent (Takara, Shiga, Japan) and cDNA was synthesized using a PrimeScript RT Reagent Kit (Perfect Real Time; Takara, Shiga, Japan) according to the manufactuer’s instruction. StepOnePlus Real-Time PCR System (Applied Biosystems, Grand Island, NY, USA) was applied to detect the expression level of target gene using the SYBR Premix Ex Taq II (Takara, Japan), and the amplified transcript level of each specific gene was normalized to that of GAPDH. The fold change of target lncRNA’s expression intensity was calculated by the 2−ΔΔCt method. Primer sequences are listed in Supplementary Table S2B.
Cell proliferation assay
Cells were seeded into in 96-well plates at an initial density of 3000 cells/well and treated with 10 μl of CCK-8 (Dojindo Molecular Technologies, Kyushu, Japan) each well for 1.5 hours at the time points of 24 h, 48 h, 72 h and 96 h after the planting. Cell absorbance was detected by scanning with a microplate reader at 450 nm.
Transwell chamber assay
After the chamber was coated with fresh Matrigel (diluted in 1:4 with serum-free medium) (BD Bioscience San Jose, CA, USA) in 24-well dishes, 2 × 105 cells transfected with lncRNA siRNAs or control siRNA and suspended in serum-free medium were plated on the top of each chamber, while medium containing 20% FBS was placed in the lower chamber and used as a chemo-attractant. After 48 h incubation, cells that did not pass through the filter were removed by a cotton swab, whereas cells on the lower surface of the filter were fixed and stained with formaldehyde and crystal violet, respectively. The cells on the lower side of the filters were defined as invasive cells.
Statistical analysis
Data were presented as the means ± SD. SPSS 22.0 software (IBM Corp., Armonk, NY, USA) was used for statistical analysis. Graphical representations were carried out with GraphPad Prism 6 (San Diego, CA, USA) and Medcalc 11.5 (Ostend, Belgium) software. For clinicopathological analysis, the chi-square test or Fisher’s exact test was performed. The survival calculations were illustrated with Kaplan-Meier curves and differences between survival curves were tested by the log-rank test. Cox proportional hazards model was used to identify the prognostic factors by univariable and multivariable analysis. The student’s t-test or Mann–Whitney U test was used for comparison between two groups depending on distribution. P values (two-sided) less than 0.05 were considered statistically significant.
Additional Information
How to cite this article: Fu, X.-L. et al. Analysis of long non-coding RNA expression profiles in pancreatic ductal adenocarcinoma. Sci. Rep. 6, 33535; doi: 10.1038/srep33535 (2016).
References
Costello, E., Greenhalf, W. & Neoptolemos, J. P. New biomarkers and targets in pancreatic cancer and their application to treatment. Nat Rev Gastroenterol Hepatol 9, 435–444, doi: 10.1038/nrgastro.2012.119 (2012).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2015. CA Cancer J Clin 65, 5–29, doi: 10.3322/caac.21254 (2015).
Chen, W. et al. Report of cancer incidence and mortality in China, 2010. Ann Transl Med 2, 61, doi: 10.3978/j.issn.2305-5839.2014.04.05 (2014).
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921, doi: 10.1158/0008-5472.can-14-0155 (2014).
Waddell, N. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501, doi: 10.1038/nature14169 (2015).
Ryan, D. P., Hong, T. S. & Bardeesy, N. Pancreatic adenocarcinoma. N Engl J Med 371, 1039–1049, doi: 10.1056/NEJMra1404198 (2014).
Brosnan, C. A. & Voinnet, O. The long and the short of noncoding RNAs. Curr Opin Cell Biol 21, 416–425, doi: 10.1016/j.ceb.2009.04.001 (2009).
Gibb, E. A., Brown, C. J. & Lam, W. L. The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10, 38, doi: 10.1186/1476-4598-10-38 (2011).
Prensner, J. R. & Chinnaiyan, A. M. The emergence of lncRNAs in cancer biology. Cancer Discov 1, 391–407, doi: 10.1158/2159-8290.cd-11-0209 (2011).
Nelson, K. M. & Weiss, G. J. MicroRNAs and cancer: past, present, and potential future. Mol Cancer Ther 7, 3655–3660, doi: 10.1158/1535-7163.mct-08-0586 (2008).
Farazi, T. A., Spitzer, J. I., Morozov, P. & Tuschl, T. miRNAs in human cancer. J Pathol 223, 102–115, doi: 10.1002/path.2806 (2011).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166, doi: 10.1146/annurev-biochem-051410-092902 (2012).
Brunner, A. L. et al. Transcriptional profiling of long non-coding RNAs and novel transcribed regions across a diverse panel of archived human cancers. Genome Biol 13, R75, doi: 10.1186/gb-2012-13-8-r75 (2012).
Qi, P. & Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol 26, 155–165, doi: 10.1038/modpathol.2012.160 (2013).
Li, J. et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 63, 1700–1710, doi: 10.1136/gutjnl-2013-305806 (2014).
Zeng, S. et al. Long Noncoding RNA in Digestive Tract Cancers: Function, Mechanism, and Potential Biomarker. Oncologist, doi: 10.1634/theoncologist.2014-0475 (2015).
Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47, 199–208, doi: 10.1038/ng.3192 (2015).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346, doi: 10.1038/nature10887 (2012).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076, doi: 10.1038/nature08975 (2010).
Zhang, J., Zhang, P., Wang, L., Piao, H. L. & Ma, L. Long non-coding RNA HOTAIR in carcinogenesis and metastasis. Acta Biochim Biophys Sin (Shanghai) 46, 1–5, doi: 10.1093/abbs/gmt117 (2014).
Kim, K. et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625, doi: 10.1038/onc.2012.193 (2013).
Yuan, J. H. et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25, 666–681, doi: 10.1016/j.ccr.2014.03.010 (2014).
Michelhaugh, S. K. et al. Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem 116, 459–466, doi: 10.1111/j.1471-4159.2010.07126.x (2011).
Zhang, X. et al. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol Dis 48, 1–8, doi: 10.1016/j.nbd.2012.06.004 (2012).
Yang, J. et al. Analysis of lncRNA expression profiles in non-small cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer 85, 110–115, doi: 10.1016/j.lungcan.2014.05.011 (2014).
Du, Z. et al. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 20, 908–913, doi: 10.1038/nsmb.2591 (2013).
Ye, Y. et al. High expression of AFAP1-AS1 is associated with poor survival and short-term recurrence in pancreatic ductal adenocarcinoma. J Transl Med 13, 137, doi: 10.1186/s12967-015-0490-4 (2015).
Huang, C. et al. Increased expression of the lncRNA PVT1 is associated with poor prognosis in pancreatic cancer patients. Minerva Med 106, 143–149 (2015).
You, L., Chang, D., Du, H. Z. & Zhao, Y. P. Genome-wide screen identifies PVT1 as a regulator of Gemcitabine sensitivity in human pancreatic cancer cells. Biochem Biophys Res Commun 407, 1–6, doi: 10.1016/j.bbrc.2011.02.027 (2011).
Wolpin, B. M. et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet 46, 994–1000, doi: 10.1038/ng.3052 (2014).
Childs, E. J. et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet 47, 911–916, doi: 10.1038/ng.3341 (2015).
Muller, S. et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer 14, 94, doi: 10.1186/s12943-015-0358-5 (2015).
Zhao, J. et al. Long non-coding RNA Linc00152 is involved in cell cycle arrest, apoptosis, epithelial to mesenchymal transition, cell migration and invasion in gastric cancer. Cell Cycle, 0, doi: 10.1080/15384101.2015.1078034 (2015).
Klein, U. et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17, 28–40, doi: 10.1016/j.ccr.2009.11.019 (2010).
Li, T. et al. Amplification of Long Noncoding RNA ZFAS1 Promotes Metastasis in Hepatocellular Carcinoma. Cancer Res 75, 3181–3191, doi: 10.1158/0008-5472.can-14-3721 (2015).
Li, T. et al. Upregulation of long noncoding RNA ZEB1-AS1 promotes tumor metastasis and predicts poor prognosis in hepatocellular carcinoma. Oncogene, doi: 10.1038/onc.2015.223 (2015).
Yao, J. et al. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumour Biol 35, 7935–7944, doi: 10.1007/s13277-014-1949-2 (2014).
Chang, T. H. et al. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics 14 Suppl 2, S4, doi: 10.1186/1471-2105-14-s2-s4 (2013).
Xue, M., Li, X. & Chen, W. [Bioinformatics analysis and identification of transcriptional regulation of human UCA1 gene]. Nan Fang Yi Ke Da Xue Xue Bao 33, 1596–1599 (2013).
Xue, M. et al. Upregulation of long non-coding RNA urothelial carcinoma associated 1 by CCAAT/enhancer binding protein alpha contributes to bladder cancer cell growth and reduced apoptosis. Oncol Rep 31, 1993–2000, doi: 10.3892/or.2014.3092 (2014).
Xie, X. J., Li, X., Wang, F. & Chen, W. [Cellular localization and tissue expression pattern of UCA1, a non-coding RNA]. Nan Fang Yi Ke Da Xue Xue Bao 30, 57–60 (2010).
Ellis, B. C., Molloy, P. L. & Graham, L. D. CRNDE: A Long Non-Coding RNA Involved in CanceR, Neurobiology, and DEvelopment. Front Genet 3, 270, doi: 10.3389/fgene.2012.00270 (2012).
Wang, Y. et al. CRNDE, a long-noncoding RNA, promotes glioma cell growth and invasion through mTOR signaling. Cancer Lett, doi: 10.1016/j.canlet.2015.03.027 (2015).
Khalil, A. M. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 106, 11667–11672, doi: 10.1073/pnas.0904715106 (2009).
Wang, F., Li, X., Xie, X., Zhao, L. & Chen, W. UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett 582, 1919–1927, doi: 10.1016/j.febslet.2008.05.012 (2008).
Han, Y. et al. UCA1, a long non-coding RNA up-regulated in colorectal cancer influences cell proliferation, apoptosis and cell cycle distribution. Pathology 46, 396–401, doi: 10.1097/pat.0000000000000125 (2014).
Huang, J. et al. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis 5, e1008, doi: 10.1038/cddis.2013.541 (2014).
Li, J. Y., Ma, X. & Zhang, C. B. Overexpression of long non-coding RNA UCA1 predicts a poor prognosis in patients with esophageal squamous cell carcinoma. Int J Clin Exp Pathol 7, 7938–7944 (2014).
Cheng, N. et al. Long non-coding RNA UCA1 induces non-T790M acquired resistance to EGFR-TKIs by activating the AKT/mTOR pathway in EGFR-mutant non-small cell lung cancer. Oncotarget 6, 23582–23593, doi: 10.18632/oncotarget.4361 (2015).
Wang, F. et al. Upregulated lncRNA-UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b and activation of FGFR1/ERK signaling pathway. Oncotarget 6, 7899–7917, doi: 10.18632/oncotarget.3219 (2015).
Zheng, Q. et al. Aberrant expression of UCA1 in gastric cancer and its clinical significance. Clin Transl Oncol 17, 640–646, doi: 10.1007/s12094-015-1290-2 (2015).
Yu, H. et al. Identification and validation of long noncoding RNA biomarkers in human non-small-cell lung carcinomas. J Thorac Oncol 10, 645–654, doi: 10.1097/jto.0000000000000470 (2015).
Lee, G. L., Dobi, A. & Srivastava, S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol 8, 123–124, doi: 10.1038/nrurol.2011.10 (2011).
Davis, M. E. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1070, doi: 10.1038/nature08956 (2010).
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747, doi: 10.1016/j.ccr.2014.04.021 (2014).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264, doi: 10.1093/biostatistics/4.2.249 (2003).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3, doi: 10.2202/1544-6115.1027 (2004).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003).
Acknowledgements
We thank all the subjects of this study for their participation. This work was supported by grants from the science and technology funds of School of Medicine, Shanghai Jiao Tong University (No. 14xJ10022) to ZJF, National Basic Research Program of the National Natural Science Foundation (No. 31271366) and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (No. 201268) to HJ; the grants from National Natural Science Foundation (No. 81502489) to JL; the grants from National High Technology Research and Development Program of China (No. 2014AA020609) to SYW.
Author information
Authors and Affiliations
Contributions
Y.-W.S., H.-Y.C. and J.H. conceived and designed the experiments, X.-L.F., D.-J.L. and T.-T.Y. performed the experiments and wrote the manuscript, H.-Y.C., R.H. and X.-L.F. analyzed the data, J.-Y.Y., M.-W.Y., J.L., Y.-M.H., W.L. and J.-F.Z. contributed clinical samples and data. Y.-W.S., H.-Y.C., R.H. and J.H. revised and edited the manuscript. All authors reviewed and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fu, XL., Liu, DJ., Yan, TT. et al. Analysis of long non-coding RNA expression profiles in pancreatic ductal adenocarcinoma. Sci Rep 6, 33535 (2016). https://doi.org/10.1038/srep33535
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33535
This article is cited by
-
Liquid biopsy techniques and pancreatic cancer: diagnosis, monitoring, and evaluation
Molecular Cancer (2023)
-
Pancreatic Cancer: Genetic Conditions and Epigenetic Alterations
Journal of Gastrointestinal Surgery (2023)
-
Prognostic value of the long noncoding RNA AFAP1-AS1 in cancers*
Oncology and Translational Medicine (2023)
-
The impact of LncRNA dysregulation on clinicopathology and survival of pancreatic cancer: a systematic review and meta-analysis (PRISMA compliant)
Cancer Cell International (2021)
-
Long non-coding RNA PART1 predicts a poor prognosis and promotes the malignant progression of pancreatic cancer by sponging miR-122
World Journal of Surgical Oncology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
