
Abstract
Benzylisoquinoline alkaloids (BIQ) are among the most structurally diverse and pharmaceutically valuable secondary metabolites. A plant-specific WRKY-type transcription factor, CjWRKY1, was isolated from Coptis japonica and identified as a transcriptional activator of BIQ biosynthesis. However, the expression of CjWRKY1 gene alone was not sufficient for the activation of genes encoding biosynthetic enzymes. Here, we report the importance of post-translational regulation of CjWRKY1 in BIQ biosynthesis. First, we detected the differential accumulation of CjWRKY1 protein in two cell lines with similar CjWRKY1 gene expression but different levels of accumulated alkaloids. Further investigation of the WRKY protein identified the phosphorylation of the WRKYGQK core domain at Y115. The CjWRKYY115E phosphorylation-mimic mutant showed loss of nuclear localization, DNA-binding activity and transactivation activity compared to wild-type CjWRKY1. Rapid degradation of the CjWRKY1 protein was also confirmed following treatment with inhibitors of the 26S proteasome and protease inhibitors. The existence of two independent degradation pathways as well as protein phosphorylation suggests the fine-tuning of CjWRKY1 activities is involved in the regulation of biosynthesis of BIQs.
Similar content being viewed by others


Introduction
Higher plants produce a large variety of low molecular weight secondary metabolites, including nitrogen-containing alkaloids, which are often used as pharmaceuticals1. Alkaloid biosynthesis, including the relevant biosynthetic enzymes and genes, has been intensively investigated2,3,4, but the regulation of alkaloid biosynthesis, especially by transcription factors, is largely unknown because only a limited number of plant species and cells produce certain types of alkaloids, making analysis difficult. Recently, however, WRKY and basic helix-loop-helix transcription factors in benzylisoquinoline alkaloids (BIQ) biosynthesis and some WRKY, bHLH and AP2/ERF proteins in the biosynthesis of nicotine and monoterpenoid indole alkaloids biosynthesis have been isolated5,6,7,8,9,10.
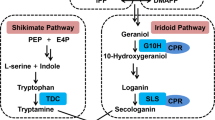
Plants-specific WRKY transcription factors are one of the most important regulators in defence responses, development and senescence11,12,13. They all contain the 60-amino acid WRKY domain, which has a highly conserved amino acid sequence, WRKYGQK, at the N-terminal end and a zinc finger motif at the C-terminal end. The WRKY domain specifically recognizes the W-box DNA motif (TTGACC/T)13. The WRKY family can be divided into three groups based on the structures; (i) Group I has two WRKY domains and a C2H2-type zinc-finger motif, (ii) Group II has a single WRKY domain and the C2H2 zinc-finger and can be further divided into five subgroups, (iii) Group III has a single WRKY domain and a zinc finger structure of C2HC13. The first WRKY identified in alkaloid biosynthesis, CjWRKY1, was isolated from C. japonica cells in the biosynthesis of BIQ-type alkaloids5 (see BIQ biosynthetic pathway, Supplementary Fig. S1) and belongs to Group IIc.
Transient RNAi of CjWRKY1 clearly decreased the expression of BIQ biosynthetic enzyme genes, including tyrosine decarboxylase (TYDC), (S)-norcoclaurine synthase (NCS), (S)-norcoclaurine 6-O-methyltransferase (6OMT), (S)-coclaurine-N-methyltransferase (CNMT), (S)-N-methylcoclaurine 3′-hydroxylase (CYP80B2), (S)-3′-hydroxy-N-methylcoclaurine-4′-O-methyltransferase (4′OMT), berberine bridge enzyme (BBE) and canadine synthase (CYP719A1), in high BIQ-producing Cj156-S cells. Over-expression of CjWRKY1 enhanced the transcript levels of the above-mentioned genes in Cj156-S cells. These results confirmed that CjWRKY1 is a specific and general transcriptional activator of BIQ biosynthesis in Cj156-S cells5. However, we also found that high expression of CjWRKY1 transcripts was not correlated with the expression of BIQ biosynthetic enzyme genes in low BIQ-producing CjY cells5. Thus, we attempted to clarify why high CjWRKY1 gene expression did not induce the expression of target biosynthetic genes in CjY cells.
Post-translational modification seems to be one of the key regulatory mechanisms of transcription factors involved in the regulation of plant processes. In fact, some WRKY proteins are reported to be post-translationally regulated by protein phosphorylation, protein-protein interaction and protein turnover14,15. Here, we report that the post-translational regulation of CjWRKY1, i.e., tyrosine phosphorylation and the 26S proteasome-mediated and non-mediated protein degradation, would play a crucial role in the regulation of CjWRKY1 activity in BIQ biosynthesis.
Results
Tyrosine phosphorylation of CjWRKY1
We first prepared specific antibodies against a CjWRKY1 peptide and characterized the expression of CjWRKY1 in both Cj156-S and CjY cells (Fig. 1). Immunoblotting analysis clearly detected the accumulation of CjWRKY1 in Cj156-S cells but not in CjY cells. Thus, we next considered the possibility of post-translational modification of CjWRKY1 in C. japonica cells.

Expression of CjWRKY1 and the biosynthetic enzyme genes in Cj156-S and CjY cells.
(a) Transcripts of CjWRKY1 and the other biosynthetic enzyme genes in 2-week-cultured cells were measured by quantitative RT-PCR. The relative expression levels were estimated by the standard curve method with three biological replicates and standardized with the α-tubulin gene as an internal control. The average value of the CjY was set as 1. The data are shown as the mean ± s.d., **p < 0.01, Student’s t-test. (b) CjWRKY1 accumulation in three independent cultures grown for 2 weeks. An asterisk indicates CjWRKY1 protein. Left axis indicates the size of reference proteins. kDa; molecular mass in kilodaltons.
We examined the modification of the CjWRKY1 protein using Cj156-S cells because CjWRKY1 in CjY cells was too unstable for analysis. Recombinant CjWRKY1-sGFP proteins expressed in Cj156-S protoplasts were recovered via co-immunoprecipitation (Co-IP) with anti-GFP antibody-bound microbeads and analysed by liquid chromatography coupled with tandem-mass spectrometry (LC-MS/MS) after trypsin digestion. MassMatrix analysis of peptide fragments of CjWRKY1 showed phosphorylation of CjWRKY1 at Tyr115, which is part of the WRKYGQK core sequence (Supplementary Table S1, Supplementary Fig. S2).
The tyrosine phosphorylation of CjWRKY1 was further confirmed with anti-phospho-tyrosine (pY) antibodies (Fig. 2a). The phosphorylation signal was detected as a slightly slower moving band during immunoblot analysis with anti-GFP antibodies after the removal of anti-pY antibodies (Fig. 2a). The relatively faint phosphorylation signal, less than 40% compared to total signal intensity (Supplementary Fig. S3), suggest that phosphorylation of CjWRKY1 was not strong in Cj156-S cells or that there was rapid turnover of phosphorylated proteins as discussed below. Calf intestine alkaline phosphatase (CIP) treatment of IP proteins confirmed that the signal was derived from the phosphorylated CjWRKY1-sGFP (Fig. 2b). These data demonstrate the tyrosine phosphorylation of the CjWRKY1 protein in C. japonica cells.

Tyrosine phosphorylation of CjWRKY1.
(a) CjWRKY1-sGFP or sGFP (as a control) protein was expressed in C. japonica 156-S protoplasts, immunoprecipitated with anti-GFP microbeads and immunodetected with anti-pY and anti-GFP antibodies. Arrows indicate the bands of tyrosine-phosphorylated CjWRKY1-sGFP. Left axis indicates the size of reference proteins. kDa; molecular mass in kilodaltons, pY; phospho-tyrosine. (b) CIP treatment of co-IP samples. Co-IP samples were treated without (−) or with (+) CIP before immunoblot analysis. (c) Immunodetection of CjWRKY1WT-sGFP or CjWRKY1Y115F-sGFP with anti-pY antibodies. (d) The signal intensity of Fig. 2c was quantified by ImageJ software. The value is the average of results from three independent experiments. The data are represented as the mean ± s.d., *p < 0.05, Student’s t-test. The experiments repeated at least three times in a and b showed similar results.
Phosphorylation of CjWRKY1 at Y115 was also characterized using mutant CjWRKY1Y115F with a Phe substitution for Y115. A much weaker signal was observed with immunoprecipitated CjWRKY1Y115F-sGFP with anti-pY antibodies compared to wild-type CjWRKY1WT-sGFP (Fig. 2c,d), confirming the phosphorylation of Y115. However, the remaining signal also suggested the possibility of phosphorylation at other Tyr residue(s) in CjWRKY1.
Functional characterization of phosphorylation of CjWRKY1
To investigate the functional role of phosphorylation of CjWRKY1 at Y115, mutant CjWRKY1Y115E with a Glu residue at Y115 to generate a phosphorylation mimic as well as another mutant, CjWRKYY115L, with a Leu substitution to generate a dephosphorylated peptide mimic were prepared. Then, their DNA-binding activities to WRKY target cis-elements (W-box) were characterized in comparison to wild-type CjWRKY1. Electrophoretic mobility shift assays (EMSA) using an oligonucleotide probe containing three tandem copies of the W-box found in the CYP80B2 gene promoter (Supplementary Fig. S4) showed decreased DNA binding activities of all mutants, while the CjWRKY1WT protein showed high binding activity (Fig. 3a). Among the mutants, however, the binding activities of CjWRKY1Y115F and CjWRKY1Y115L were obviously higher than that of CjWRKY1Y115E (Supplementary Fig. S5).

The phosphorylation of CjWRKY1 at Tyr 115 affected the DNA binding and nuclear localization of the CjWRKY1 protein.
(a) EMSA of GST-CjWRKY1 mutated at Tyr115. Arrows indicate the shifted bands corresponding to the protein-DNA complexes. (b) Subcellular localization of WT and mutant CjWRKY1-sGFP proteins in Cj156-S protoplasts. Scale bars represent 50 μm.
Mutant CjWRKY1Y115E-sGFP was transiently expressed in Cj156-S protoplasts to determine the subcellular localization of Tyr-phosphorylated CjWRKY1. CjWRKY1Y115E-sGFP protein clearly showed a loss of nuclear localization, in contrast to the wild-type CjWRKY1WT-sGFP protein, which was localized in the nucleus (Fig. 3b). On the other hand, the CjWRKY1Y115F-sGFP protein showed nuclear localization similar to CjWRKY1WT (Fig. 3b).
Transcriptional activation of the CjWRKY1 mutants was assayed using reporter constructs containing the promoter region of the CYP80B2 gene linked to the Photinus pyralis luciferase (LUC) gene (CYP80B2pro::PpLUC) in Cj156-S protoplasts. The transactivation activity of CjWRKY1Y115E was much less than CjWRKY1WT (Fig. 4a). On the other hand, CjWRKY1Y115F and CjWRKY1Y115L exhibited considerable transactivation activities compared to CjWRKY1Y115E, although their activities were slightly lower than that of CjWRKY1WT. Similar results were also observed when the expression of endogeneous BIQ biosynthetic genes was measured in CjWRKY1-expressed Cj156-S protoplasts (Supplementary Fig. S6).

The phosphorylation of CjWRKY1 at Tyr 115 affected the transcriptional activity of the CjWRKY1 protein.
(a) Transactivation activity of mutant CjWRKY1 proteins detected by dual-LUC reporter assays. The top panel shows the gene organization of the effector and reporter constructs. The bottom panel indicates the relative activity level standardized using the value of the VC as 1. The values are the average of medians of five independent experiments with three biological transfections. The data are represented as the mean ± s.d., *p < 0.05, **p < 0.01, n.s., not significant, Student’s t-test. (b) Transactivation activity of GAL4DB-fused mutant CjWRKY1 proteins detected as described in (a). The values are the average of medians of three independent experiments with three biological transfections. The data are represented as the mean ± s.d., *p < 0.05, **p < 0.01, Student’s t-test.
Transcriptional activity using mutants fused with the yeast GAL4 DNA-binding domain (GAL4DB) and a reporter construct with 5 × GAL4 target elements linked to the LUC gene to equalize the DNA binding activities of mutant CjWRKY1s, however, indicated that all CjWRKY1 mutants exhibited nearly equal transactivation activities compared to CjWRKY1WT (Fig. 4b). These results suggest that the phosphorylation of CjWRKY1 at Y115 reduced its transcriptional activity by reducing DNA binding to the W-box elements and also via the modification of subcellular localization.
Finally, the effect of tyrosine phosphorylation inhibition on the transactivation activity of CjWRKY1 was examined using three tyrosine phosphorylation inhibitors (genistein and AG18 as specific inhibitors for tyrosine phosphorylation and staurosporine as a broad kinase inhibitor) and genistein was the only inhibitor that did not affect protoplast viability (Fig. 5a). As expected, genistein treatment resulted in higher transcriptional activity of CjWRKY1 than DMSO treatment (Fig. 5b). Based on the above results, we also examined the effect of genistein in cultured Cj156-S and CjY cells. However, the expression levels of the genes encoding BIQ biosynthetic enzymes were not changed (Supplementary Fig. S7), suggesting that other types of post-translational regulation of CjWRKY1 are involved in the transcriptional activity of CjWRKY1 in intact cells.

A tyrosine phosphorylation inhibitor enhanced the transactivation activity of CjWRKY1.
(a) Effect of kinase inhibitors on tyrosine phosphorylation of CjWRKY1. Cj156-S protoplasts were treated with 100 μM genistein, 25 μM AG18 and 0.5 μM staurosporine and WRKY proteins were co-immunoprecipitated with anti-GFP microbeads. (b) Effects of genistein on the transcriptional activation activity of CjWRKY1. A dual-LUC reporter assay was carried out with 0.05% DMSO or 100 μM genistein treatment for 24 h. The relative activity was standardized to the value of each vector control (VC) as 1. The values are the averages of three biological transfections. The above results were confirmed by at least three independent experiments. The data are represented as the mean ± s.d., **p < 0.01, Student’s t-test.
Degradation of CjWRKY1 and protein phosphorylation
Because the Cj156-S and CjY cells showed obvious differences in CjWRKY1 accumulation, we also examined the accumulation of CjWRKY1 mutants in Cj156-S protoplasts. Interestingly, the accumulation levels of mutant CjWRKY1 proteins were higher than those of CjWRKY1WT; CjWRKY1Y115E in particular showed the highest accumulation (Fig. 6a, Supplementary Fig. S8). Higher accumulation of mutant CjWRKY1 proteins, however, did not correlate with increased transcriptional activities in the transient reporter assay described above. The different stabilities of CjWRKY1 mutants clearly suggested that post-translational modification was involved in the turnover of CjWRKY1 proteins. A decrease in the accumulation of CjWRKY1 proteins after prolonged incubation also indicated that CjWRKY1 proteins were actively degraded even when post-translational modification was blocked (Fig. 6a).

The stability of the CjWRKY1 protein in C. japonica 156-S protoplasts.
(a) The different stabilities of WT and mutant CjWRKY1 proteins. CjWRKY1 proteins were detected using CjWRKY1-specific antibodies. (b) A proteasome inhibitor, MG132 (50 μM), stabilized the CjWRKY1 proteins for 48 h. (c) Synergistic effects of MG132, protease inhibitors and protein phosphatase inhibitors on the accumulation of CjWRKY1 proteins. Cj156-S protoplasts were treated with 50 μM MG132, 1 mg/ml complete EDTA-free protease inhibitor cocktail, and/or 5 mg/ml phosSTOP phosphatase inhibitor cocktail. An acrylamide gel containing 50 μM Phos-tag was used to detect phosphorylated WRKY1. Arrows indicate the shifted bands corresponding to phosphorylated CjWRKY1. DMSO (0.1%) was used for mock treatments in (b,c). All experiments were repeated at least three times with similar results.
Treatment with MG132, an inhibitor of the 26S proteasome, indicated that CjWRKY1 was degraded by the ubiquitin-proteasome system (UPS) (Fig. 6b), although the transcript levels of CjWRKY1 in protoplasts incubated for 48 h were reduced to one-fifth of the levels in protoplasts incubated for 16 h (Supplementary Fig. S9). Because MG132 treatment did not affect the abundance of phosphorylated CjWRKY1, tyrosine phosphorylation may be not involved in the degradation of CjWRKY1 via the UPS or rapid dephosphorylation may occur.
When a phosphatase inhibitor cocktail was added, however, a marked decrease in CjWRKY1 was observed compared to control protoplasts without MG132 and protease inhibitors and also those with MG132. Addition of a protease inhibitor cocktail reduced the degradation of CjWRKY1 enhanced by the phosphatase inhibitors and resulted in a slower migrating band (Fig. 6c). These results suggest that phosphorylated CjWRKY1 protein can be degraded by proteases in the cytosol or in vacuoles.
Whereas MG132 and/or protease inhibitor treatment increased the stability of CjWRKY1 in Cj156-S protoplasts/cells, little enhancement of target biosynthetic gene expression was observed (Supplementary Fig. S10). As discussed below, transcriptional and post-transcriptional regulation of BIQ biosynthesis may be more complicated than expected or these inhibitors may be harmful to diverse processes related to BIQ biosynthesis in C. japonica protoplasts/cells.
Discussion
In this report, we characterized the post-translational modification of CjWRKY1 in the BIQ biosynthesis in C.japonica. Our functional characterization of the modification of CjWRKY1 is still not complete, but our data suggest that two types of modification modulate the functional activity of CjWRKY1 in BIQ biosynthesis: Tyr phosphorylation and protein degradation.
In animal cells, protein tyrosine kinases play a crucial role in signal transductions as serine/threonine kinases, whereas the importance of tyrosine phosphorylation has been rather unclear in plant cells because no protein tyrosine kinase homologue was found in Arabidopsis thaliana or rice genome16,17. However, recent phosphoproteome analyses showed comparable tyrosine phosphorylation in plant cells as animal cells, suggesting that tyrosine phosphorylation is also important in plant processes18,19,20. Our results that tyrosine phosphorylation clearly modifies DNA binding activity and subcellular localization of CjWRKY1 in C. japonica cells provide the novel evidence on the physiological role of tyrosine phosphorylation in plant cells.
Whereas our results clearly showed the phosphorylation of CjWRKY1 at Y115, the remaining signals detected in immunoprecipitated mutant CjWRKY1Y115F-sGFP protein by immunoblot analysis with anti-pY antibodies suggest additional phosphorylation. Recent phosphoproteome analysis indicated that tyrosine phosphorylation preferentially occurred in the conserved domain of phosphoproteins and multiple phosphorylation of tyrosine residues were found in more than 75% of phosphopeptides18. Since CjWRKY1 contains nine tyrosine residues except for Y115, additional experiment is needed to study the involvement of other tyrosine residue(s) in the phosphorylation and the regulation of WRKY activity.
On the other hand, several groups have reported that serine/threonine phosphorylation of WRKY proteins by mitogen-activated protein kinases (MAPKs) is important in plant immune responses14,21,22. Nicotiana benthamiana WRKY8 was phosphorylated by tobacco MAPKs such as wound-inducible protein kinase (WIPK) and salicylic acid (SA)-inducible protein kinase (SIPK) and the phosphorylation of NbWRKY8 enhanced its DNA binding and trans-activation activities21. AtWRKY33, the closest homologue to NbWRKY8 in A. thaliana, was also a substrate of AtMPK3/MPK6 and the phosphorylation of AtWRKY33 activated camalexin biosynthesis22. Intriguingly, these WRKY proteins belong to Group I WRKY family and have conserved clustered proline-directed serines (SP cluster) at the N-terminal end, the targets of multiple phosphorylation by MAPKs. In addition, Nicotiana tabacum WRKY1 and AtWRKY25, which were also phosphorylated by MAPKs in vitro, belong to Group I WRKYs23,24. In contrast, the tyrosine phosphorylation of WRKYGQK core domain was found in CjWRKY1, which belongs to Group IIc. This is first observation of phosphorylation of tyrosine residue comprising WRKYGQK core domain and it is interesting to know whether tyrosine phosphorylation of WRKY domain commonly occurs to inactivate its transcription factor function in Group IIc WRKYs.
On the other hand, UPS-dependent degradation of transcription factors including WRKYs is common in diverse aspects of plant environmental responses and development25,26,27,28. For example, Oryza sativa WRKY45, which functions in SA signaling in the blast-resistance29, was reported to be degraded by the 26S proteasome in the nucleus and to interact with Panicle blast1 (Pb1) protein to enhance plant resistance27,30,31. Considering the alterations in subcellular localization and accumulation levels of mutant CjWRKY1 proteins, especially CjWRKY1Y115E, UPS-dependent degradation likely occurs mainly in the nucleus and regulates CjWRKY1 activity in BIQ biosynthesis.
Our results suggest that tyrosine phosphorylation of CjWRKY1 inactivates the expression of genes encoding BIQ biosynthetic enzymes through the reduction of binding activity to promoter sequences and translocation to the cytosol, followed by degradation by proteases in the cytosol or vacuoles (Supplementary Fig. S11). Even without phosphorylation, CjWRKY1 proteins would be rapidly degraded in the nucleus by the 26S proteasome, probably due to the high number of PEST residues32. One remaining question is how we can control the turnover of CjWRKY1 to improve BIQ productivity in C. japonica cells because the accumulation of CjWRKY1 proteins in CjY cells treated with any inhibitor was also quite low (Supplementary Fig. S10). No additional experiments were successfully able to overcome the low accumulation of CjWRKY1 protein in CjY cells, which show comparable transcript levels in CjWRKY1 gene and Cj156-S cells2. The identification of two CjWRKY1 degradation pathways indicates that BIQ biosynthesis is intensively regulated at a post-translational level by more factors than previously believed. More recently, Nemoto and colleagues reported that A. thaliana calcium-dependent protein kinase-related protein kinases (CRKs) can phosphorylate tyrosine residues of β-tubulin proteins as well as several transcription factors including AtWRKY1433. Since our expressed sequence tag (EST) prepared from C. japonica cells contains several candidate genes encoding protein kinases including a CRK homologue and E3 ligases, it is of interest to identify the interacting partners involved in the phosphorylation and degradation of CjWRKY1 and apply our findings to metabolic engineering of BIQ biosynthesis.
Materials and Methods
Plant materials
Suspension-cultured C. japonica 156-S cells over-expressing the (S)-scoulerine-9-O-methyltransferase (SMT) gene34,35 were grown in Linsmaier-Skoog (LS) medium (pH 5.7) containing 3% sucrose, 10 μM 1-naphthylacetic acid (NAA) and 10 nM benzyladenine (BA) on a gyratory shaker (90 rpm) at 23 °C in the dark. Non-selected CjY cells were grown in LS medium (pH 5.7) containing 3% sucrose, 100 μM NAA and 1 μM BA under the same conditions as Cj156-S cells.
Vector construction
In addition to a Cauliflower mosaic virus (CaMV) 35S::CjWRKY1 vector constructed in a previous report5, the 35S::CjWRKY1-sGFP vector was constructed as follows: the SalI/NcoI fragment of CjWRKY1 obtained by PCR was inserted between the 35S promoter and the NOS terminator in a pUC18-sGFP vector36. Mutant-CjWRKY1 and mutant-CjWRKY1-sGFP vectors were generated by inverse PCR-based site-directed mutagenesis using a KOD-plus DNA polymerase (Toyobo) and the constructs were confirmed by nucleotide sequencing.
Protein extraction from cultured cells
Approximately 1–5 g (FW) of Cj156-S and CjY cells were inoculated in 20 or 100 ml of culture medium and cultured for 1–2 weeks. The grown cells were collected and ground in liquid nitrogen. An equal volume of extraction buffer [50 mM Tris-HCl (pH 7.5), 1% SDS, 10% glycerol, 2 mM DTT, 1 mM EDTA, 50 μM MG132 (Millipore), complete EDTA-free protease inhibitor cocktail (Roche) and phosSTOP phosphatase inhibitor cocktail (Roche)] to the cell weight was added to powdered samples and placed on ice for 10 min. After centrifugation at 15,000 × g at 4 °C for 20 min, supernatants were recovered and used as total protein fractions. Protein concentrations were measured using Bio-Rad protein assay dye reagent concentrate (Bio-Rad). Equal amounts of protein samples were separated by 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and the separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking the membrane with 5% skim milk in TTBS buffer [20 mM Tris-HCl (pH 7.6), 137 mM NaCl and 0.1% Tween-20], the WRKY1 proteins were detected using anti-CjWRKY1 peptide antibodies (prepared by MBL, 1:1,000) and a horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:15,000 dilution) in TTBS buffer. The immunoblot signal was detected with ECL Prime Western Blotting Detection Reagent (GE Healthcare) and an ImageQuant LAS4010 (GE Healthcare) or a Chemidoc Touch Imaging System (Bio-Rad). After the first round of immunoblotting, the membrane was stripped and re-used for a second immunoblot assay using an anti-α-tubulin antibody (T5168, Sigma, 1:1,000 dilution) to correct for sample loading.
PCR analysis
Total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen) and cDNAs were synthesized from 0.2–1 μg of total RNA using a Super Script III (Invitrogen) or a PrimeScript RT reagent kit with gDNA Eraser (TaKaRa). RT-PCR was performed using Go-Taq Master Mix (Promega) under the following conditions: 95 °C for 4 min, followed by the appropriate number of cycles at 95 °C for 15 s, 60 °C for 20 s and 72 °C for 20 s. Real-time PCR was performed using iQ SYBR Green Supermix and a CFX96 Real-Time PCR Detection System (Bio-Rad) under the following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 20 s and 72 °C for 20 s. The data were quantified by a standard curve method or the ∆∆Ct method and the relative expression levels were standardized by the amplification of ATPase, α-tubulin or β-actin as internal controls. The sequences of specific primers are shown in Supplementary Table S2.
Co-immunoprecipitation (IP) assay
Protoplasts were isolated from Cj156-S cells cultured for 2–3 weeks and transformed using polyethylene glycol (PEG) as previously described with minor modifications37. Approximately 80–100 μg of CjWRKY1-sGFP plasmids was introduced into approximately 106 protoplasts with 20% (v/v) PEG 4000 solution. After 24 h of culturing on a gyratory shaker (40 rpm) at 24 °C in the dark, the protoplasts were collected at 500× g for 3 min and lysed in 500 μl of lysis buffer [50 mM Tris-HCl (pH 7.5), 500 mM NaCl, 10 mM KCl, 10 mM MgCl2, 1% Triton X-100, 10% glycerol, 2 mM EDTA, 1 mM DTT and complete protease inhibitor cocktail, EDTA-free] with two gentle rounds of sonication. After 1 h incubation on a rotator at 4 °C, cell lysate (500 mM NaCl) was diluted to 1/3 with lysis buffer and incubated with 50 μl of anti-GFP microbeads (Miltenyi Biotech) on a rotator for 1 h at 4 °C. The lysate-microbeads mixture was applied to μColumns and eluted after washing according to the manual from the μMACS GFP Tag Protein Isolation Kit (Miltenyi Biotech). Co-immunoprecipitated proteins were analysed with 10% SDS-PAGE and immunoblotted on a PVDF membrane. After blocking the membrane with 5% BSA, phosphorylated proteins were detected using an anti-pY antibody (4G10 platinum, Millipore, 1:1,000 dilution) and a HRP-conjugated anti-mouse IgG (1:10,000–15,000 dilution) antibody in TTBS buffer. The immunoblot signal was detected with ECL Prime Western Blotting Detection Reagent or EzWest Lumi (Atto) and an ImageQuant LAS4010. The same membrane was re-used for the immunoblot assay using anti-GFP antibodies (A6455, Life Technologies, 1:1,000 dilution).
CIP treatment was performed on a μColumn for 40 min at 37 °C before elution. The immunoblot signals were quantified using ImageJ software (http://rsb.info.nih.gov/ij/). Phosphorylation inhibitor treatments were performed in the protoplast culture medium and lysis buffer with 100 μM genistein, 25 μM AG18, or 0.5 μM staurosporine.
EMSA
GST-CjWRKY1 recombinant proteins were expressed in Escherichia coli BL21 (DE3) from full-length cDNAs of CjWRKY1 or its mutants cloned into pGEX-6P-1 (GE Healthcare) at the BamHI and NotI sites. Protein expression was induced with 0.1 mM isopropyl-β-D-thiogalactoside (IPTG) for 6 h at 28 °C. GST-CjWRKY1 proteins were extracted from E. coli cells and purified using Glutathione Sepharose 4B (GE Healthcare).
The double-stranded DNA probes for EMSA were prepared from biotin-labelled sense (5′-CTGCTGACTAGCTGACTAGCTGACTAGATGG-3′) and antisense (5′-CCATCTAGTCAGCTAGTCAGCTAGTCAGCAG-3′) nucleotides annealed in TEN buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA and 0.1 mM NaCl] for 5 min at 95 °C and cooled at room temperature.
For EMSA, biotin-labelled probes (20 fmol) and the purified recombinant proteins (3 μg) were mixed with 1× binding buffer [1 mM MgCl2, 0.5 mM EDTA, 0.05 μg/μl poly (dI-dC)] from a LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific). After a 20 min incubation at 4 °C, the reaction mixtures were separated on a 5% polyacrylamide gel and transblotted on Hybond-N+ nylon transfer membranes. The shifts in biotinylated probes were detected using a Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific) and an ImageQuant LAS4010.
Subcellular localization
Approximately 105 protoplasts isolated from 2- to 3-week-old Cj156-S cells were transfected with 10 μg of 35S::CjWRKY1-sGFP plasmids and localization of the sGFP fusion proteins was detected using a BZ-9000 microscope (Keyence).
LUC reporter assay
Transactivation activities of CjWRKY1 mutants were analysed with CYP80B2 promoter::PpLUC (reporter) and 35S::Renilla reniformis LUC (reference) vectors as reported previously5. In brief, 4 μg of WRKY1 plasmids, 1 μg of reporter plasmids and 0.025 μg of reference plasmids were co-transfected into protoplasts isolated from 2- to 3-week-cultured Cj156-S cells and luciferase activities were detected using a Dual-Luciferase Reporter Assay System (Promega) and a Lumat LB9507 luminometer (Berthold) after incubation for 24 h at 24 °C in the dark.
The 35S::GAL4DB-CjWRKY1 constructs were prepared by insertion of the CjWRKY1 and mutated CjWRKY1 fragments into 35S::GAL4DB at the SmaI and SalI sites. Transactivation activity of the 35S::GAL4DB-CjWRKY1 constructs was assayed after transfection of 5 μg of effector plasmids, 4 μg of reporter plasmids and 0.025 μg of reference plasmids into Cj156-S protoplasts. All experiments were performed with three biological transfections.
Protein extraction from protoplasts
The stabilities of wild-type and mutant CjWRKY1 proteins were evaluated in Cj156-S protoplasts transfected with 10 μg of 35S::CjWRKY1 plasmids. After 20–48 h culture at 24 °C in the dark, the protoplasts were harvested at 500× g for 3 min and treated with 50 μl of SDS sample buffer for 5 min at 95 °C. Total protein extracts were separated on a 15% SDS-PAGE gel or 10% SDS-PAGE gel containing 50 μM Phos-tag (Wako). Immunoblot analysis was performed using anti-CjWRKY1 and anti-α-tubulin antibodies as described above. The effects of protease inhibitors and/or phosphatase inhibitors were measured with 50 μM MG132, complete EDTA-free protease inhibitor cocktail, and/or phosSTOP phosphatase inhibitor cocktail.
Mass spectrometric analysis
Co-IP samples of sGFP and CjWRKY1-sGFP proteins were obtained as described above. Co-immunoprecipitated samples were separated by SDS-PAGE and silver stained (Supplementary Fig. S2). Protein bands were extracted and proteins were digested in-gel with MS grade modified trypsin (Promega). The extracted peptides were subjected to LTQ LC/MS/MS (Thermo Fisher Scientific). The collected ion spectrum data were analysed with the help of Mr. Y. Watanabe (Proteomic Facility Lab., Grad. Sch. Biostudies, Kyoto Univ.) using MassMatrix version 2.4.2 (www.massmatrix.net/)38.
Additional Information
How to cite this article: Yamada, Y. and Sato, F. Tyrosine phosphorylation and protein degradation control the transcriptional activity of WRKY involved in benzylisoquinoline alkaloid biosynthesis. Sci. Rep. 6, 31988; doi: 10.1038/srep31988 (2016).
References
Facchini, P. J. ALKALOID BIOSYNTHESIS IN PLANTS: Biochemistry, cell biology, molecular regulation and metabolic engineering applications. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 29–66 (2001).
Sato, F. Characterization of plant functions using cultured plant cells and biotechnological applications. Biosci. Biotechnol. Biochem. 77, 1–9 (2013).
Dewey, R. E. & Xie, J. Molecular genetics of alkaloid biosynthesis in Nicotiana tabacum. Phytochemistry 94, 10–27 (2013).
Zhu, X., Zeng, X., Sun, C. & Chen, S. Biosynthetic pathway of terpenoid indole alkaloids In Catharanthus roseus. Front. Med. 8, 285–293 (2014).
Kato, N. et al. Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol. 48, 8–18 (2007).
Yamada, Y. et al. Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-type transcription factor in Coptis japonica. Plant Cell Physiol. 52, 1131–1141 (2011).
Yamada, Y., Koyama, T. & Sato, F. Basic helix-loop-helix transcription factors and regulation of alkaloid biosynthesis. Plant. Signal. Behav. 6, 1627–1630 (2011).
Yamada, Y. & Sato, F. Transcription factors in alkaloid biosynthesis. Int. Rev. Cell. Mol. Biol. 305, 339–382 (2013).
Yamada, Y., Motomura, Y. & Sato, F. CjbHLH1 homologs regulate sanguinarine biosynthesis in Eschscholzia californica cells. Plant Cell Physiol. 56, 1019–1030 (2015).
Zhou, M. & Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. (2016).
Eulgem, T., Rushton, P. J., Robatzek, S. & Somssich, I. E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206 (2000).
Eulgem, T. & Somssich, I. E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 10, 366–371 (2007).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Ishihama, N. & Yoshioka, H. Post-translational regulation of WRKY transcription factors in plant immunity. Curr. Opin. Plant Biol. 15, 431–437 (2012).
Chi, Y. et al. Protein-protein interactions in the regulation of WRKY transcription factors. Mol. Plant. 6, 287–300 (2013).
de la Fuente van Bentem, S. & Hirt, H. Protein tyrosine phosphorylation in plants: More abundant than expected? Trends Plant Sci. 14, 71–76 (2009).
Ghelis, T. Signal processing by protein tyrosine phosphorylation in plants. Plant. Signal. Behav. 6, 942–951 (2011).
Sugiyama, N. et al. Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol. Syst. Biol. 4, 193 (2008).
Grimsrud, P. A. et al. Large-scale phosphoprotein analysis in Medicago truncatula roots provides insight into in vivo kinase activity in legumes. Plant Physiol. 152, 19–28 (2010).
van Wijk, K. J., Friso, G., Walther, D. & Schulze, W. X. Meta-analysis of Arabidopsis thaliana phospho-proteomics data reveals compartmentalization of phosphorylation motifs. Plant Cell 26, 2367–2389 (2014).
Ishihama, N., Yamada, R., Yoshioka, M., Katou, S. & Yoshioka, H. Phosphorylation of the Nicotiana benthamiana WRKY8 transcription factor by MAPK functions in the defense response. Plant Cell 23, 1153–1170 (2011).
Mao, G. et al. Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell 23, 1639–1653 (2011).
Menke, F. L. et al. Tobacco transcription factor WRKY1 is phosphorylated by the MAP kinase SIPK and mediates HR-like cell death in tobacco. Mol. Plant Microbe Interact. 18, 1027–1034 (2005).
Andreasson, E. et al. The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J. 24, 2579–2589 (2005).
Duek, P. D., Elmer, M. V., van Oosten, V. R. & Fankhauser, C. The degradation of HFR1, a putative bHLH class transcription factor involved in light signaling, is regulated by phosphorylation and requires COP1. Curr. Biol. 14, 2296–2301 (2004).
Qin, F. et al. Arabidopsis DREB2A-interacting proteins function as RING E3 ligases and negatively regulate plant drought stress-responsive gene expression. Plant Cell 20, 1693–1707 (2008).
Matsushita, A. et al. Nuclear ubiquitin proteasome degradation affects WRKY45 function in the rice defense program. Plant J. 73, 302–313 (2013).
Zhai, Q. et al. Phosphorylation-coupled proteolysis of the transcription factor MYC2 is important for jasmonate-signaled plant immunity. PLoS Genet. 9, e1003422 (2013).
Shimono, M. et al. Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 19, 2064–2076 (2007).
Hayashi, N. et al. Durable panicle blast-resistance gene Pb1 encodes an atypical CC-NBS-LRR protein and was generated by acquiring a promoter through local genome duplication. Plant J. 64, 498–510 (2010).
Inoue, H. et al. Blast resistance of CC-NB-LRR protein Pb1 is mediated by WRKY45 through protein-protein interaction. Proc. Natl. Acad. Sci. USA 110, 9577–9582 (2013).
Rogers, S., Wells, R. & Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234, 364–368 (1986).
Nemoto, K., Takemori, N., Seki, M., Shinozaki, K. & Sawasaki, T. Members of the plant CRK superfamily are capable of trans-/auto- phosphorylation of tyrosine residues. J. Biol. Chem. 290, 16665–16677 (2015).
Sato, F. & Yamada, Y. High berberine-producing cultures of Coptis japonica cells. Phytochemistry 23, 281–285 (1984).
Sato, F. et al. Metabolic engineering of plant alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 98, 367–372 (2001).
Niwa, Y., Hirano, T., Yoshimoto, K., Shimizu, M. & Kobayashi, H. Non-invasive quantitative detection and applications of non-toxic, S65T-type green fluorescent protein in living plants. Plant J. 18, 455–463 (1999).
Yamada, Y. et al. Identification of regulatory protein genes involved in alkaloid biosynthesis using a transient RNAi system. Methods Mol. Biol. 643, 33–45 (2010).
Xu, H. & Freitas, M. A. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 9, 1548–1555 (2009).
Acknowledgements
We thank Dr. Yasuo Niwa for the sGFP vector, Dr. Masaru Ohme-Takagi and Dr. Tomotsugu Koyama for the 35S::GAL4DB and 5 × GAL4-LUC vectors and Dr. Kazuhumi Yazaki for the CjY cultured cells. We also thank Mr. Yuzo Watanabe (Proteomics Facility Lab., Grad. Sch. Biostudies, Kyoto Univ., Japan) for his assistance with the LC-MS/MS analysis. This research was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) [Grant-in-Aid for Scientific Research (S); 26221201 to F.S.]
Author information
Authors and Affiliations
Contributions
Y.Y. and F.S. designed the research, Y.Y. performed the experiments and Y.Y. and F.S. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yamada, Y., Sato, F. Tyrosine phosphorylation and protein degradation control the transcriptional activity of WRKY involved in benzylisoquinoline alkaloid biosynthesis. Sci Rep 6, 31988 (2016). https://doi.org/10.1038/srep31988
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31988
This article is cited by
-
Interplay of transcription factors orchestrating the biosynthesis of plant alkaloids
3 Biotech (2022)
-
Genome-wide identification of AP2/ERF transcription factor-encoding genes in California poppy (Eschscholzia californica) and their expression profiles in response to methyl jasmonate
Scientific Reports (2020)
-
Tyrosine phosphorylation of the GARU E3 ubiquitin ligase promotes gibberellin signalling by preventing GID1 degradation
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
