
Abstract
While aberrant JAK/STAT signaling is crucial to the development of gastric cancer (GC), its effects on epigenetic alterations of its transcriptional targets remains unclear. In this study, by expression microarrays coupled with bioinformatic analyses, we identified a putative STAT3 target gene, NR4A3 that was downregulated in MKN28 GC daughter cells overexpressing a constitutively activated STAT3 mutant (S16), as compared to an empty vector control (C9). Bisulphite pyrosequencing and demethylation treatment showed that NR4A3 was epigenetically silenced by promoter DNA methylation in S16 and other GC cell lines including AGS cells, showing constitutive activation of STAT3. Subsequent experiments revealed that NR4A3 promoter binding by STAT3 might repress its transcription. Long-term depletion of STAT3 derepressed NR4A3 expression, by promoter demethylation, in AGS GC cells. NR4A3 re-expression in GC cell lines sensitized the cells to cisplatin and inhibited tumor growth in vitro and in vivo, in an animal model. Clinically, GC patients with high NR4A3 methylation, or lower NR4A3 protein expression, had significantly shorter overall survival. Intriguingly, STAT3 activation significantly associated only with NR4A3 methylation in low-stage patient samples. Taken together, aberrant JAK/STAT3 signaling epigenetically silences a potential tumor suppressor, NR4A3, in gastric cancer, plausibly representing a reliable biomarker for gastric cancer prognosis.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is the third leading cause of cancer death worldwide1. About 90% of GCs are adenocarcinomas, which can be classified into poorly differentiated diffuse, well-differentiated intestinal and mixed types2. Due to the lack effective therapy, the prognosis of patients with GC remains poor with a 5-year overall survival of less than 25%3. Infection by Helicobacter pylori (H. pylori), a gram-negative bacillus, is considered to be the strongest risk factor for gastric carcinogenesis4. Patients infected with cytotoxin-associated gene A (CagA)-positive H. pylori have a much higher risk for atrophic gastritis, as well as gastric cancer5,6,7. Once the stomach epithelial cells are infected by H. pylori, the CagA protein is injected into the host cells, resulting in multiple signaling pathway dysregulation, including activation of JAK/STAT signaling8,9,10,11. Moreover, infection of H. pylori associates with increased cytokine expression in particularly, interleukin-6, (IL-6) and robust inflammatory response, in gastric cancer12,13, thus suggesting that activation of IL-6-JAK/STAT3 signaling pathways may be crucial for GC development.
JAK/STAT signaling is involved in host defense as well as cancer development14,15,16. Several studies now indicate that STAT3 activation is crucial for GC initiation and progression17,18. Upon binding of IL-6 to its transmembrane receptor, the cytoplasmic tyrosine kinase, Janus kinase (JAK), is activated, followed by phosphorylation and dimerization of STAT319. P-STAT3 then translocates to the nucleus and binds to specific DNA sequence to regulate transcription of specific target genes. Other studies have also demonstrated that STAT3 activation is more prominent in GC patients infected with CagA-positive H. pylori20, although the exact role of JAK/STAT signaling in GC is not fully understood.
Epigenetic modifications including DNA methylation, are essential gene-regulatory event21. In the human genome, methylation takes place at the 5′ position of cytosine in CG dinucleotides resulting in the formation of 5-methylcytosine (5mC) which is initiated and maintained by DNA methyltransferases (DNMTs). Between 60 to 90% of the cytosine at CG dinucleotides are methylated under physiological conditions. In contrast, cytosines that are found in a cluster of CpG rich regions, or “CpG islands”, within the promoter regions of tumor suppressor genes, are typically protected from methylation in normal somatic cells22. However, numerous studies including our own, have demonstrated that tumor suppressor genes are frequently silenced by promoter hypermethylation in human cancers, including GC23,24,25,26,27,28, leading to this event being considered a “hallmark of cancer”21. However, the mechanism of how aberrant promoter methylation arises is not fully understood.
Relatedly, we recently found that dysregulation of TGF-β signaling may lead to epigenetic silencing of its targets in ovarian cancer22,29. To examine the existence of such “epigenetic-mediated transcriptional silencing” in GC, we investigated how STAT3 activation influences the epigenetic silencing of STAT3 targets. To that end, we performed gene expression microarray data, together with bioinformatic analysis, to identify STAT3 targets that are epigenetically silenced by promoter methylation in GC. Our results showed that a nuclear receptor, NR4A3, was epigenetically silenced by promoter DNA methylation in GC cells with constitutive STAT3 activation. The clinical significance of P-STAT3-mediated methylation of NR4A3, in GC patients, was also investigated.
Results
Generation and characterization of a constitutively activated STAT3 gastric cancer cell line
To examine the effect of STAT3 on epigenetic alteration of its downstream targets, we first established a constitutively activated STAT3 gastric cancer (GC) model using MKN28 human GC cell lines24. Either an empty vector control or vector containing Stat3c, a constitutively activated mouse Stat3 mutant30, were then transfected into MKN28 GC cells. After several rounds of selection, Stat3c (S16) stable transfectants and empty vector control (C9) cells, were obtained (Fig. 1A).

Integrated expression microarray and bioinformatic analyses identifies NR4A3 as an epigenetically silenced target of STAT3 in gastric cancer.
(A) Schematic diagram showing the experimental scheme of this study. Empty vector (pcDNA 3.1) or vector expressing a constitutively activated mouse Stat3 mutant (pcDNA3.1-stat3c) were transfected into MKN28 gastric cancer (GC) cells. Stable transfectants were selected from cells expressing the empty vector control (C9) or the Stat3 mutant (S16). Total RNA from C9 and S16 cells was then extracted for expression microarray analysis using an Agilent Human Whole Genome 44 K Expression Microarray, with data presented as a scatter plot. Each dot of the scatter plot represents the fluorescence signal (i.e., the mRNA expression level) of each gene on the array in C9 vs. S16 cells. Genes outside of the black dotted lines denote expression changes of ≥1.5-fold. In addition, bioinformatic analysis was performed to identify potential STAT3 targets by filtering genes with at least one STAT3-binding site within 5 kb of the promoter CpG island, revealing 526 putative STAT3 target genes. One such downregulated STAT3 target in S16 cells, NR4A3, was selected for further analysis. (B) Western blot was performed to investigate the activation status of STAT3 in C9, S16 and the parental MKN28 GC cells. AGS GC cells showing constitutive STAT3 activation were used as a positive control. Stat3c stable transfectants (S16) were confirmed by hybridization of an anti-FLAG antibody. (C) STAT3 activation in S16 cells was confirmed by the expression of STAT3 targets MMP7, Bcl-2 and Mac-2bp, using qRT-PCR (***P < 0.001; **P < 0.01; *P < 0.05). Both S16 and AGS cells showed MMP7 upregulation, compared to C9 control and MKN28 GC cells. (D) STAT3C overexpression slightly increased cell growth in MKN28 cells. S16 and C9 cell numbers were measured by a haemocytometer at each designated day, revealing increased proliferation of S16 cells at day 6 of the experiment, as compared to C9 cells (*P < 0.05). (E) Expression levels of the STAT3 target, NR4A3, were examined in GC cancer cell lines and transfectants and compared to those of MKN28 cells, set to 1.0 as a positive control (***P < 0.0001). Each bar represents mean± SD of duplicate experiments.
The successful stable transfection of Stat3c in S16 GC cells was confirmed by increased expression of total STAT3 and the presence of FLAG (Fig. 1B). Likewise, hyperphosphorylation of Stat3 was observed in S16, but not in C9 vector control or MKN28 GC parental cells, suggesting that STAT3 signaling is constitutively activated in S16 cells. This phenomenon can also be observed in AGS GC cells, in which constitutive activation of STAT3 signaling has previously been reported20,24.
To examine whether Stat3 was functionally active in S16 cells, we examined the expression of the STAT3 upregulated targets, MMP731, Bcl-232 and downregulated target, Mac-2bp33 in S16 and C9 cells. Upregulation of MMP7 and Bcl-2 while down-regulation of Mac-2BP mRNA was observed in S16 cells as compared to C9 cells (Fig. 1C). Interestingly, upregulation of MMP7 was also observed in AGS (Supplementary Figure S1). In addition, S16 cells also showed a slight but significant increase in cell growth (Fig. 1D). Taken together, we successfully established a stable clone with constitutively active Stat3 signaling, via gene overexpression, in MKN28 GC cells.
Combined expression microarray and bioinformatic analyses identify NR4A3 as an epigenetically silenced STAT3 target
To identify genes differentially expressed after Stat3 constitutive activation, gene expression microarray analysis was performed to compare the expression profiles of S16 and C9 cells (Fig. 1A). To further identify differentially expressed genes that were regulated by STAT3, we performed bioinformatic analyses for genome-wide CpG island promoters containing STAT3-binding sites (Fig. 1A, Supplementary Table S3). Combining the results of the expression arrays and bioinformatic analyses of S16 cells, we found 49 upregulated genes with STAT3-binding sites in their promoter CpG islands and 23 downregulated genes, respectively (Fig. 1A).
Our previous ovarian cancer study demonstrated that aberrant TGF-β/SMAD4 signaling may lead to epigenetic silencing of its downstream targets29,34. To identify if aberrant JAK/STAT signaling plays a similar role in GC, we focused on downregulated genes in S16 cells. One downregulated target, NR4A3 (nuclear receptor subfamily 4, group A, member 3), was chosen for further investigation. NR4A3 was previously found to function as a tumor suppressor in acute myeloid leukemia cells35. To confirm our result, RT-PCR showed that NR4A3 mRNA was downregulated in S16 cells having constitutively activated STAT3, as compared to C9 or MKN28 cells (Fig. 1E). It was interesting to note that decreased expression of NR4A3 was also observed in AGS GC cells, in which STAT3 was also constitutively activated (Fig. 1E).
NR4A3 is epigenetically silenced by promoter DNA methylation in gastric cancer
To investigate whether epigenetic modifications contribute to NR4A3 downregulation in S16 GC cells, NR4A3 mRNA expression was analyzed after demethylation treatment with the DNA methyltransferase inhibitor, 5-aza-2′-deoxyglucose (5-aza). 5-aza derepressed NR4A3 expression, as validated by RT-PCR (Fig. 2B). Bisulphite pyrosequencing (Fig. 2C) and methylation-specific PCR (Fig. 2D) also clearly demonstrated NR4A3 promoter hypermethylation in its CpG island “shore”36 (Fig. 2A) in S16, but neither in C9 nor the parental MKN28 cells. To exclude the possibility of incomplete bisulphite conversion, or the presence of 5-hydroxymethylcytosine (5hmC)37. we performed MBD (methyl-binding domain) immunoprecipitation38, followed by PCR, showing that methylated cytosine was enriched in the promoters of S16, but not C9, cells (Fig. 2E).

NR4A3 is repressed by promoter DNA methylation in S16 and other gastric cancer cells.
(A) Schematic diagram showing the genomic map of the NR4A3 promoter, with corresponding locations of CpG sites and a putative STAT3-binding element (SBE, red box). CpG sites interrogated by bisulphite prosequencing (PyroSeq) and MSP are indicated by the red line and black arrows, respectively. (B) NR4A3 expression in S16 cells, following DNA demethylation treatment with 0.5 μM 5-aza-2′-deoxycytidine (5-aza) or DMSO control, was examined by qRT-PCR. As depicted, 5-aza treatment significantly restored NR4A3 expression (*P < 0.05). (C) Bisulphite pyrosequencing was performed to quantitatively examine the methylation levels of 14 CpG sites within the NR4A3 promoter CpG island in C9, S16 and other gastric cancer (GC) cell lines. The percent methylation of each CpG site (circle) is indicated by the intensity of the blue color. NR4A3 promoter methylation was also examined by (D) methylation-specific PCR (MSP) and (E) methyl-binding protein DNA capture (MBDcap) coupled to PCR. For MSP, bisulphite-modified DNA was PCR-amplified using specific primers. “M” and “U” indicate the presence of methylated and unmethylated alleles, respectively. IVD (in vitro methylated DNA) was a positive control for methylation and NB (normal blood) was a negative control for methylation. Water (H2O) was used as a negative control for PCR. In MBDcap-PCR, methylated DNA fragments were immunoprecipitated by MBD protein followed by qPCR (**P < 0.001). (F) Relative expression of NR4A3 in GC lines and (G) 5-aza-treated MKN45 GC cells, as determined by qRT-PCR. Each bar represents mean± SD of dupliate experiments.
We then investigated possible epigenetic silencing of NR4A3 in other GC cell lines. Except for immortalized gastric epithelial (GES) and SNU16 GC cells, promoter hypermethylation was found in all other GC cell lines (Fig. 2C). Results from RT-PCR were consistent with promoter DNA methylation, except for GES cells, in which the NR4A3 promoter was devoid of methylation (Fig. 2F). Robust restoration of NR4A3 expression was observed in MKN45 GC cells treated with 5aza or the histone deacetylase (HDAC) inhibitor trichostatin A (TSA), separately or in combination (Fig. 2G). Taken together, our results suggested that NR4A3 is epigenetically silenced by promoter hypermethylation in GC cell lines.
Activation of STAT3 suppresses NR4A3 expression by promoter hypermethylation
Having demonstrated that NR4A3 was epigenetically silenced in GC, we next investigated whether NR4A3 expression was directly regulated by STAT3. Luciferase reporter assays of the NR4A3 promoter, with or without its putative STAT3-binding site, were performed in AGS GC cells (Fig. 3A). Robust luciferase activity was observed in the promoter containing a putative STAT3-binding site (598 bp fragment). As expected, promoters without the putative STAT3-binding site (413 bp fragment) showed significantly upregulated luciferase activity (Fig. 3A). Surprisingly, transient STAT3 knockdown or treatment with JAK inhibitor, AG490 did not result in significant changes in luciferase activity (Supplementary Figure S2). These results suggest that other co-repressors may be involved in NR4A3 suppression in a long term manner.

Binding of STAT3 represses NR4A3 expression by promoter methylation in AGS GC cells.
(A) NR4A3 promoter regions with (598-bp fragment) or without (413-bp fragment) a putative STAT3 binding site were cloned into pGL3 luciferase-expressing vectors and transfected into AGS GC cells. 24 hours after transfection, luciferase reporter activities were determined. The promoter fragment lacking a STAT3-binding site (pGL3-413) showed significantly (**P < 0.001; ***P < 0.0001) higher luciferase activity than that with a STAT3-binding site (pGL3-598). (B) ChIP-PCR showing that STAT3 binding was significantly enriched in the NR4A3 promoter of S16 cells, as compared to C9 cells. (C) As compared to an unrelated control (GAPDH promoter), STAT3 binding was significantly enriched in the NR4A3 promoter in AGS cells. Expression of (D) STAT3 and (E) NR4A3, in AGS cells, following long-term depletion of STAT3 by lentiviral shRNA knockdown, at each indicated cell passage number. (F) NR4A3 promoter methylation, at each indicated passage number, following long-term STAT3 depletion, as determined by bisulphite pyrosequencing. shGFP, negative control.
As compared to C9 cells, ChIP-PCR showed that STAT3 binding to the NR4A3 promoter was significantly higher in S16 cells, with lower NR4A3 expression (Fig. 3B). Also as expected, STAT3 binding was observed in the NR4A3 promoter, but not in a negative control region (GAPDH), in AGS GC cells, with similarly low NR4A3 expression (Fig. 3C).
Since the above experiments suggested that STAT3 might suppress NR4A3 expression by promoter methylation in GC, we examined NR4A3 expression in AGS cells depleted of STAT3. Lentiviral knockdown, in AGS cells, resulted in STAT3 downregulation for at least 15 passages (Fig. 3D). However, STAT3 expression was gradually restored from passage 20 onward, probably due to methylation of the LTR-driven promoter of the lentiviral shRNA vector (i.e., “position effect variegation”)39. To our surprise, expression of NR4A3 was not restored immediately, but only after 15 passages of STAT3 knockdown (Fig. 3E).
We then investigated NR4A3 promoter methylation during the course of STAT3 knockdown. While NR4A3 promoter methylation in the control cells remained high, it began to decrease at passage 15 in STAT3-knockdown cells and remained more of less the same until the end of the experiments (Fig. 3F). Such multiple passage number dependence of NR4A3 promoter demethylation resembles the progressive loss of DNA methylation (i.e. passive DNA demethylation) through inhibition of DNMT1, by 5-aza incorporation, during DNA replication40. Taken together, these results strongly suggest that STAT3 expression and activation can suppress NR4A3 expression, in a long-term and stable manner through promoter DNA hypermethylation.
Ectopic expression of NR4A3 inhibits tumor growth in vitro and in vivo
Given that NR4A3 is a tumor suppressor in acute myelocytic leukemia35, our results demonstrated that NR4A3 is epigenetically silenced in GC. Consequently, we investigated the functional role of NR4A3 in GC cell lines. Ectopic expression of NR4A3 (Supplementary Figure S3) inhibited growth of AGS (Fig. 4A) and MKN45 GC cells (Fig. 4B,C and Supplementary Figure S4). The lack of a significant sub-G1 population in NR4A3-overexpressing cells suggested that NR4A3 might not act through conventional apoptotic pathways (Fig. 4D). However, expression of NR4A3 significantly sensitized MKN45 GC cells to cisplatin (Supplementary Figure S5) by restoring G2/M arrest (Fig. 4D and Supplementary Figure S6). Finally, nude mice subcutaneously injected with NR4A3-overexpressing MKN45 cells grew tumors of significantly less volume, as compared to mice injected with MKN45 cells with empty vector only (Fig. 4E). Taken together, these in vitro and in vivo results indicate NR4A3 to be a potential tumor suppressor in gastric cancer.

Ectopic expression of NR4A3 inhibits tumor growth in vitro and in vivo in a xenograft mouse model.
Ectopic expression of NR4A3 inhibited tumor growth, as determined by colony formation assay. AGS or MKN45 GC cells transfected with empty or NR4A3 expression vector were selected for further experiments (Supplementary Figure S1). (A) AGS or (B) MKN45 GC cells overexpressing NR4A3 had significantly fewer colonies than the control. Right panel, quantitative analysis of the colony formation assay. (C) Cell growth of MKN45 GC cells, with or without NR4A3 expression, was determined by cell counting. Ectopic expression of NR4A3 significantly inhibited cell growth in MKN45 GC cells. (D) Flow cytometry analysis of MKN45 GC cells with or without cisplatin (1 μg/ml) for 24 hr. DNA fluorescence histogram shows that NR4A3 overexpression enhanced G2/M arrest in cisplatin-treated cells (please also refer to Supplementary Figure S4). (E) Effect of NR4A3 overexpression on in vivo tumor growth in a nude mouse model. MKN45 cells stably transfected with NR4A3 or empty vector (control) were injected subcutaneously into both flanks of athymic nude mice. Tumor volumes were measured daily. Representative examples of tumors formed in nude mice are also shown (right panel). **P < 0.01; *P < 0.05.
Epigenetic suppression of NR4A3 is associated with poor prognosis in gastric cancer patient samples
The above experiments demonstrated NR4A3 as a potential tumor suppressor that is epigenetically silenced by promoter methylation, upon STAT3 activation. To investigate the clinical significance of NR4A3 methylation in gastric carcinogenesis, we performed quantitative methylation-specific PCR (qMSP) to analyze NR4A3 methylation in human gastritis and primary gastric tumor samples (Supplementary Table S1). NR4A3 methylation levels in tumor tissues were significantly higher than those found in samples from adjacent normal or gastritic tissue specimens (Fig. 5A). NR4A3 methylation also progressively increased according to advancement in tumor stage (Fig. 5B), grade (Fig. 5C) and lymph node metastasis (Supplementary Table S4). Patients with higher NR4A3 methylation had significantly shorter survival than those with lower NR4A3 methylation (Fig. 5D, Table 1). Interestingly, patients with high STAT3 activation also had shorter survival (Fig. 5E, Table 1), while Cox regression analysis demonstrated that NR4A3 methylation could serve as an independent prognostic factor for overall survival (Table 1, multivariate analysis). We also analyzed the relationship between STAT3 activation and NR4A3 methylation in this sample cohort. Intriguingly, STAT3 activation significantly associated only with NR4A3 methylation in early-stage patient samples (Supplementary Table S5).

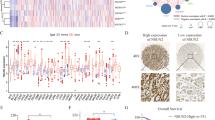
NR4A3 promoter hypermethylation and increased STAT3 activity correlate with poor survival in gastric cancer patients.
Quantitative real time MSP (qMSP) was performed to determine NR4A3 methylation levels in gastritis (n = 9) and primary gastric cancer (GC) patient samples (n = 88). NR4A3 methylation was significantly higher in (A) GC tumor samples and matched adjacent normal tissues, as compared to gastritis. NR4A3 methylation in tumor samples also showed a progressive increase with (B) stage and (C) grade. (D) Kaplan-Meier analysis of NR4A3 methylation in tumor tissues for overall survival of gastric cancer patients. GC patients with higher NR4A3 methylation demonstrated shorter overall survival than patients with lower methylation (log-rank test, **P = 0.003). (E) Kaplan-Meier analysis also showed that patients with higher STAT3 nuclear staining (“STAT3 IHC high,” red line, vs. “STAT3 IHC low, black line) had significantly (P = 0.0382) shorter overall survival than patients with lower STAT3 nuclear staining (representative STAT3 IHC images shown in right panel). (F) NR4A3 IHC, as performed on another independent cohort of 128 GC patient tumors, using tissue microarrays. Similar to high STAT3 IHC, Kaplan-Meier analysis also showed that patients with lower NR4A3 staining (“NR4A3 IHC low,” red line, vs. “NR4AC IHC low,” black lines) had shorter (P = 0.0272) overall survival than those with higher NR4A3 staining. Representative NR4A3 IHC images are shown in the right panel.
As NR4A3 methylation associated with GC patient shorter survival, we performed immunohistochemistry (IHC) to investigate NR4A3 expression in tissue microarrays of another independent cohort containing 128 GC tumor samples (Supplementary Table S1). In this second cohort, patients with low NR4A3 expression did not associate with any clinical parameters (Supplementary Table S6), except shorter overall survival (Fig. 5F, Supplementary Table S7).
Discussion
DNA methylation, an epigenetic modification, is a genome-wide biological phenomenon which plays an essential role in gene dosage regulation in diverse cellular processes, including genomic imprinting and X chromosome inactivation and also governs cell fate commitment. Several studies have shown that dysregulated DNA methylation participates in the development of numerous disease states, including human cancers41. However, how DNA methylation goes awry in human cancer is not fully understood.
Several studies have already demonstrated that activation of JAK/STAT signaling is crucial to gastric carcinogenesis with particular regard to H. pylori-infected tissues8,17,42; however, its role in epigenetic silencing of its target genes has not been fully explored. Since we previously demonstrated that aberrant TGF-β/SMAD4 signaling in ovarian cancer led to epigenetic silencing of its downstream targets29,34, we further examined whether such “signaling-mediated epigenetic silencing”22 exists in gastric cancer (GC), through aberrant activation of JAK/STAT signaling. By establishing hyperactivated STAT3 signaling in MKN28 GC cells, we showed that epigenetic silencing of NR4A3, a nuclear receptor and putative STAT3 target, could be derepressed. Interestingly, epigenetically silenced NR4A3 could also be observed in AGS GC cells having constitutive, endogenous activation of STAT3 signaling. However, it is noteworthy to point out that NR4A3 methylation was not observed in immortalized gastric epithelial GES cells, which do not express NR4A3. Thus, the role of other epigenetic modifications, such as histone acetylation/methylation, in regulating NR4A3 transcription, cannot be excluded.
Our results from promoter luciferase assays and ChIP-PCR suggest that STAT3 might act as a transcriptional repressor of NR4A3. Importantly, depletion of STAT3 in AGS GC cells resulted in NR4A3 promoter demethylation and re-expression. It is noteworthy that transient STAT3 knockdown or treatment with JAK inhibitor, AG490 did not result in a significant increase of promoter luciferase activity in AGS cells. While derepression and presumably, demethylation of NR4A3 could only be achieved after long-term depletion of STAT3 (≥15 passages). Taken together, it is thus suggested that NR4A3 repression is established in a stable, STAT3 concentration-dependent manner, in AGS cells. Repression of NR4A3 was rapidly restored after STAT3 re-expression, indicating that STAT3 may be a key initiator in the transcriptional repression of NR4A3. However, involvement of other co-repressors cannot be excluded.
Several groups have previously demonstrated that aberrant activation of transcription factors can induce epigenetic silencing of their target genes. For example, Di Croce et al. discovered that activation of the oncogenic fusion protein, PML-RARα, resulted in promoter hypermethylation and transcriptional repression of its downstream target gene, RARβ, by recruitment of a DNA methyltransferase (DNMT)43. Wasik and colleagues further demonstrated that malignant T lymphocytes, with persistent activation of STAT3, could directly bind and recruit DNMT1, as well as histone deacetylase-1 (HDAC1), to the promoter of the tyrosine phosphatase gene SHP1, resulting in its promoter DNA hypermethylation and epigenetic silencing44. Our current study, however, is the first to demonstrate that activation of STAT3 could result in promoter hypermethylation and epigenetically silence another target, NR4A3, in gastric cancer. Thus, the involvement of certain corepressors, such as the Daxx45, in the recruitment of DNMT to the NR4A3 promoter, deserves further investigation.
NR4A3, also known as NOR-1, is a nuclear receptor and transcription factor involved in various cellular, metabolic and tumor suppressor processes46,47,48,49. Mullican et al., using NR4A1/NR4A3 double-knockout mice, reported that loss of these two genes can result in the development of acute myelocytic leukemia (AML), due to uncontrolled expansion of myeloid progenitor cells35. Consistent with those results, our in vitro and in vivo functional studies also showed that overexpression of NR4A3 inhibited tumor growth (Fig. 4). Moreover, GC patients with higher methylation or lower expression of NR4A3 demonstrated significantly shorter overall survival, indicating a potential tumor suppressor role (Fig. 5C). Given that activation of STAT3 signaling resulted in epigenetic silencing of NR4A3 in GC, we expected to observe a positive correlation between STAT3 activation and NR4A3 methylation in our sample cohort. However, such a correlation could only be observed in early-stage tumor samples (Supplementary Table S5, P = 0.01). This phenomenon might be due to the fact that STAT3-mediated epigenetic silencing of NR4A3 is initiated during early gastric carcinogenesis; however, this must be confirmed in a larger sample cohort.
Activation of STAT3 was previously found to associate with drug resistance in GC50 and derepression of NR4A3 sensitized gastric cancer cells to cisplatin. In this regard, the shorter overall survival we observed in patients with NR4A3 methylation may likely be due to chemo-resistance-related tumor recurrence. Thus, NR4A3 methylation might be an effective prognostic marker for predicting therapeutic response.
Several studies has previously demonstrated that targeted inhibition of STAT3 could be a novel therapeutic strategy against GC51,52. Our study may provide a mechanistic reason such that inhibition of STAT3 may reverse the epigenome and thus restoring the expression of tumor suppressor genes. Combination of STAT3 inhibitor together with low dose epigenetic modifiers such as 5aza, might result in a more specific reversal of the epigenome and derepression of tumor suppressors, thus deserves further clinical trial in GC.
There are several limitations in our present study. First, as a retrospective study, selection bias may exist and cannot be totally avoided, even after multivariate analysis adjustment. Second, no association between H. pylori infection and methylation of NR4A3 was found in this tumor sample cohort. This shortcoming may be attributed to the relatively small sample size of H. pylori-positive samples (n = 6). Therefore, prospective studies, with a more balanced sample-distribution, should be conducted to confirm any hypothesized associations.
Taken together, our results reveal that constitutive activation of JAK/STAT3 signaling can lead to transcriptional silencing of a STAT3 target, NR4A3, through aberrant promoter DNA methylation. Overexpression of NR4A3 suppressed tumor growth in vitro and in vivo, strongly suggesting that it may be a novel tumor suppressor in GC, as increased NR4A3 hypermethylation associated with shorter overall survival. In conclusion, NR4A3 methylation may be able to serve as a diagnostic and prognostic biomarker for GC. The therapeutic potential of targeted inhibition of STAT3 in GC deserves further investigation.
Methods
Study subjects and tissue samples collection
Genomic DNA from 88 gastric tumor and matched adjacent normal samples were collected from the Changhua Christian Hospital, Chang-Hua, Taiwan (clinical-pathological data summarized in Supplementary Table S1). For tissue microarrays, another independent cohort, containing 128 gastric tumor specimens, was also procured from the same hospital. For a negative control, genomic DNA from nine non-cancer gastritis patients was collected at the Prince of Wales Hospital, Shatin, Hong Kong. All human subject assessments were approved by the Institutional Review Board (IRB) of Changhua Christian Hospital, Chang-Hua, Taiwan and the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee. The study was carried out in strict accordance with approved guidelines. Informed consent was obtained from all participants.
Cell culture and epigenetic treatment
Six gastric cancer cell lines (AGS, MKN28, MKN45, KATOIII, SNU1 and SNU16) and an immortalized gastric epithelial cell line, GES (a kind gift from Dr. Jun Yu, The Chinese University of Hong Kong, Hong Kong), were maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA). The transformed human embryonic kidney cell line (HEK293T) was maintained in DMEM (Invitrogen). All the culture media used was supplemented with 10% fetal bovine serum (FBS, Invitrogen), 50 units/ml of penicillin/streptomycin (Invitrogen) and cells were incubated at 37 °C under a humidified atmosphere containing 5% CO2. For DNA demethylation treatment, cells were seeded in 60-mm plates and treated with 0.5 μM 5′-aza-2′-deoxycytidine (5aza, Sigma, St. Louis, MO, USA) for 72 hr, with or without 0.5 μM of the histone deacetylase (HDAC) inhibitor trichostatin A (TSA, Sigma) for 12 hr. Culture media and drugs were replenished every 24 hr. Following the various treatments, the cells were lysed and harvested for DNA or RNA analyses. Experiments were repeated twice.
mRNA expression microarray
Gene expression microarray analysis was performed by Welgene Biotech (Taipei, Taiwan), using Agilent (Santa Clara, CA, USA) Human Whole Genome 44 K expression microarrays and expression-varied (1.5-fold up- or down-regulated) genes were chosen for further assessment. The microarray data has been deposited in the Gene Expression Omnibus database (accession number: GSE78714).
Real-time quantitative methylation-specific PCR (qMSP)
For measuring DNA methylation, DNA was bisulphite-modified (resulting in deamination of unmethylated cytosine to uracil) using an EZ DNA methylationTM kit (ZYMO Research, Irvine, CA, USA), according to the manufacturer’s protocol and subjected to quantitative methylation-specific PCR (qMSP), as previously described53. PCR qMSP amplification was performed using the StepOne Real-Time PCR instrument (Applied Biosystems, Carlsbad, CA, USA), with ACTB used to normalize the input DNA. The absolute amount of methylated NR4A3 was deter ed by the threshold PCR cycle number (Ct), for each sample, using a standard curve generated by qMSP of an SssI-treated cloned DNA fragment. The relative percentage of NR4A3 methylation was calculated as the NR4A3 vs ACTB ratio for each sample, divided by the same ratio for SssI-treated sperm DNA (positive DNA methylation control, Millipore, Billerica, MA, USA) and multiplied by 100. Experiments were repeated twice.
Bisulphite pyrosequencing
Bisulphite pyrosequencing was performed as described previously54. In brief, bisulphite-modified DNA was subjected to PCR amplification, using a tailed reverse primer and a biotin-labeled universal primer (Supplementary Table S2). PCR and sequencing primers, for a 194-bp fragment (+2338 to +2532) of the NR4A3 promoter CpG island “shore”36, were designed using the PyroMark Assay Design 2.0 instrument (Qiagen, Hercules, CA, USA) and pyrosequencing performed using PyroMark Q24 (Qiagen) and Pyro Gold Reagents (Qiagen), according to the manufacturer’s protocol. Methylation levels of 14 CpG sites, within the 194-bp sequence, were measured and methylation percentages for each cytosine were determined by their fluorescence intensities divided by the sum total fluorescence intensities of cytosines and thymines (converted from uracil by PCR), at each CpG site.
Cell cycle analysis
MKN45/control or MKN45/NR4A3 (S16) GC cells were harvested, washed with cold PBS, fixed with 100% cold methanol and incubated overnight at 4 °C. The cells were subsequently treated with 50 μg/mL RNaseA (Invitrogen) and 40 μg/mL propidium iodide (Sigma-Aldrich) for 30 min. DNA content was analyzed using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA), with the cell cycle percentages of the cells calculated using ModFit software (Verity Software House, Topsham, MA, USA). Experiments were repeated twice.
Immunohistochemical (IHC) analysis
Paraffin-embedded GC tissue samples from the above-mentioned patients were obtained from the Department of Pathology at the Changhua Christian Hospital, following Institutional Review Board approval. Tissue sections were dewaxed in xylene and rehydrated in alcohol. Antigen retrieval was performed by heating each section at 100 °C for 20 min in 10 mM sodium citrate buffer (pH 6.0). IHC followed a standard protocol using an anti-STAT3 monoclonal antibody (1:100, Cell Signaling, Danvers, MA, USA) and an anti-NR4A3 antibody (Cell Signaling), using the NovolinkTM Min Polymer Detection System (Leica, Wetzlar, Germany). IHC scores were calculated using the formula: intensity x percentage of cells showing positive staining. The STAT3 percent activation score was calculated using the formula: (nuclear IHC score)/(nuclear IHC score + cytosolic IHC score). The results were then assessed by two pathologists independently (CYL and KTY).
In vivo tumorigenicity assay
To investigate the effect of NR4A3 on GC growth in vivo, 1×107 of MKN45/control or MKN45/NR4A3 (S16) cells were injected subcutaneously into the flanks of 5-week-old female nude mice (BALB/cByJNarl, n = 4), obtained from the National Laboratory Animal Center, Taiwan. To reduce the number of animals used, the control and experimental groups used both sides of the same mouse. Tumor size was measured daily with calipers as length (L) and width (W) and tumor volume calculated using the formula (L x W2/2). At the end of the experiment, all mice were sacrificed by cervical dislocation. All mice were kept under specific pathogen-free conditions using a laminar airflow rack, with free access to sterilized food and autoclaved water. All experiments were approved by the Animal Experimentation Ethics Committee of National Chung Cheng University, Taiwan. This study was performed in accordance with the approved guidelines and regulations of National Chung Cheng University.
Bioinformatic and statistical analysis
Bioinformatic analysis was performed to identify predicted STAT3-binding sites using the UCSC table browser (https://genome.ucsc.edu/cgi-bin/hgTables). Statistical significance was determined using GraphPad Prism Version 5.0 software packages for Windows (GraphPad Software, La Jolla, CA, USA). The Student’s t test or the Mann–Whitney U test was used to compare parameters of different groups. Overall survival (OS) was assessed by Kaplan-Meier analysis using log-rank test. Univariate and multivariate survival analysis was determined using a Cox proportional hazards model. Overall survival was defined as the duration from day of diagnosis to death. A DNA methylation level of NR4A3 at 1% (methylation level in gastritis) was used as a cutoff and for IHC scores for STAT3 and NR4A3, 50% was used as a cutoff. P < 0.05 was considered statistically significant.
Additional Materials and Methods can be found in supplementary information.
Additional Information
How to cite this article: Yeh, C.-M. et al. Epigenetic silencing of the NR4A3 tumor suppressor, by aberrant JAK/STAT signaling, predicts prognosis in gastric cancer. Sci. Rep. 6, 31690; doi: 10.1038/srep31690 (2016).
References
Torre, L. A. et al. Global cancer statistics, 2012. CA Cancer J Clin 65, 87–108 (2015).
Lauren, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand 64, 31–49 (1965).
Camargo, M. C. et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: an international pooled analysis. Gut 63, 236–243 (2014).
Polk, D. B. & Peek, R. M., Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10, 403–414 (2010).
Kuipers, E. J., Perez-Perez, G. I., Meuwissen, S. G. & Blaser, M. J. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst 87, 1777–1780 (1995).
Blaser, M. J. et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55, 2111–2115 (1995).
Parsonnet, J., Friedman, G. D., Orentreich, N. & Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40, 297–301 (1997).
Bronte-Tinkew, D. M. et al. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res 69, 632–639 (2009).
Lee, I. O. et al. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem 285, 16042–16050 (2010).
Higashi, H. et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295, 683–686 (2002).
Odenbreit, S. et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500 (2000).
Yamaoka, Y., Kita, M., Kodama, T., Sawai, N. & Imanishi, J. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 110, 1744–1752 (1996).
Yamaoka, Y. et al. Relation between cytokines and Helicobacter pylori in gastric cancer. Helicobacter 6, 116–124 (2001).
Schneider, M. R. et al. Interleukin-6 stimulates clonogenic growth of primary and metastatic human colon carcinoma cells. Cancer letters 151, 31–38 (2000).
Wu, C. W. et al. Serum interleukin-6 levels reflect disease status of gastric cancer. Am J Gastroenterol 91, 1417–1422 (1996).
Kabir, S. & Daar, G. A. Serum levels of interleukin-1, interleukin-6 and tumour necrosis factor-alpha in patients with gastric carcinoma. Cancer letters 95, 207–212 (1995).
Jenkins, B. J. et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med 11, 845–852 (2005).
Kanda, N. et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene 23, 4921–4929 (2004).
O’Shea, J. J., Gadina, M. & Schreiber, R. D. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109 Suppl, S121–131 (2002).
Jackson, C. B. et al. Augmented gp130-mediated cytokine signalling accompanies human gastric cancer progression. J Pathol 213, 140–151 (2007).
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734 (2011).
Chou, J. L., Chen, L. Y., Lai, H. C. & Chan, M. W. TGF-beta: friend or foe? The role of TGF-beta/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert Opin Ther Targets 14, 1213–1223 (2010).
Cheng, A. S. et al. Helicobacter pylori Causes Epigenetic Dysregulation of FOXD3 to Promote Gastric Carcinogenesis. Gastroenterology 144, 122–133 e129 (2013).
To, K. F. et al. Constitutional activation of IL-6-mediated JAK/STAT pathway through hypermethylation of SOCS-1 in human gastric cancer cell line. Br J Cancer 91, 1335–1341 (2004).
Yang, W. et al. Epigenetic silencing of GDF1 disrupts SMAD signaling to reinforce gastric cancer development. Oncogene (2015).
Yoshida, S. et al. Epigenetic inactivation of FAT4 contributes to gastric field cancerization. Gastric Cancer (2016).
Esteller, M., Corn, P. G., Baylin, S. B. & Herman, J. G. A gene hypermethylation profile of human cancer. Cancer Res 61, 3225–3229 (2001).
Byun, D. S., Lee, M. G., Chae, K. S., Ryu, B. G. & Chi, S. G. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res 61, 7034–7038 (2001).
Yeh, K. T. et al. Aberrant TGFbeta/SMAD4 signaling contributes to epigenetic silencing of a putative tumor suppressor, RunX1T1 in ovarian cancer. Epigenetics 6, 727–739 (2011).
Bromberg, J. F. et al. Stat3 as an oncogene. Cell 98, 295–303 (1999).
Yuan, G. et al. HER2-dependent MMP-7 expression is mediated by activated STAT3. Cell Signal 20, 1284–1291 (2008).
Liu, A. et al. XZH-5 inhibits STAT3 phosphorylation and enhances the cytotoxicity of chemotherapeutic drugs in human breast and pancreatic cancer cells. PloS one 7, e46624 (2012).
Park, Y. P. et al. Role of STAT3 as a negative regulator in Mac2- binding protein expression. The Korean journal of laboratory medicine 28, 230–238 (2008).
Chan, M. W. et al. Aberrant transforming growth factor beta1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia 10, 908–919 (2008).
Mullican, S. E. et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med 13, 730–735 (2007).
Irizarry, R. A. et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature genetics 41, 178–186 (2009).
Penn, N. W., Suwalski, R., O’Riley, C., Bojanowski, K. & Yura, R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. The Biochemical journal 126, 781–790 (1972).
Hsu, H. K., Weng, Y. I., Hsu, P. Y., Huang, T. H. & Huang, Y. W. Detection of DNA methylation by MeDIP and MBDCap assays: an overview of techniques. Methods Mol Biol 1105, 61–70 (2014).
Svoboda, J., Hejnar, J., Geryk, J., Elleder, D. & Vernerova, Z. Retroviruses in foreign species and the problem of provirus silencing. Gene 261, 181–188 (2000).
Momparler, R. L. Epigenetic therapy of cancer with 5-aza-2′-deoxycytidine (decitabine). Semin Oncol 32, 443–451 (2005).
Egger, G., Liang, G., Aparicio, A. & Jones, P. A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463 (2004).
Zhao, J. et al. Helicobacter pylori-induced STAT3 activation and signalling network in gastric cancer. Oncoscience 1, 468–475 (2014).
Di Croce, L. et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295, 1079–1082 (2002).
Zhang, Q. et al. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA 102, 6948–6953 (2005).
Muromoto, R. et al. Physical and functional interactions between Daxx and STAT3. Oncogene 25, 2131–2136 (2006).
Pearen, M. A. & Muscat, G. E. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol 24, 1891–1903 (2010).
Safe, S. et al. Nuclear receptor 4A (NR4A) family - orphans no more. J Steroid Biochem Mol Biol 157, 48–60 (2016).
Pearen, M. A. et al. The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology 149, 2853–2865 (2008).
Beard, J. A., Tenga, A. & Chen, T. The interplay of NR4A receptors and the oncogene-tumor suppressor networks in cancer. Cell Signal 27, 257–266 (2015).
Huang, S. et al. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer letters 315, 198–205 (2012).
Cafferkey, C. & Chau, I. Novel STAT 3 inhibitors for treating gastric cancer. Expert opinion on investigational drugs 1–9 (2016).
Kim, M. J. et al. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer letters 335, 145–152 (2013).
Chou, J. L. et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest 90, 414–425 (2010).
Yeh, C. M. et al. Methylomics analysis identifies ZNF671 as an epigenetically repressed novel tumor suppressor and a potential non-invasive biomarker for the detection of urothelial carcinoma. Oncotarget 6, 29555–29572 (2015).
Acknowledgements
The authors would like to thank Dr. Curt Balch for manuscript editing. This study was supported by research grants from the Ministry of Science and Technology, Taiwan (NSC97-2320-B-194-002-MY3, MOST 104-2320-B-194-003 and MOST 104-2314-B-194-001-MY3).
Author information
Authors and Affiliations
Contributions
C.M.Y., L.Y.C., S.H.L., H.Y.H., L.H.Z., C.D., C.Y.L. and J.L.C. performed experiments. S.Y.C., H.W.W. and J.M.L. performed bioinformatic analysis. E.K.N., A.S.C., J.L., S.F.W. and K.T.Y. provided samples and materials. Y.T.H., A.S.C., J.L., K.T.Y. and M.W.C. contributed to discussion. L.Y.C., H.Y.H. and M.Y.C. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yeh, CM., Chang, LY., Lin, SH. et al. Epigenetic silencing of the NR4A3 tumor suppressor, by aberrant JAK/STAT signaling, predicts prognosis in gastric cancer. Sci Rep 6, 31690 (2016). https://doi.org/10.1038/srep31690
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31690
This article is cited by
-
Potential Roles of Nr4a3-Mediated Inflammation in Immunological and Neurological Diseases
Molecular Neurobiology (2024)
-
The possibilities of LOXL4 as a prognostic marker for carcinomas
Amino Acids (2023)
-
Matriptase-2/NR4A3 axis switches TGF-β action toward suppression of prostate cancer cell invasion, tumor growth, and metastasis
Oncogene (2022)
-
Functional association of NR4A3 downregulation with impaired differentiation in myeloid leukemogenesis
Annals of Hematology (2022)
-
Identification of circRNA–miRNA–mRNA networks contributes to explore underlying pathogenesis and therapy strategy of gastric cancer
Journal of Translational Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
