
Abstract
Boron suboxide B6O, the hardest known oxide, has an R m crystal structure (α-B6O) that can be described as an oxygen-stuffed structure of α-boron, or, equivalently, as a cubic close packing of B12 icosahedra with two oxygen atoms occupying all octahedral voids in it. Here we show a new ground state of this compound at ambient conditions, Cmcm-B6O (β-B6O), which in all quantum-mechanical treatments that we tested comes out to be slightly but consistently more stable. Increasing pressure and temperature further stabilizes it with respect to the known α-B6O structure. β-B6O also has a slightly higher hardness and may be synthesized using different experimental protocols. We suggest that β-B6O is present in mixture with α-B6O, and its presence accounts for previously unexplained bands in the experimental Raman spectrum.
m crystal structure (α-B6O) that can be described as an oxygen-stuffed structure of α-boron, or, equivalently, as a cubic close packing of B12 icosahedra with two oxygen atoms occupying all octahedral voids in it. Here we show a new ground state of this compound at ambient conditions, Cmcm-B6O (β-B6O), which in all quantum-mechanical treatments that we tested comes out to be slightly but consistently more stable. Increasing pressure and temperature further stabilizes it with respect to the known α-B6O structure. β-B6O also has a slightly higher hardness and may be synthesized using different experimental protocols. We suggest that β-B6O is present in mixture with α-B6O, and its presence accounts for previously unexplained bands in the experimental Raman spectrum.
Similar content being viewed by others
Introduction
Superhard materials are used in many applications, from cutting, grinding and drilling tools to wear-resistant coatings1,2,3. However, most superhard materials4, such as diamond5 and cubic-BN6, are synthesized at high pressure, which makes them expensive, but some (boron allotropes, B6O, B4C) are thermodynamically stable already at ambient conditions. The hardness of α-B6O7 was reported to be in the range 30–45 GPa8,9, making it the hardest known oxide9,10,11.
Objects with icosahedral symmetry (Ih) bear a special fascination because of incompatibility of fivefold symmetry with crystalline periodicity. The discovery of multiply-twinned particles B6O, an icosahedral packing of B12 icosahedra with Ih symmetry, had aroused interest7. Here we report the prediction of a new phase of B6O, with space group Cmcm, which we name β-B6O. This structure is energetically almost degenerate with α-B6O (and slightly more stable), is predicted to have a higher hardness, and actually corresponds to twinned α-B6O structure.
Results
Discovery of β-B6O at ambient conditions
Our variable-composition evolutionary searches expectedly find B2O3 and B6O to be the only stable compounds in the B-O system. Interestingly, there are also several compounds very close to stability - B2O7 (this is a 2D-form of B2O3 intercalated with oxygen molecules) and oxygen-deficient versions of B6O with B6O-like structures and compositions between B and B6O. To our surprise, Cmcm-B6O (β-B6O, see Table 1 for structural parameters), instead of the well-known R m-B6O (α-B6O)7,12,13,14, turned out to be the most stable structure at ambient pressure, as shown in Fig. 1; phonon calculations have confirmed its dynamical stability. Transmission electron microscopy recently confirmed its existence15. Structural parameters and some of the physical properties of β-B6O are shown in Table 1, in comparison with α-B6O and two related forms of pure boron.
m-B6O (α-B6O)7,12,13,14, turned out to be the most stable structure at ambient pressure, as shown in Fig. 1; phonon calculations have confirmed its dynamical stability. Transmission electron microscopy recently confirmed its existence15. Structural parameters and some of the physical properties of β-B6O are shown in Table 1, in comparison with α-B6O and two related forms of pure boron.

(a) Convex hull of the B-O system at ambient pressure. The solid (hollow) points represent the stable (metastable) structures. (b) Enthalpy difference between β-B6O and α-B6O, including zero-point energy.
In order to further compare the stability of β-B6O and α-B6O, we calculated their enthalpies as a function of pressure, as shown in Fig. 1b. We found that the enthalpy of β-B6O is lower than that of α-B6O at ambient pressure, but the energy difference is only about 1.8 meV/formula within the GGA (and almost degenerate within the HSE06 hybrid functional). As pressure increases, β-B6O becomes progressively more favorable than α-B6O, indicating that β-B6O might be more easily synthesized under pressure. The enthalpies of the two structures are so close that it makes us think: will the two structures coexist? what is their relationship? how to synthesize β-B6O? In order to answer these questions, we perform a detailed comparison of their structure, Raman spectra and phonon densities of states (PDOS).
Comparison of crystal structures of α-B6O and β-B6O
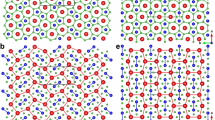
β-B6O structure has hexagonal close packing of B12 icosahedra (ABAB… stacking), while α-B6O is based on the cubic close packing (ABCABC… stacking) of B12 icosahedra, as shown in Fig. 2d,c. As is the case of hcp and fcc metals, twinning of α-B6O can produce local β-B6O stackings. It may also be possible to obtain β-B6O-like stacking faults by deformation of α-B6O, through plane slips. Most properties of these two phases are very similar: e.g. predicted volume per formula (V(α-B6O = 51.71 Å3/formula; V(Cmcm-B6O) = 51.69 Å3/formula), hardness (Hv (α-B6O) = 38 GPa; Hv (Cmcm-B6O) = 39 GPa), elastic moduli (Table 1), DFT band gaps (α-B6O has a 1.85 eV direct band gap, while Cmcm-B6O has a 1.81 eV indirect band gap).

Crystal structures of (a) α-B, (b) Cmcm-B, (c) α-B6O, (d) Cmcm-B6O, and their local structures, (e,f) B12 icosahedra. Green (large) and red (small) spheres denote B and O atoms, respectively.
Another interesting aspect is that if we remove the oxygen atoms from α-B6O and Cmcm-B6O, they turn into α-B16 and Cmcm-B, respectively (Fig. 2a,b). α-B and Cmcm-B are energetically nearly degenerate structures of boron at low pressure17, while Cmcm-B is a newly predicted structure18,19. As shown in Fig. 2b, displacements of layers I and II can transform this structure into α-B, and vice versa; Cmcm-B6O and α-B6O have a similar relationship (Fig. 2d). Furthermore, we found that the conversion of Cmcm-B6O and α-B6O will cause a deflection of B-O bond by 36°, as shown in their local structure (Fig. 2e,f).
However, it should be pointed out that it is not easy to change the stacking in covalent systems, though some examples are known – lonsdaleite (metastable “hexagonal diamond”) is formed in shocked cubic diamond. To obtain multiple polytypes, methods like physical vapor transport (PVT), also known as seeded sublimation growth, can be used: e.g., different polytypes of SiC were obtained using the PVT method20.
Comparison of Raman spectra
As mentioned above, β-B6O and α-B6O are energetically almost degenerate at zero pressure, structurally related and can coexist. To test the latter, we calculated their Raman spectra and compared with the experimental results12,13,21. In Fig. 3, the topmost curve is the Raman spectrum reported by Solozhenko et al.12; below it are the Raman frequencies reported by Werheit and Kuhlmann21 and marked by vertical bars (|). The two curves below are our Raman spectra of β-B6O and α-B6O. The experimental data correspond to normal isotopic abundance, and so do our calculations. Correspondingly, the atomic mass of boron in the calculations adopt the weighted average value, i.e. 10.811, which is based on the isotopic abundance of boron. As one could expect, the Raman spectra of β-B6O and α-B6O are similar and match perfectly with the experimental spectra. For example, the Raman modes at 374, 499, 541, 737, 785, 833, 1088, 1119, 1141 cm−1 are consistent with Solozhenko’s results12. However, there are four Raman modes which are unique for β-B6O. The first two are computed to be at 516 and 533 cm−1 (marked by letter A in Fig. 3), and Werheit and Kuhlmann indeed observed these two modes at 519 and 534 cm−1. The third mode is predicted to be at 630 cm−1 (marked with letter B), and Werheit and Kuhlmann have indeed observed a Raman-active phonon at 627 cm−1. We note that α-B6O does not have Raman-active modes between 570 and 700 cm−1, thus the one observed by Werheit and Kuhlmann at 627 cm−1 cannot come from α-B6O, but β-B6O. The fourth mode has the theoretical Raman frequency of 896 cm−1 (marked with letter C), while α-B6O has no Raman-active modes between 850 and 1000 cm-1. Thus, this mode is also unique to the β-B6O structure, and again seen in experiments: Solozhenko et al. observed Raman active phonon at 889 cm−1, and Werheit and Kuhlmann observed a Raman-active phonon at 902 (909) cm−1. Moreover, Wang et al. also observed similar Raman spectra in their B6O samples (see Fig. 1 in ref. 13). This analysis clearly shows that experimental samples contain β-B6O.

Comparison of PDOSs and Gibbs free energy
Comparing phonon densities of states (PDOSs) of α-B6O and β-B6O at ambient pressure (Fig. 4), we once again see a great degree of similarity. In order to further confirm the stability of β-B6O, we have calculated the Gibbs free energy of β-B6O and α-B6O as a function of temperature, shown in Supplementary Fig. S2. We conclude that β-B6O remains more stable than α-B6O also when temperature is taken into account – and is even slightly stabilized by thermal effects.

For clarity, the PDOS of α-B6O was shifted.
Having found that β-B6O should be more stable than α-B6O, and demonstrated that the two phases actually coexist in experimental samples, we ask a question: why is the synthetic compound mostly α-B6O, instead of the more stable β-B6O? While there can be no definitive answer at this point, we suggest that this may be because of the use of α-rhombohedral-B (R m)9 or β-rhombohedral-B (R
m)9 or β-rhombohedral-B (R m)22 as a starting material (together with B2O3) for synthesis of rhombohedral-B6O. Hubert et al.23 used amorphous-B as the starting material, but they got not only B6O but also B6O twinning particles and some amorphous phases. And they observed a stacking of ABAB… around the planar defect, which is the same as β-B6O (We speculate it probably is β-B6O). To obtain β-orthorhombic-B6O, one would need to crystallize it from the melt (preferably at high pressure, to increase its thermodynamic advantage over α-B6O), or use other phases of boron as precursors. Using Cmcm-B would be ideal, but this phase (just 11 meV/atom higher in energy than α-B19) remains hypothetical, though likely to be eventually synthesized. Alternatively, the PVT method20 could be used.
m)22 as a starting material (together with B2O3) for synthesis of rhombohedral-B6O. Hubert et al.23 used amorphous-B as the starting material, but they got not only B6O but also B6O twinning particles and some amorphous phases. And they observed a stacking of ABAB… around the planar defect, which is the same as β-B6O (We speculate it probably is β-B6O). To obtain β-orthorhombic-B6O, one would need to crystallize it from the melt (preferably at high pressure, to increase its thermodynamic advantage over α-B6O), or use other phases of boron as precursors. Using Cmcm-B would be ideal, but this phase (just 11 meV/atom higher in energy than α-B19) remains hypothetical, though likely to be eventually synthesized. Alternatively, the PVT method20 could be used.
In summary, to our big surprise, ab initio structure prediction calculations discovered a new ground state for the widely studied superhard compound B6O – our predicted β-B6O is more stable than experimentally known α-B6O. The two phases are polytypes and have nearly the same densities (β-B6O is slightly denser), energies (slightly lower for β-B6O), band gaps (slightly smaller and indirect, rather than direct, for β-B6O), hardnesses (β-B6O is slightly harder) and phonon densities of states, but have important differences in Raman spectra. By comparing calculated and experimental Raman spectra, we demonstrated that the experimental samples are actually a mixture of α-B6O and β-B6O. The discovery of β-B6O opens up new possibilities, in view of its greater stability, hardness and indirect band gap. Our findings also indicate possibilities of tuning the properties of B6O by obtaining phase-pure samples (probably not obtained to date), and the possibility of metastable oxygen-deficient compounds based on α-B6O or β-B6O - these can be obtained at high temperatures (where disordered oxygen vacancies will stabilize the structure) and low chemical potentials of oxygen.
Methods
We used the ab initio evolutionary algorithm USPEX24,25,26,27 to search for thermodynamically stable B-O compounds and their structures at ambient pressure. This methodology has shown its predictive power in many studies (e.g., ref. 17 and 27, 28, 29). All structures were relaxed; structure relaxations and total energy calculations were done using density functional theory (DFT) within the generalized gradient approximation (GGA)30 as implemented in the VASP code31, with the projector-augmented wave method32. We used plane-wave kinetic energy cutoff of 600 eV, and sampled the Brillouin zone with uniform Γ-centered meshes of is 2π*0.07 Å−1 resolution within structure search, and 2π*0.04 Å−1 for subsequent highly precise relaxations and properties calculations. In order to confirm the relative stability of α-B6O and β-B6O, we used local density approximation (LDA)33 and HSE06 hybrid functional34. Phonon spectra was computed by PHONOPY35 and VASP, and Raman spectra were calculated using the Fonari-Stauffer method36. Hardness was calculated with Chen model37 and Lyakhov-Oganov model38. Elastic moduli were computed using Voigt-Reus-Hill averaging39.
Additional Information
How to cite this article: Dong, H. et al. Prediction of a new ground state of superhard compound B6O at ambient conditions. Sci. Rep. 6, 31288; doi: 10.1038/srep31288 (2016).
References
Kaner, R. B., Gilman, J. J. & Tolbert, S. H. Designing Superhard Materials. Science 308, 1268–1269 (2005).
Kurakevych, O. O. Superhard phases of simple substances and binary compounds of the B-C-N-O system: from diamond to the latest results (a Review). J. Superhard Mater. 31, 139–157 (2009).
Wang, S. et al. Novel superhard B-C-O phases predicted from first principles. Phys. Chem. Chem. Phys. 18, 1859–1863 (2016).
Solozhenko, V. L. & Gregoryanz, E. Synthesis of superhard materials. Mater. Today 8, 44–51 (2005).
Bundy, F. P., Hall, H. T., Strong, H. M. & Wentorf, R. H. Man-Made Diamonds. Nature 176, 51–55 (1955).
Tian, Y. et al. Ultrahard nanotwinned cubic boron nitride. Nature 493, 385–388 (2013).
Hubert, H. et al. Icosahedral packing of B12 icosahedra in boron suboxide (B6O). Nature 391, 376–378 (1998).
McMillan, P. F. New materials from high-pressure experiments. Nat. Mater. 1, 19–25 (2002).
He, D. W. et al. Boron suboxide: As hard as cubic boron nitride. Appl. Phys. Lett. 81, 643–645 (2002).
Mukhanov, V. A., Kurakevych, O. O. & Solozhenko, V. L. Thermodynamic model of hardness: Particular case of boron-rich solids. J. Superhard Mater. 32, 167–176 (2010).
Veprek, S., Zhang, R. F. & Argon, A. S. Mechanical properties and hardness of boron and boron-rich solids. J. Superhard Mater. 33, 409–420 (2011).
Solozhenko, V. L., Kurakevych, O. O. & Bouvier, P. First and second-order Raman scattering of B6O. J. Raman Spectrosc. 40, 1078–1081 (2009).
Wang, Z. W., Zhao, Y. S., Lazor, P., Annersten, H. & Saxena, S. K. In situ pressure Raman spectroscopy and mechanical stability of superhard boron suboxide. Appl. Phys. Lett. 86, 041911 (2005).
Kurakevych, O. O. & Solozhenko, V. L. Experimental study and critical review of structural, thermodynamic and mechanical properties of superhard refractory boron suboxide B6O. J. Superhard Mater. 33, 421–428 (2011).
An Q., Reddy K. M., Dong H., Chen M.-W., Oganov A. R. & Goddard W. A. Nanotwinned Boron Suboxide (B6O): New Ground State of B6O. Nano Lett. 10.1021/acs.nanolett.6b01204 (http://dx.doi.org/10.1021/acs.nanolett.6b01204) (2016).
Decker, B. F. & Kasper, J. S. The crystal structure of a simple rhombohedral form of boron. Acta Crystallogr. B 12, 503–506 (1959).
Oganov, A. R. et al. Ionic high-pressure form of elemental boron. Nature 457, 863–867 (2009).
Zhu, Q., Oganov, A. R., Lyakhov, A. O. & Yu, X. Generalized evolutionary metadynamics for sampling the energy landscapes and its applications. Phys. Rev. B 92, 024106 (2015).
Pickard, C. J. & Needs, R. J. Ab initio random structure searching. J. Phys.: Condens. Matter. 23, 053201 (2011).
Hofmann, D. et al. SiC-bulk growth by physical-vapor transport and its global modelling. J. Cryst. Growth 174, 669–674 (1997).
Werheit, H. & Kuhlmann, U. FTIR and FT Raman Spectra of B6O. J. Solid State Chem. 133, 260–263 (1997).
Solozhenko, V. L., Kurakevych, O. O., Turkevich, V. Z. & Turkevich, D. V. Phase Diagram of the B−B2O3 System at 5 GPa: Experimental and Theoretical Studies. J. Phys. Chem. B 112, 6683–6687 (2008).
Hubert, H. et al. High-Pressure, High-Temperature Synthesis and Characterization of Boron Suboxide (B6O). Chem. Mater. 10, 1530–1537 (1998).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704 (2006).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How Evolutionary Crystal Structure Prediction Works—and Why. Acc. Chem. Res. 44, 227–237 (2011).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 184, 1172–1182 (2013).
Zhu, Q., Oganov, A. R., Glass, C. W. & Stokes, H. T. Constrained evolutionary algorithm for structure prediction of molecular crystals: methodology and applications. Acta Crystallogr. B 68, 215–226 (2012).
Zhang, W. W. et al. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 342, 1502–1505 (2013).
Dong, H., Oganov, A. R., Zhu, Q. & Qian, G.-R. The phase diagram and hardness of carbon nitrides. Sci. Rep. 5, 9870 (2015).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558(R) (1993).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079 (1981).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 124, 219906 (2006).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78, 134106 (2008).
Fonari, A. & Stauffer, S. vasp_raman.py (https://github.com/raman-sc/VASP/, 2013).
Chen, X.-Q., Niu, H., Li, D. & Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 19, 1275–1281 (2011).
Lyakhov, A. O. & Oganov, A. R. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and TiO2 . Phys. Rev. B 84, 092103 (2011).
Hill R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. London A 65, 349 (1952).
Zhou, X. F., Tian, Y. & Wang, H. T. Large shear strength enhancement of gamma-boron by normal compression. J. Superhard Mater. 33, 401–408 (2011).
Kurakevych, O. O. & Solozhenko, V. L. Thermoelastic equation of state of boron suboxide B6O up to 6 GPa and 2700 K: Simplified Anderson-Grüneisen model and thermodynamic consistency. J. Superhard Mater. 36, 270–278 (2014).
Acknowledgements
H. F. D. and Q. G. W. gratefully acknowledges financial support from the National Natural Science Foundation of China (Grant No. 11547174 and No. 11504004) and the PhD Start-up Fund of Natural Science Foundation of Guangdong Province, China (Grant No. 2016A030310352). A. R. O. thanks the Government of Russian Federation (grant No. 14.A12.31.0003), and Foreign Talents Introduction and Academic Exchange Program (No. B08040). Calculations were carried out in part at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575.
Author information
Authors and Affiliations
Contributions
H.F.D. performed all the calculations presented in this article with help from Q.Z. and X.F.Z. Research was designed by A.R.O., H.D. and A.R.O. wrote the first draft of the paper and A.R.O., Q.G.W., S.N.W., Z.H.W., J.Z., M.M.D.E., F.G.W. and Q.Z. contributed to revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dong, H., Oganov, A., Wang, Q. et al. Prediction of a new ground state of superhard compound B6O at ambient conditions. Sci Rep 6, 31288 (2016). https://doi.org/10.1038/srep31288
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31288
This article is cited by
-
First-principles study of novel icosahedral-based B12CN and B13CN structures
Science China Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
