
Abstract
The exceptionally conserved metazoan MAB21 proteins are implicated in cell fate decisions and share considerable sequence homology with the cyclic GMP-AMP synthase. cGAS is the major innate immune sensor for cytosolic DNA and produces the second messenger 2′-5′, 3′-5′ cyclic GMP-AMP. Little is known about the structure and biochemical function of other proteins of the cGAS-MAB21 subfamily, such as MAB21L1, MAB21L2 and MAB21L3. We have determined the crystal structure of human full-length MAB21L1. Our analysis reveals high structural conservation between MAB21L1 and cGAS but also uncovers important differences. Although monomeric in solution, MAB21L1 forms a highly symmetric double-pentameric oligomer in the crystal, raising the possibility that oligomerization could be a feature of MAB21L1. In the crystal, MAB21L1 is in an inactive conformation requiring a conformational change - similar to cGAS - to develop any nucleotidyltransferase activity. Co-crystallization with NTP identified a putative ligand binding site of MAB21 proteins that corresponds to the DNA binding site of cGAS. Finally, we offer a structure-based explanation for the effects of MAB21L2 mutations in patients with eye malformations. The underlying residues participate in fold-stabilizing interaction networks and mutations destabilize the protein. In summary, we provide a first structural framework for MAB21 proteins.
Similar content being viewed by others

Introduction
The male abnormal 21 (MAB21) like proteins form a sequence related subfamily within the large and diverse family of nucleotidyltransferase (NTase) fold proteins1. NTases generally catalyze the transfer of a nucleoside monophosphate (NMP) from a donor nucleoside triphosphate (NTP) to an acceptor hydroxyl. A prominent member of the MAB21 like protein family is cyclic GMP-AMP synthase (cGAS), a sensor of cytosolic DNA in the innate immune system2,3. cGAS detects cytosolic DNA arising from intracellular bacteria, damaged mitochondria, DNA viruses and retroviruses and triggers a type I interferon response4,5,6. Upon DNA recognition, cGAS produces the second messenger, 2′-5′, 3′-5′ cyclic GMP-AMP (2′,3′-cGAMP) by two sequential NTase reactions from substrate ATP and GTP7,8.
Besides cGAS (also known as MAB21 domain-containing protein 1 - MB21D1), MAB21 like proteins in humans include MB21D2, MAB21-like protein 1 (MAB21L1), MAB21L2 and MAB21L31. Although cGAS or cGAS-like activities are present in insects, vertebrates and possibly sea anemone, the MAB21 protein was first discovered in C. elegans as a factor important for cell fate determination9,10,11. Mutations in C. elegans MAB21 cause posterior-to-anterior homeotic transformation of sensory ray 6 in the male tail. The fundamental and crucial role of MAB21 during development is emphasized by additional pleiotropic phenotypes previously described and the fact that, the null allele of MAB21 is lethal prior to hatching. MAB21 is unusually conserved throughout species and vertebrate MAB21 proteins have almost identical primary sequences.
Despite comprehensive characterization of MAB21 phenotypes and an emerging role in cell fate decisions, the molecular function remains unknown. In vertebrates, MAB21L1 and MAB21L2 have a 94% identical amino acid sequence and exhibit similar expression patterns12,13,14,15,16. In agreement with the observations in C. elegans, mutations of MAB21L2 result in drastic phenotypes and defects that lead to death in mid-gestation17,18,19. Studies on MAB21L1 revealed relatively mild phenotypes in tissues where the expression of MAB21L1 differs from that of MAB21L2, namely in the lens and in reproductive organs20. MAB21L2 antagonizes the effects of BMP4 in Xenopus via interactions with the transcription factor SMAD1 and is therefore involved in regulation of the TGF-β pathway21,22. Moreover, MAB21L3, which only shares a sequence identity of 25%, was recently shown to be acting downstream of the Notch pathway in cell fate specifications of multiciliate cells and ionocytes23.
Recent current whole exome sequencing projects identified four different missense mutations in the single-exon gene MAB21L2 in eight individuals with major eye malformations24. In particular, an Arg51 mutation to cysteine (R51C) led to bilateral anophtalmia, intellectual disability, and rhizomelic skeletal dysplasia25. A mutation of the same residue to histidine (R51H) in a large multiplex family with colobomatous microphtalmia, as well as the mutation Glu49Lys was linked to coloboma. A homozygous mutation altering a different region of the protein Arg247Gln was also associated with retinal coloboma. The underlying molecular pathology of these mutations is unclear and both protein stabilizing and destabilizing effects have been proposed24,26.
Understanding the pivotal role of MAB21 proteins during development, their pathophysiology in human disease, and their evolutionary connection to the cGAS innate immune DNA sensor is hampered because the mechanistic function, biochemical activity and interaction partners, ligands or substrates of MAB21 proteins remain elusive. Here, we provide the first crystal structure of a MAB21 protein and the structure of MAB21L1 bound to CTP. The human MAB21L1 structure exhibits a high degree of structural conservation with cGAS but features a number of differences. Our study shows that MAB21L1 would require an activating conformational change in order to possess its potential NTase activity and unveils a potential binding site for an activating ligand of this poorly understood nucleotidyltransferase. Intriguingly, the NTase contains the residues to recognize a donor NTP and active site magnesium ions, but lacks some residues that are important for NTase activity in cGAS. Finally, we provide a structure-based explanation for the effects of mutations in MAB21L2 associated with eye malformations and show that these mutations lead to destabilization of the fold.
Results
Crystal Structure of Human MAB21L1
We crystallized native MAB21L1 in space group C2 with unit cell constants a = 167.7 Å, b = 177.6 Å, c = 115.6 Å, β = 126.5°. The native crystals contained 5 molecules in the asymmetric unit and diffracted to a limiting resolution of 3.05 Å. In order to determine experimental phases, we also crystallized selenomethionine substituted MAB21L1 (space group P21, unit cell constants a = 122.9 Å, b = 181.0 Å, c = 131.3 Å, β = 96.5° and 10 molecules per asymmetric unit). Single anomalous diffraction (SAD) data to 3.4 Å followed by phase improvement produced an interpretable electron density map that was used to build initial models for all ten copies in the asymmetric unit using non-crystallographic symmetry restrains. This initial model was used to phase the native dataset by molecular replacement. Further model building and refinement resulted in a final atomic model with good R-factors and stereochemistry (Table 1, Materials and Methods).
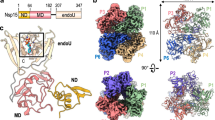
MAB21L1 is a two lobed, globular protein of a mixed α/β topology with approximate molecular dimensions of 64 Å × 35 Å × 48 Å. (Fig. 1a). The N-terminal lobe possesses the NTase subdomain with a five-stranded, antiparallel and highly twisted β-sheet (β1–β9) that is flanked on the surface by two long α-helices (α1/α4). The C-terminal lobe is formed by a conical four-helix bundle (α8–α13). This bi-lobal structure is stabilized by a long N-terminal ‘spine’ helix (α1), reaching across and spanning both lobes, as well as a central linker region composed of two helices (α5, α6). In general, the overall topology is highly similar to cGAS in its inactive form8,27,28,29. A notable difference with respect to cGAS is the lack of the zinc-thumb region which is important for DNA binding and dimerization27,30,31. Similarities and differences to cGAS and other members of the NTase fold protein family are described in more detail below.

(a) Front and side views of the human MAB21L1 crystal structure. The model is shown as cartoon representation with annotated domains and secondary structure. (b) Sequence alignment of MAB21, MAB21L1 and MAB21L2 amino acid sequences (abbreviations: Homo sapiens: hs, Drosophila melanogaster: dm, Caenorhabditis elegans: ce). Darker shadings indicate higher conservation (BLOSUM62 conservation score). The respective secondary structure elements for the human homolog are shown on top of the alignment with dots marking the conserved active site residues usually involved in metal coordination. Red stars denote residues that have been identified in disease related mutations.
Oligomerization and Interaction Interfaces
The crystal packing of MAB21L1 is unusual and consists of an intimate assembly of ten copies of MAB12L1 into a decamer, formed from two back-to-back stacked pentameric rings. The decamer has a D5 symmetry (one five-fold and five perpendicular two-fold symmetry axes) (Fig. 2a, Supplementary Fig. S1). Whereas the MAB21L1 protomers within the decamer are intimately packed, the decamers themselves pack less dense in the lattice and the interactions resemble crystal lattice interactions (Fig. 2a). The highly symmetric packing of MAB21L1 raises the possibility that oligomerization is part of MAB21L1’s function – similar to e.g. cGAS - and it might be worthwhile to describe the packing within the decamer in more detail. The N-terminal lobes of each protomer are partially surface accessible and located on the outside of the pentameric rings, whereas the C-terminal lobes are to a large extent buried and mediate the contacts within the pentamer. The pentamer interface is composed of hydrophobic and some polar interactions between the N-terminal helix α1 of one monomer and the C-terminal helices α11 and α12 of the neighboring molecule (Fig. 2a). Y10 stacks with W343 and interacts with a hydrophobic patch of other nearby residues (L305, I2, A6), while N20 hydrogen-bonds with R314/E336. A citrate molecule from the crystallization condition stabilizes this interface (Fig. 2a). The citrate is coordinated by residues R344 and K340 from one monomer together with K14 and K354 from the next monomer in a very basic surface patch. In total, each pentamer buries a surface area of approximately 2500 Å2.

(a) Front and side views of the MAB21L1 pentamer and decamer formation in the crystallographic asymmetric unit. One MAB21L1 monomer is colored in purple. The left inset shows a close-up of the hydrophobic interface between MAB21L1 molecules (purple, grey) in the pentameric ring. The right inset shows the interaction between two pentameric rings in the decameric assembly which is mainly composed of stacking interactions between highly conserved residues. (b) Measured SAXS data for MAB21L1 (red curve shows the measured scattering data) in comparison with theoretical scattering curves (calculated with CRYSOL) of monomeric, pentameric and decameric MAB21L1 assemblies (purple, green and blue curves, respectively) support that MAB21L1 is monomeric in solution. (c) Size-exclusion chromatography coupled static right-angle light scattering (SEC-RALS) of MAB21L1 shows a single peak with a constant molecular mass of Mw = 47 kDa. (d) Solvent accessible electrostatic surface representations of the MAB21L1 decamer interface, decamer (front view and side view), monomer (front view with NTase core and back view with ligand binding pocket) colored by charge (blue = 5 kT/e to red = −5 kT/e). The positively-charged ligand binding pocket is indicated with an arrow.
The interface between the two back-to-back stacked pentamers is in the range of 5000–6000 Å2, i.e. the decamer contains approx. 10000 Å buried surface. The pentamer stacking is mediated by contacts between opposing lobes and the linker region, both of which are highly conserved regions of MAB21L1 and L2 (Figs 1b and 2a). Salt bridges and hydrogen bonds are formed between the linker helices α6 and α5, β3 and the loop between β2–β3 as well as between the small helices α2 and α3 from the opposing monomer. Hydrophobic interactions are formed between loops connecting β2 with β3 and loop α2–α3 (Fig. 2a). The same double-pentameric packing is observed in all three of our crystals despite of changes in space groups and unit cell constants (Supplementary Fig. S1).
In order to address the oligomeric state of MAB21L1 in solution, we performed size exclusion chromatography coupled to static light scattering (SEC-RALS) as well as small angle X-ray scattering (SAXS) experiments. Both methods show that MAB21L1 is present as a monomer in solution, at least under the assayed conditions (Fig. 2b,c). MAB21L1 elutes as a single peak with a molecular mass determined by SEC-RALS of Mw = 47 kDa, in good agreement with the theoretical mass of a monomer (Mw = 41 kDa). Molecular weight determined from the SAXS data (Mw = 41 kDa, Rg = 2.4 nm) as well as the shape of the scattering curve are only compatible with a monomeric MAB21L1 molecule in solution. In order to rule out oligomerization in the crystal due to higher salt concentrations, we performed analytical size-exclusion experiments using buffers containing different salt concentrations up to 1 M. However, MAB21L1 still remained monomeric (Supplementary Fig. S1). Although MAB21L1 is monomeric in solution, it is possible that ligand interactions may promote oligomerization, such as in the case of cGAS30,31.
It has been proposed that MAB21L2 binds ssRNA24. Consistent with this, the solvent accessible electrostatic surface of the MAB21L1 monomer shows several positively-charged regions. One notable positive surface patch is a pocket at the surface adjacent to the ‘spine’ helix at the side of the double-pentamer (Fig. 2d). The corresponding region binds the DNA backbone in the case of cGAS. Another notable and quite extended patch is located around the five-fold symmetry axis of the pentamer (Fig. 2d). In order to test MAB21L1’s affinity for oligonucleotides, we performed electrophoretic mobility shift assays (EMSAs) with dsDNA/RNA as well as ssDNA/RNA (Fig. 3a,b). MAB21L1 has a preference for ssRNA in comparison to dsRNA and ss/dsDNA in good agreement with Rainger et al.24 (Fig. 3a), as can be estimated from the concentrations at half-maximal binding (ssRNA = 3.3 μM, ssDNA = 6.7 μM; dsRNA = 5 μM, dsDNA = 6 μM). The variations in the total binding of MAB21L1 to ssRNA and ssDNA are due to weak binding constants and therefore no binding curves and respective KD values were calculated. However, the affinities for these generic DNA or RNA sequences are considerably lower in our hands than those for DNA binding to cGAS (Fig. 3b). We could also not detect any enzymatic NTase activity of MAB21L1 either in the absence or presence of these nucleic acids.

(a) Quantified electrophoretic mobility shift assays of 1 μM 50mer dsRNA, dsDNA and 0.5 μM, ssRNA or ssDNA with MAB21L1 (protein concentrations of 0, 0.5, 1, 2, 5, 7, 10, 15 μM for binding to dsDNA/RNA and 0, 1, 2, 10, 14, 20, 30 μM for binding to ssDNA/RNA). Intensities of unbound oligonucleotides (relative shift) were plotted as triplicates against protein concentration (upper plot: ssRNA-red, ssDNA-black; lower plot: dsRNA-red, dsDNA-black). The half-maximal binding of ssRNA is at 3.3 μM protein concentration, whereas for ssDNA it is 6.7 μM, dsRNA = 5 μM and dsDNA = 6 μM, showing that MAB21L1 has a preference for ssRNA. (b) Corresponding gels of electro mobility shift assays of MAB21L1 in comparison to cGAS (same conditions as before). cGAS clearly has a higher affinity for generic nucleic acids than MAB21L1.
Structure of MAB21L1 Bound to CTP Reveals Possible Ligand Binding Site
In order to identify potential nucleotide substrates of MAB21L1, we performed thermal shift assays and isothermal titration calorimetry experiments using various nucleoside phosphates. We observed large increase in the melting temperature of MAB21L1 in the presence of CTP, CDP, ATP and ADP of ∆Tm = 8 °C (Supplementary Fig. S2). Other nucleotides tested, such as GTP and UTP, did not stabilize the protein and did not bind in isothermal titration calorimetry experiments (Supplementary Fig. S2). However, isothermal titration calorimetry experiments reveal strong binding of CTP, ATP and ADP with dissociation constants of KD(CTP) = 0.41 μM, KD(ATP) = 0.34 μM and KD(ADP) = 3.1 μM. These data indicate a preference for a triphosphate over a diphosphate (Fig. 4c).

(a) Front views of the MAB21L1-CTP complex crystal structure (left) and the crystal structure of the cGASMAB21-DNA-ATP-GTP complex (right, PDB code 4KB6). (b) Close-up view of the CTP binding site with the bound CTP. Key residues interacting with the CTP are depicted and annotated. The CTP is bound in a positively charged pocket via interactions with the NB-loop. (c) Titration of MAB21L1 with different nucleotides by ITC [KD(CTP) = 0.41 μM, KD(ATP) = 0.34 μM and KD(ADP) = 3.1 μM] suggest that MAB21L1 has a preference for nucleotide triphosphate coordination at this site.
In order to test for Mg2+ -binding we performed a thermal stability shift assay in the absence and presence of magnesium. Magnesium did not stabilize MAB21L1 and this might be explained by the fact that MAB21L1 is present in an inactive conformation (Supplementary Fig. S2).
To further address the nucleotide binding, we co-crystallized MAB21L1 with both ATP and CTP in the crystallization condition and determined the resulting crystal structure of MAB21L1 to 2.55 Å resolution. In this structure each protomer shows clear density for a single CTP moiety (Supplementary Fig. S3), and we did not see any density for ATP. Unexpectedly, the CTP moiety is not localized at the NTase active site, but rather bound at the positively-charged pocket on the ‘platform’ side of the molecule adjacent to the spine helix (Fig. 4a). The pocket coincides with a positively-charged surface patch on the ‘side’ of the double-pentamer (Fig. 2d). We denote this pocket ‘ligand binding pocket’. CTP is well coordinated in a partially hydrophobic, partially positively-charged pocket. In the loop connecting α1-β1, R62 stacks with the cytosine base in a planar conformation (Fig. 4b). This loop is better resolved in the MAB21L1:CTP structure, compared to apo MAB21L1, arguing for some sort of induced fit. Further contacts of MAB21L1 with CTP include hydrophobic interactions between I31, I27, V68 and A28 and carbons of the base and sugar. Backbone interaction between L66 and Y63 and the amino group at C4 of the cytosine explain the preference of MAB21L1 for CTP and ATP, compared to UTP and GTP where no binding affinity was detected. The negative charge of the phosphate groups of the nucleotide is well and specifically coordinated by residues from α1 and α8. The α-phosphate is coordinated by K255 and R23, the β-phosphate builds salt bridges with K255 and S252 and R23, while the γ-phosphate is held in place by K24 and K248. During model building of the MAB21L1 structure with no nucleotides added, we observed an additional electron density not associated with the polypeptide chain in the ligand binding pocket (Supplementary Fig. S3). However, we were not able to identify by structural or mass spectrometric methods the nature of this low molecular weight ligand, which may be a buffer component or have co-purified with the protein from E. coli.
In summary, CTP binding did not obviously affect the overall conformation of MAB21L1 and the superposition of apo MAB21L1 (crystallized without added nucleotides) and MAB21L1 in complex with CTP reveals no major conformational changes (RMSD 0.94 Å) (Supplementary Fig. S3). Thus, it is unlikely that CTP is the physiological ligand that can induce a potential activating conformational change (see below).
Comparison of MAB21L1 and cGAS Active Sites
To reveal differences and similarities between MAB21L1 and cGAS in the NTase active sites, we superposed the structures of N-terminally truncated porcine apo cGAS (RMSD 2.2 Å, PDB code 4JLX) and porcine cGASMAB21-DNA-ATP-GTP complex (RMSD 2.8 Å, PDB code 4KB6) with the structure of the MAB21L1:CTP complex (Fig. 5). MAB21L1 shows a high degree of structural conservation to cGAS and the conserved MAB21-domain fold strongly resembles other human nucleotidyltransferases, such as OAS1, and the bacterial DncV protein (Supplementary Fig. S4). In general, many active site residues located on the α-helical C-terminal lobe – responsible for binding of the ‘donor’ nucleotide in cGAS and other NTases, such as OAS1 - are highly conserved (Supplementary Fig. S4). For instance, Y272 in MAB21L1 corresponds to Y413 in sscGAS and to Y436 to hscGAS, where it stacks with the adenine base (Fig. 5). Further conserved residues essential for correctly positioning of ATP in the active site of cGAS are sscGAS E360/hscGAS E383 (MAB21L1 E238), sscGAS K416/hscGAS K439 (MAB21L1 K275) and sscGAS K391/hscGAS K414 (MAB21L1 K255). Thus, at least from sequence conservation, the structure of MAB21L1 suggests that MAB21-like proteins may bind an NTP at the NTase donor position.

(a) Close-up views of the NTase active sites of human MAB21L1 (purple) superposed with porcine cGAS (blue, PDB code 4JLX). (b) The corresponding part of the porcine cGAS-DNA-ATP-GTP (blue, PDB code 4KB6) and (c) human MAB21L1 structure in the same orientation. Residues responsible for magnesium ion coordination in the cGAS-DNA-ATP-GTP structure (PDB code 4KB6) Q200 and N202 are mutated in the figure to E200 and D202, respectively, and a second magnesium ion was added to the catalytic site of cGAS for reason of clarity. Selected residues of MAB21L1, which are conserved to cGAS residues implicated in catalysis, are annotated. (d) Close-up view of nucleotide binding sites in the cGAS-DNA-ATP-GTP and MAB21L1-CTP complexes. CTP binding to MAB21L1 prevents the NB-loop rearrangements required for activation and therefore access to the catalytic pocket.
The N-terminal lobe binds the Mg2+ -triphosphate group of the donor nucleotide and specifically recognizes and orients the ‘acceptor’ nucleotide GTP in cGAS. Consequently, the Watson-crick and Hoogsteen edges of the guanine base are within hydrogen bonding distance of the residues sscGAS T186, S355, R353 and S357 (hscGAS T211, S378, R376 and S380) respectively. Whereas the donor site is partially conserved between MAB21L1 and cGAS, these residues, which confer cGAS specificity for the guanine base, cannot be assigned, such as sscGAS T186 on the NB-loop, or are missing in MAB21L1 (A235, V231 and Q233, respectively).
A yet unanswered question is whether MAB21 proteins have any NTase activity, like e.g. cGAS or OAS. The catalytic triad on lobe I, which coordinates the two Mg2+ ions and polarizes the acceptor hydroxyl group in the active site of cGAS is only partially conserved in MAB21 proteins. While the presence of the Mg2+ ion coordinating MAB21L1 E73 and E75 (sscGAS: E200 and D202/hscGAS E225 and E227) suggest the existence of a functional Mg2+ and perhaps triphosphate coordinating site, MAB21L1 has a glutamine (Q169) at the position of sscGAS D296 (hscGAS D319). The carboxylate of D296 coordinates a second Mg2+ and helps to polarize the acceptor hydroxyl. Alanine mutations in either of the three residues or a double E200Q, D202N mutation in sscGAS lead to inactive proteins in vitro or in cellular assays8. It is unclear whether a more conserved asparagine at position hscGAS D319, similar to the Q169 of MAB21L1 still allows NTase activity.
However, in case MAB21L1 is an NTase, it would likely require a conformational change for activation. cGAS and OAS undergo conformational changes upon activation and especially part of the β1–β2 hairpin becomes a α-helical turn, which then constitutes the so called NB-loop32. In structures of inactive cGAS or OAS1 the loop connecting β1 and β2 is disordered in the crystal structures. Although the equivalent region of MAB21L1 is ordered in our crystal structure, it notably does not adopt the α-helical NB-loop conformation. CTP binding to MAB21L1 appears to stabilize the current, presumably inactive, conformation (Fig. 5d).
In summary, the structure of MAB21L1 exhibits similarities and differences with the structures of cGAS and OAS1, explaining why we currently fail to see NTase activity. Overall, the active site conformation observed in the crystal structure is inconsistent with catalytic activity. Residues that bind the donor NTP in the active site of NTases as well as coordinate the di-metal center are to a considerable extent conserved to cGAS, OAS proteins and other 3′-specific nucleotidyltransferases, such as poly(A) polymerase (PAP) and CCA-adding enzyme (CCA)32.
Human Missense Mutations of MAB21L2 Mapped on MAB21L1 Structure are Responsible for Protein Stability
A whole exome sequencing project revealed four different MAB21L2 missense mutations in patients with ocular coloboma. Out of 38 candidate genes for eye malformations, MAB21L2 was the only one altered by mutations. Previous studies attempted to explain the drastic phenotype of these mutations based on alignments of MAB21L2 with cGAS24. However, only the mutation R247Q, located in the C-terminal lobe on α8, is structurally conserved in cGAS and even OAS1 (Supplementary Fig. S4) and previous work disagreed whether the patient derived mutations increased or decreased protein stability24,26.
All of the described amino acids mutated in MAB21L2 are conserved in MAB21L1 (Fig. 1b), so our crystal structure offers a good model to reveal the molecular basis for this disease. We therefore mapped the mutations R51C/H, E49K and R247Q on our MAB21L1 structure, in order to shed light on the possible effects on the protein stability (Fig. 6a). In our MAB21L1 structure R247 forms a salt bridge with E288 on the α10-turn. This salt bridge is disrupted by the mutation of R247 to glutamine and consequently may lead to decreased stability of the protein. Residues E49 and R51 (Mutated to K and C/H, respectively) are located on the beginning of loop α1-β1. They are unique to MAB21 proteins and are highly conserved (Fig. 1b). Based on our MAB21L1 structure, E49 and R51 participate in a network of salt bridges with E115 (Fig. 6a). E49 and R51 stabilize the large loop between β4 and α4 by anchoring the loop via interactions to E115. This loop and E115 are a highly conserved element in MAB21L1 and L2, indicating that destabilization caused by the mutations of E49 to lysine and R51 to cysteine or histidine inactivate MAB21L2.

(a) Amino acids of MAB21L2 mutated in patients with eye malformations are depicted in blue (R247Q) and red (E49K, R51C/H) in the MAB21L1 structure. The insets show close-up views of the salt bridges formed by the mutated residues. R247 forms a salt bridge with E288, while E49 and R51 build a network of salt bridges with E115. Mutation of these stabilizing interactions will affect the correct MAB21L1 fold. (b) Thermal shift assay derivative melt curve plots of MAB21L1 (black), MAB21L1 R51C (red) and MAB21L1 R247Q (blue). The disease related mutations in MAB21L1 all show a decreased melting temperature supporting the idea that the mutations destabilize the protein.
To experimentally address the effects of the mutations on the protein stability we performed comparative fluorescence thermal shift assays. The thermal stability of MAB21L1 was tested in comparison to MAB21L1 R247Q and MAB21L1 R51C. MAB21L1 R51C and MAB21L1 R247Q showed significantly decreased melting temperatures, which indicates a decreased stability of the mutated protein (Fig. 6b). The mutant MAB21L1 R247Q showed an even more drastic decrease in melting point stability loss as R51C. The fact that R247Q is also conserved in cGAS (sscGAS R383 and hscGAS R406) suggests that this residue plays a more fundamental role in protein stability (Supplementary Fig. S4). In summary, we conclude that the MAB21L2 mutations associated with ocular coloboma lead to a protein with reduced stability because critical fold-stabilizing bonding networks are disrupted.
Discussion
MAB21 family proteins are implicated in cell fate decision processes in metazoans. Whereas MAB21 is required for pattern formation of the male tail in nematodes, MAB21 is sufficient and required for development of the dorso-ventral axis in vertebrates10,33,34. MAB21 proteins act downstream of the TGF-β signaling pathway by antagonizing the effect of BMP4 in Xenopus embryo ventralization and in C. elegans MAB21 is negatively regulated by cet-1 (vertebrate BMP4)22. Direct interactions of MAB21L2 with SMAD1, a nuclear transducer of the TGF-β pathway, suggest that MAB21L1 could be a transcriptional repressor21. Several studies in the model organisms Xenopus, zebrafish and mouse revealed that both genes have similar expression patterns in the developing eye, mid- and hindbrain, neural tube and branchial arches13,14,15,35. In particular, recent studies in zebrafish detected MAB21L2 transcripts in the ciliary marginal zone of the retina. Similarly, expression of MAB21L3 in Xenopus was specific for multiciliate cells and ionocytes associating MAB21 proteins with cell fate specification of cilia23,26.
The biochemical activity and physiological role of MAB21 proteins remain to be established. As with many other NTases there are three ligand or substrate molecules to be considered, which make identification of the biochemical activity and biological function of these types of molecules challenging. NTases transfer a NMP from a donor NTP onto a hydroxyl group of an acceptor molecule. A third molecule required for development of catalytic activity is a potential activating ligand. In the case of cGAS, these molecules are ATP, GTP and double-stranded DNA. In the case of MAB21 proteins, neither the substrates of a NTase reaction nor a potential activating ligand have been described, if such molecules exist at all. Our structural studies show that MAB21L1 resembles cGAS in the unliganded inhibited state and show that any NTase activity would require a conformational change similar to that DNA induces in cGAS8,27. It is of course possible that MAB21 proteins exert their functions via a predominantly structural role, e.g. by binding other proteins or nucleic acids such as mRNAs or micro-RNAs, and do not possess any catalytic activity at all. For instance, it has previously been proposed that MAB21L2 interacts with mRNA24. We also see some preferential but moderate binding to generic ssRNA with MAB21L1 and the positive electrostatic surface potential would be consistent with nucleic acid binding. Generic nucleic acids bind, however, with considerably less affinity to MAB21L1 than to cGAS27. This reduced affinity suggests that MAB21 binds perhaps a specific sequence or structure, if any. Of note in this context, potential functional link to micro-RNAs in the case of MAB21L3 has been proposed, although there are no experimental data for a functional or physical interaction at this point23. Apart from nucleic acids, it is also possible that MAB21 proteins interact with other protein partners such as the SMAD proteins21.
MAB21 proteins could also confer their function through a catalytic NTase activity. As stated above, MAB21L1 has an inactive conformation in the crystal structure, but a cGAS-like conformational change could in principle form a proper active site. If such a hypothetical conformational change takes place, our structure shows that residues typically associated with donor NTP binding are conserved, in particular at the C-terminal lobe 2. These include Y272, which stacks with the base, and E238, which binds to the sugar OH groups. Finally, donor NTP binding also requires the catalytic triad carboxylates on lobe I, which coordinate together with the triphosphate moiety the two active site magnesium ions. However, residues coordinating the acceptor nucleotide GTP in cGAS are not found in MAB21L1 and residues located on the NB-loop cannot be assigned in our conformation, due to the lack of a MAB21L1 structure in an active conformation. Furthermore, in MAB21L1 and L2, two out of the three catalytic triad residues are conserved (E73, E75), whereas the third triad residue (Q169) is altered. In the case of cGAS the aspartate equivalent to Q169 (hscGAS D319) coordinates a second active site Mg2+ and could help to polarize the attacking acceptor OH. A glutamine at this position could still coordinate this magnesium ion, but may result in a reduced but perhaps not entirely abolished activity. As such, we do not want to rule out the possibility that MAB21 proteins still possess NTase activity.
An unexpected observation was that CTP was not bound at the NTase active site, even though it binds to MAB21L1 with 0.41 μM affinity and leads to a substantial increase in thermal stability. In previous studies by ourselves and other groups with related enzymes (cGAS, OAS, MiD51), co-crystallization with NTPs or soaking of crystals with NTPs resulted in binding of the NTPs at the NTase active site, although the NTPs were often bound in a catalytically inactive conformation and not sufficient to switch the enzyme from an inactive to an active conformation27,32,36,37. In our structure, CTP is bound to a site that recognizes activating ligands in the case of related NTases (cGAS, OAS1), and it is tempting to speculate that this site is a ligand-binding site of MAB21 proteins as well. Obviously CTP did not switch the protein into an active conformation, so CTP is unlikely a physiological activating ligand. It is possible however, that CTP mimics part of a physiological ligand. It should be noted that the sugar and base moieties of CTP are bound by the NB-loop in its inactive conformation and thus would argue against an activating structural switch in MAB21, while the triphosphate binds into a highly positively-charged pocket that may not be affected by a conformational switch. The fact that the structure of MAB21L1 without any nucleotides added shows density for an unidentified molecule in the ligand binding pocket may support the suggestion that this positively-charged site acts as a binding site for an acidic ligand. The explicit preference of MAB21L1 for a nucleoside triphosphate (over a diphosphate) at this site suggests that MAB21L1 could bind, for instance, the 5′ triphosphate of an RNA such as mRNA or precursor miRNA. This positively-charged pocket in MAB21L1 could act as an activation site but also as an inactivation site, such as the equivalent site on the bacterial nucleotidyltransferase DncV that binds 5MTHFGLU238. In the folate-bound conformation of DncV residues Y117 and Q116 sterically hinder binding of nucleotides in the active site.
In our structure, MAB21L1 packs into a decamer with D5 symmetry. This highly symmetric packing is quite unusual for generic crystal lattice interactions. Although we cannot rule out that this packing is purely a result of the high concentrations in the crystal, it may have a biological function, even though it was not detected in vitro in solution. cGAS, for instance, was shown to cooperatively bind dsDNA through two distinct interaction sites, which induced oligomerization required for activation30,31. The interaction interfaces between protomers in the double pentamer are rather small and distinct from those of the oligomers of related proteins, such as cGAS and MiD5130,36. The interface area between two pentameric rings, however, is substantial and mediated by a highly conserved region in MAB21L1 and L2 comprising amino acids G96-Q138 (Fig. 1b). The same sequence was shown to be involved in protein-protein interactions in related MAB21 domain proteins. Drp1 recruitment to MiD51 was abolished when a single amino acid was mutated in this region36. In addition, residues differing between the 94% identical MAB21L1 and MAB21L2 proteins do not affect the predicted oligomer interface. The extreme surface conservation of MAB21 proteins throughout species further argues for potential oligomerization and/ or interaction partner binding features. Therefore, the oligomerization of MAB21 could be of biological importance and should be considered in future studies.
Finally, our structure allows a molecular interpretation of the MAB21L2 mutations in ocular coloboma since the underlying residues and their interaction partners are conserved between L1 and L2. Interestingly, we find that the mutated residues are not located on the surface but rather stabilize the structure of MAB21L1/L2. Consistently, the mutations reduce the thermal stability of MAB21L1 in vitro, suggesting that the disease is caused by destabilized MAB21L2. Of note, the mutations E49K and R51C/H help fold a highly conserved area in MAB21L1/L2 that in our structure mediates the pentamer-pentamer interactions. In MiD51 the same loop is responsible for protein-protein interactions36. Therefore, the phenotype caused by mutations E49K and R51C/H could be due to disruption of possible protein-protein interactions with a yet unknown partner, that might be required for MAB21L1 to fulfill its function. Furthermore, these mutations were previously shown to disrupt MAB21L2 ability to bind ssRNA, consistent with the locations of the affected residues close to the RNA binding cleft in OAS124.
In line with the mutations observed for MAB21L2, a recent study reports severe effects of a the Cys246Leufs*18 frameshift mutation in MAB21L139. This mutation leads to a potentially disrupted structural integrity of the whole alpha helical arrangement in the C-terminal lobe, also affecting the mapped intramolecular interaction R247:E288 in MAB21L2 mutation R247Q. The fact that the MAB21L1 mutation causes an additional phenotype compared to MAB21L2 argues for an important role of both proteins in the organism.
In summary, we provide a first structural framework for the highly conserved MAB21 family of nucleotidyltransferase fold proteins.
Methods
Protein Expression and Purification
The full length human MAB21L1 gene, purchased from GENEART with codon optimization for expression in E. coli, was cloned into a modified pET21 vector with an N-terminal His-MBP-tag. Site-directed mutagenesis was performed using the Quikchange (Stratagene) protocol with the respective primers. Expression was performed in E. coli BL21 Rosetta (DE3) and B834 (DE3) strains for native and selenomethionine labeled proteins, respectively. Cells were induced with 0.1 mM IPTG after reaching an OD600 0.5–0.6 and proteins were expressed at 18 °C for 18 h. Harvested cells were resuspended in lysis buffer (50 mM Tris, 300 mM NaCl, 25 mM imidazole, 2 mM β-mercaptoethanol, 5% v/v glycerol) and disrupted by sonication. MAB21L1 was purified by nickel-affinity chromatography and the His-MBP-tag was subsequently removed by TEV-protease cleavage (ratio 1:30) at 4 °C over night in buffer A (30 mM Tris-HCl, 100 mM NaCl, 2 mM DTT, pH 7.0). The protein was then further purified by cation exchange chromatography step using a HiTrap SP HP column (GE Healthcare) at pH 7.0. The protein was eluted with a linear gradient up to 50% buffer B (30 mM Tris, 1 M NaCl, 2 mM DTT, pH 7.0). Fractions containing MAB21L1 were pooled and loaded on a Superdex 75 size exclusion chromatography column (GE Healthcare) using 20 mM Tris-HCl pH 7.5, 150 mM NaCl and 2 mM DTT as running buffer. MAB21L1 fractions were concentrated (2–3 mg/ml) before being flash-frozen in liquid nitrogen for storage at −80 °C. MAB21L1 mutants were expressed and purified accordingly. The final crystallization buffer for selenomethionine derivatized protein contained 2 mM TCEP (Tris(2-carboxyethyl) phosphine).
Crystallization of MAB21L1
Co-crystallization of MAB21L1 with a mixture of CTP/ATP (5 mM final concentration) was performed by hanging drop vapor diffusion in 0.1 M MES pH 5.5 and 1 M tri-sodium citrate. 2 μL protein/nucleotide mix were added to 1 μL reservoir solution from a total reservoir volume of 250 μL in the well. Native crystals grew for one week at 20 °C before being mounted on a Free Mounting System (FMS, Proteros Biostructures GmbH). Liquid surrounding the crystal was removed in the humidified gas stream with a glass capillary. TMAO (trimethylamine oxide) was supplied as a cryo-protectant to the protein crystal with the PicoDropper during a humidity gradient. The crystal was then flash-frozen by the quick rotational replacement of the humidity nozzle with the cryo nozzle and finally stored in liquid nitrogen (for more details see also40). Selenomethionine derivatized protein crystals grew in 0.2 M Tri-potassium citrate pH 7.8 and 16% PEG 3350 and were cryo protected by soaking in reservoir solution containing 30% (v/v) glycerol prior to flash freezing.
Data Collection and Structure Determination
X-ray diffraction data were collected at beamline SLS X06SA (Swiss Light Source, Villigen, Switzerland) and PETRAIII beamline P13 (EMBL/DESY, Hamburg, Germany). Diffraction data were processed using XDS and XSCALE41. We determined the structure of MAB21L1 using a high-redundant single anomalous diffraction (SAD) dataset measured at the Se-peak wavelength. Three finely-sliced datasets of translations on one needle-shaped crystal were merged (10800 × 0.1° frames) to provide enough phase information. The self-rotation function calculated with MOLREP42 of the CCP4 package43 suggested the presence of an additional five-fold symmetry axis and in combination with the Matthews’ coefficient we assumed either 10 or 15 copies. Phasing and density modification using the PHENIX suite44 located 70 Se sites for the 10 copies of MAB21L1 in the ASU leading to an initial low resolution map at 3.4 Å. This map allowed partial automatic model building using BUCCANEER45 followed by manual model building in COOT46 and alternate refinement steps in PHENIX. The initial starting model could be used to phase the two better diffracting native crystals using PHASER47, leading to readily interpretable electron density. Extensive manual model building in COOT and further iterative refinements with PHENIX resulted in a final model with reasonable statistics. Data collections and refinement statistics are listed in Table 1. Figures were created with PyMOL48.
Thermal Shift Assay
The thermal stability of MAB21L1 in presence of different ligands was analyzed by fluorescence thermal shift assays. 20 μM MAB21L1 was incubated with 5 mM of the respective ligand in 25 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2. After addition of SYPRO Orange (final concentration of 5×, Invitrogen) the fluorescence signal was detected during a temperature gradient between 5 °C to 100 °C at a rate of 0.5 K/30 s with one scan per 0.5 K in a real-time thermal cycler using the FRET mode (CFX96 touch/Biorad).
Isothermal Titration Calorimetry
Isothermal Titration Calorimetry was carried out at 23 °C using a VP-ITC Microcal calorimeter (Microcal, GE Healthcare). Proteins were dialyzed overnight against 20 mM HEPES pH 8.0, 150 mM NaCl, 1 mM TCEP and nucleotides were resuspended in dialysis buffer. 1.44 mL of protein in the cell with concentrations of 5–10 μM were titrated with nucleotides with 10× higher concentrations. 8 μL nucleotides was injected 25–30 times with 3.5 min intervals between injections. A background curve for each titration consisting of the titration of the nucleotide into buffer without protein was subtracted to account for heat dilution. The ITC data were analyzed using the Origin version 7 software package of the ITC instrument (Microcal).
Analytical Size-exclusion chromatography
Analytical size-exclusion chromatography with MAB21L1 was performed with a Superdex 200 increase 10/300 (GE Healthcare) in 20 mM HEPES pH 8.0, 2 mM DTT with varying concentrations of NaCl (150 mM, 500 mM, 750 mM and 1 M).
Size-exclusion chromatography coupled static light scattering
Size-exclusion coupled static right-angle light scattering (SEC-RALS) was performed using an AEKTAmicro system (GE Healthcare Life Sciences) equipped with a refractive index detector (VE 3580) and a TD270- RALS –device (Viscotek/Malvern Instruments) with a Superdex 200 10/300 size-exclusion column (GE Healthcare). BSA (66 kDa) was used as standard protein for system calibration. Analysis of data was performed with Software OmniSEC (Viscotek/Malvern Instruments).
Small-Angle X-ray Scattering
Small-angle X-ray scattering (SAXS) experiments were performed at PETRAIII beamline P12 (EMBL/DESY, Hamburg, Germany). Protein samples were purified by size-exclusion chromatography (SEC) and centrifuged prior to the measurements. All samples were monodisperse as judged from the SEC- chromatograms and dynamic light-scattering size distributions. The scattering of the SEC running buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 2 mM DTT) was used for buffer correction of the MAB21L1 samples which were measured at different concentrations (1.3–6.5 mg/mL). The samples did not show signs of radiation damage and the scattering data were processed in the ATSAS suite49 as e.g. described in50. Guinier-plot (ln I(s) vs s2) analysis did not show signs of aggregation of the sample and provided a radius of gyration for MAB21L1 of Rg = 2.4 nm. By extrapolation to zero angle scattering intensity (I0) in the Guinier region s*Rg < 1.3 the molecular weight of MAB21L1 in solution was determined Mw(I0) = 49.8 kDa using BSA (66 kDa) as a I0-reference. This is in good agreement with the molecular weight determined from the Porod volume Mw(porod) = 41 kDa. Theoretical scattering curves of crystallographic coordinates were calculated using CRYSOL51.
Electrophoretic Mobility Shift Assays
1 μM of 50mer ds/ssDNA or ds/ssRNA (5′-GGATACGTAACAACGCTTATGCATCGCCCCGCTACATCCCTGAGCTGAC-3′), RNA same sequence as DNA) was incubated with the indicated increasing concentration of purified protein for 30 min on ice. As reaction buffer 20 mM Tris pH 8.0 and 200 mM NaCl was used. Samples were separated by a 1% agarose gel supplemented with Gel-Red (Biotium) for staining as suggested by the manufacturer instructions.
Additional Information
Accession codes: MAB21L1 coordinates and structure factors have been deposited in the Protein Data Bank with the accession codes 5EOG (apo-form) and 5EOM (CTP-bound), respectively.
How to cite this article: de Oliveira Mann, C. C. et al. Structural and biochemical characterization of the cell fate determining nucleotidyltransferase fold protein MAB21L1. Sci. Rep. 6, 27498; doi: 10.1038/srep27498 (2016).
References
Kuchta, K., Knizewski, L., Wyrwicz, L. S., Rychlewski, L. & Ginalski, K. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic acids research 37, 7701–7714 (2009).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Gao, D. et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 (2013).
Li, X.-D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Ablasser, A. et al. cGAS produces a 2 [prime]-5 [prime]-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Gao, P. et al. Cyclic [G (2′, 5′) pA (3′, 5′) p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Kranzusch, P. J. et al. Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2′, 3′ cGAMP Signaling. Molecular cell 59, 891–903 (2015).
Baird, S. E., Fitch, D., Kassem, I. & Emmons, S. W. Pattern formation in the nematode epidermis: determination of the arrangement of peripheral sense organs in the C. elegans male tail. Development 113, 515–526 (1991).
Chow, K. L., Hall, D. H. & Emmons, S. W. The mab-21 gene of Caenorhabditis elegans encodes a novel protein required for choice of alternate cell fates. Development 121, 3615–3626 (1995).
Mariani, M. et al. Two murine and human homologs of mab-21, a cell fate determination gene involved in Caenorhabditis elegans neural development. Human molecular genetics 8, 2397–2406 (1999).
Wong, Y.-M. & Chow, K. L. Expression of zebrafish mab21 genes marks the differentiating eye, midbrain and neural tube. Mechanisms of development 113, 149–152 (2002).
Mariani, M. et al. Mab21, the mouse homolog of a C. elegans cell-fate specification gene, participates in cerebellar, midbrain and eye development. Mechanisms of development 79, 131–135 (1998).
Kudoh, T. & Dawid, I. B. Zebrafish mab21l2 is specifically expressed in the presumptive eye and tectum from early somitogenesis onwards. Mechanisms of development 109, 95–98 (2001).
Cederlund, M. L. et al. mab21l2 transgenics reveal novel expression patterns of mab21l1 and mab21l2, and conserved promoter regulation without sequence conservation. Developmental Dynamics 240, 745–754 (2011).
Lau, G. T. et al. Embryonic XMab21l2 expression is required for gastrulation and subsequent neural development. Biochemical and biophysical research communications 280, 1378–1384 (2001).
Yamada, R., Mizutani-Koseki, Y., Koseki, H. & Takahashi, N. Requirement for Mab21l2 during development of murine retina and ventral body wall. Developmental biology 274, 295–307 (2004).
Kennedy, B. N. et al. Zebrafish rx3 and mab21l2 are required during eye morphogenesis. Developmental biology 270, 336–349 (2004).
Yamada, R. et al. Cell-autonomous involvement of Mab21l1 is essential for lens placode development. Development 130, 1759–1770 (2003).
Baldessari, D., Badaloni, A., Longhi, R., Zappavigna, V. & Consalez, G. G. MAB21L2, a vertebrate member of the Male-abnormal 21 family, modulates BMP signaling and interacts with SMAD1. BMC cell biology 5, 48 (2004).
Morita, K., Chow, K. L. & Ueno, N. Regulation of body length and male tail ray pattern formation of Caenorhabditis elegans by a member of TGF-beta family. Development 126, 1337–1347 (1999).
Takahashi, C., Kusakabe, M., Suzuki, T., Miyatake, K. & Nishida, E. mab21-l3 regulates cell fate specification of multiciliate cells and ionocytes. Nature communications 6 (2015).
Rainger, J. et al. Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. The American Journal of Human Genetics 94, 915–923 (2014).
Horn, D. et al. A Novel Oculo-Skeletal syndrome with intellectual disability caused by a particular MAB21L2 mutation. European journal of medical genetics 58, 387–391 (2015).
Deml, B. et al. Mutations in MAB21L2 Result in Ocular Coloboma, Microcornea and Cataracts. PLoS genetics 11, e1005002–e1005002 (2015).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Kranzusch, P. J., Lee, A. S.-Y., Berger, J. M. & Doudna, J. A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell reports 3, 1362–1368 (2013).
Kato, K. et al. Structural and functional analyses of DNA-sensing and immune activation by human cGAS. PloS one 8, e76983 (2013).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell reports 6, 421–430 (2014).
Lohöfener, J. et al. The Activation Mechanism of 2′-5′-Oligoadenylate Synthetase Gives New Insights Into OAS/cGAS Triggers of Innate Immunity. Structure 23, 851–862 (2015).
Sridharan, J., Haremaki, T., Jin, Y., Teegala, S. & Weinstein, D. C. Xmab21l3 mediates dorsoventral patterning in Xenopus laevis. Mechanisms of development 129, 136–146 (2012).
Wong, R. L. Y. & Chow, K. L. Depletion of Mab21l1 and Mab21l2 messages in mouse embryo arrests axial turning, and impairs notochord and neural tube differentiation. Teratology 65, 70–77 (2002).
Cederlund, M. L. et al. mab21l2 transgenics reveal novel expression patterns of mab21l1 and mab21l2, and conserved promoter regulation without sequence conservation. Developmental Dynamics 240, 745–754 (2011).
Losón, O. C. et al. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 22, 367–377 (2014).
Richter, V. et al. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. The Journal of cell biology 204, 477–486 (2014).
Zhu, D. et al. Structural biochemistry of a Vibrio cholerae dinucleotide cyclase reveals cyclase activity regulation by folates. Molecular cell 55, 931–937 (2014).
Bruel, A. L. et al. Autosomal recessive truncating MAB21L1 mutation associated with a syndromic scrotal agenesis. Clinical Genetics (2016).
Kiefersauer, R., Grandl, B., Krapp, S. & Huber, R. IR laser-induced protein crystal transformation. Acta Crystallographica Section D: Biological Crystallography 70, 1224–1232 (2014).
Kabsch, W. Xds. Acta Crystallographica Section D: Biological Crystallography 66, 125–132 (2010).
Vagin, A. & Teplyakov, A. MOLREP: an automated program for molecular replacement. Journal of applied crystallography 30, 1022–1025 (1997).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D:Biological Crystallography 67, 235–242 (2011).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallographica Section D:Biological Crystallography 68, 352–367 (2012).
Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallographica Section D:Biological Crystallography 62, 1002–1011 (2006).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D:Biological Crystallography 60, 2126–2132 (2004).
McCoy, A. J. et al. Phaser crystallographic software. Journal of applied crystallography 40, 658–674 (2007).
DeLano, W. L. The PyMOL molecular graphics system. (2002).
Petoukhov, M. V. et al. New developments in the ATSAS program package for small-angle scattering data analysis. Journal of applied crystallography 45, 342–350 (2012).
Putnam, C. D., Hammel, M., Hura, G. L. & Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly reviews of biophysics 40, 191–285 (2007).
Svergun, D., Barberato, C. & Koch, M. CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. Journal of applied crystallography 28, 768–773 (1995).
Acknowledgements
We thank the staffs of PETRA3 P12/P13/P14 at EMBL Hamburg/DESY and of SLS X06SA Villingen, Switzerland for excellent onsite support and beamtime allocation, Katja Lammens and Dirk Kostrewa for support with X-ray data, Claire Basquin for help with the ITC experiments, Robert Byrne for comments on the manuscript and the Hopfner group for discussions. This work was funded by grant GRK1721 (Deutsche Forschungsgemeinschaft) to G.W. and K.-P.H. and by DFG HO2489/8 to K.-P.H. C.C.O.M. is supported by GRK1721.
Author information
Authors and Affiliations
Contributions
C.C.d.O.M. conducted all experiments, and evaluated data. R.K. optimized cryo conditions on the Free Mounting Device. G.W. participated in structural analysis and SAXS evaluation. G.W. and K.-P.H. designed research. C.C.d.O.M., G.W. and K.-P.H. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.K. declares that he is an employee of Proteros Biostructures GmbH. R.K. has invented and further developed the FMS and the PicoDropper.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
de Oliveira Mann, C., Kiefersauer, R., Witte, G. et al. Structural and biochemical characterization of the cell fate determining nucleotidyltransferase fold protein MAB21L1. Sci Rep 6, 27498 (2016). https://doi.org/10.1038/srep27498
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27498
This article is cited by
-
Bacterial cGAS senses a viral RNA to initiate immunity
Nature (2023)
-
DNA-methylome-assisted classification of patients with poor prognostic subventricular zone associated IDH-wildtype glioblastoma
Acta Neuropathologica (2022)
-
cGAS-like receptors sense RNA and control 3′2′-cGAMP signalling in Drosophila
Nature (2021)
-
Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia
Human Genetics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
