
Abstract
Rare copy number variations (CNVs) are a known genetic etiology in neurodevelopmental disorders (NDD). Comprehensive CNV analysis was performed in 287 Chinese children with mental retardation and/or development delay (MR/DD) and their unaffected parents. When compared with 5,866 ancestry-matched controls, 11~12% more MR/DD children carried rare and large CNVs. The increased CNV burden in MR/DD was predominantly due to de novo CNVs, the majority of which (62%) arose in the paternal germline. We observed a 2~3 fold increase of large CNV burden in the mothers of affected children. By implementing an evidence-based review approach, pathogenic structural variants were identified in 14.3% patients and 2.4% parents, respectively. Pathogenic CNVs in parents were all carried by mothers. The maternal transmission bias of deleterious CNVs was further replicated in a published dataset. Our study confirms the pathogenic role of rare CNVs in MR/DD, and provides additional evidence to evaluate the dosage sensitivity of some candidate genes. It also supports a population model of MR/DD that spontaneous mutations in males’ germline are major contributor to the de novo mutational burden in offspring, with higher penetrance in male than female; unaffected carriers of causative mutations, mostly females, then contribute to the inherited mutational burden.
Similar content being viewed by others
Introduction
Rare copy number variations (CNVs) are well established as a risk factor in a range of neurodevelopmental disorders (NDD)1,2,3,4. Chromosome microarray (CMA) has been recommended as the first-tier diagnostic test for newborns with developmental delay and congenital anomalies5. Technically, it has evolved from detection of submicroscopic chromosome changes6, to recent high-resolution analysis of single gene disruptions7,8,9. The clinical utilities of CMA results on patient management have also been demonstrated10,11.
Despite its widespread use in molecular diagnosis, challenges remain for implementing the CMA workflow. One is technological: how to maximize the sensitivity for variants discovery? Compared with array comparative genomic hybridization (CGH), single nucleotide polymorphism (SNP) arrays may not be optimized for signal-to-noise ratio reflecting copy number changes12,13. But the disadvantage could be offset by increased probe densities and exploiting the allelic specific information14. Nevertheless, accurate discovery of small CNVs has been demonstrated difficult for most platforms15. The improvements in both analytical methods and technology will be needed.
The second issue is interpretational: how to distinguish pathogenic from benign CNVs16,17? The inheritance status may not be used as a single criterion for evaluation: de novo occurrence may not necessarily imply pathogenicity18; and the role of inherited CNVs should not be underestimated19, but their interpretations are complicated by the incomplete penetrance and/or variable expressivity. For clinical interpretation, an evidence-based review approach has been recommended20. For this task, it is crucial to incorporate the findings from latest research.
Most of the published CNV studies in NDD were conducted in European populations. Studies in additional population can be valuable. They allow an assessment of potential population difference in global CNV burden and locus-specific associations, which are still not fully clear. As many rare variants including CNVs are population specific, findings from non-European populations may also help prioritize candidate genes by discovering new mutations21.
In the present study, we performed a comprehensive analysis of SNP array data of 287 Chinese children of mental retardation and/or developmental delays (MR/DD) and their unaffected parents. To shed light on the pathogenic roles of de novo and inherited CNVs, the CNV burdens in both children and their unaffected parents were compared against ~6,000 ancestry-matched controls. Pathogenic CNVs were identified by carefully reviewing evidence from databases and literature. We compared the findings with previous reports in European populations; highlighted gender differences; and documented several cases of pathogenic and benign CNVs that may help refine the genome-wide dosage sensitivity map.
Results
We analyzed CNVs in 287 children (189 males, 98 females) referred to our department for DNA testing with a general diagnosis of MR/DD. Their phenotypes encompassed a wide range of NDD including congenital malformation, craniofacial dysmorphology, cardiac/neurological defects, epilepsy/seizure, autism, hyperactivity, etc. All of them showed normal karyotype and were negative for the test of subtelomeric aberrations. Whenever possible, the DNA samples from their parents were also genotyped; and a total of 510 of them passed quality control (QC) for CNV analysis (Supplementary Table S1). CNVs identified in this clinical cohort were compared to 5,866 adult controls not ascertained for NDD.
We mainly focused on ultra-rare CNVs with population frequency <0.1%. A total of 255 CNVs (124 gains and 131 losses) were identified in 173 (60.2%) affected children, and 368 CNVs (177 gains and 191 losses) in 300 (58.4%) parents. 203 CNVs (88 gains and 115 losses) identified in parents were not found in patients. The proportions of male and female carriers of ultra-rare CNVs were similar to the gender composition (P > 0.2, chi-square test) (Supplementary Table S2). The sizes of CNV in patients ranged from <10 kb to >30 Mb (median 137 kb, inter-quartile range (IRQ): 62–380 kb), significantly larger than CNVs found in parents only (median 84 kb, IRQ: 40–167 kb; P = 2.2E-05, Mann-Whitney). The full list of annotated ultra-rare CNVs was given in Supplementary Table S8 and S9. The inheritance for more than 80% ultra-rare CNVs in patients could be determined (Fig. 1A, Supplementary Table S4). We identified and validated 41 de novo CNVs in 39 patients, including 2 patients each carrying 2 de novo CNVs.

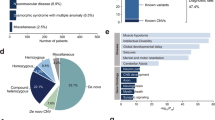
(A) A total of 255 ultra-rare CNVs were identified in 287 patients with MR/DD; the proportions of different inheritance status are shown in a pie chart. “Not from one parent”: in parent-offspring pairs, the child’s CNV was not found in the available parent; “unknown”: undetermined inheritance as neither parent was available for testing. (B) Pathogenicity of CNVs was evaluated based on three criteria: associated with known genomic disorders, deleting haplo-insufficient (HI) or duplicate triplo-sensitive (TS) genes, and affecting large number of genes (details given Materials and Methods). A total of forty CNVs in patients were evaluated as pathogenic. The number of CNVs fulfilling those overlapping criteria is displayed as a Venn diagram.
CNV Burdens
We first compared the burden of ultra-rare and large CNVs between MR/DD patients and controls (Table 1). All comparisons were restricted to CNVs in autosomes. Overall, 18.5% of the patients carried CNVs >= 500 kb compared to 6.4% in controls (OR: 3.32, P = 1.69E-11, Fisher’s Exact Test). The increased CNV burden was mainly due to large deletions, which were carried by 10.1% patients compared to 1.1% controls (OR: 10.18, P = 4.38E-17); whereas the burden of duplication was only modest (8.4% vs. 5.3%, OR: 1.64, P = 0.032). The CNV burden was slightly higher in female (20.4%) than male patients (17.5%), but not statistically different (OR: 1.21, P > 0.5). Likewise, CNVs overlapping the coding sequences (CDS) of at least 8 genes were also significantly enriched in patients (12.5%) compared to controls (1.7%; OR = 8.52, P = 1.04E-18), with a higher enrichment of deletions (OR = 19.7) than duplications (OR = 3.61), and similar between female (13.3%) and male patients (12.2%). The 10~12% increase in CNV burden of patients compared to controls was consistent across different CNV size thresholds (Supplementary Table S6).
As some previous studies19,22,23 defined rare CNVs by frequency of <1%, we also tested the burden of low frequency but not ultra-rare (0.1~1% population frequency) CNVs (Supplementary Table S7). There was a deficit of CNVs in this frequency range as compared with ultra-rare CNVs, because most rare variants including CNVs are private. The proportion of individuals carrying CNV >500 kb was similar between patients (3.1%) and controls (3.5%). Only one such CNV affecting at least 8 genes was found in patients (0.3%), not statistically deviating from the control rate of 0.7% (P > 0.5). The lack of enrichment of low frequency CNVs suggests that most high-penetrant large pathogenic CNVs should be found in the ultra-rare CNVs.
If a portion of large pathogenic CNVs found in patients were inherited, then we would expect to find an increased burden in their parents. We then analyzed the CNV burden in the parents of MR/DD patients. Overall, 7.1% of the parents carried CNVs >500 kb and 2.4% carried CNVs affecting at least 8 genes, both of which were slightly higher but not statistically different from controls (6.4% with CNV >500 kb, 1.7% with CNVs >= 8 genes). The result suggests that the increased CNV burden observed in MR/DD patients are predominantly due to de novo ones. Indeed, most large CNVs found in patients occurred de novo (Fig. 2A; Supplementary Figure S6). E.g., Over 90% of CNVs larger than 1 Mb or affecting at least 10 genes were de novo. And larger deletions were more likely occurred de novo than duplications of similar sizes or similar number of affected genes; suggesting that large deletions are less tolerated. Using parental genotypes and allelic-specific signals, we inferred the parental of origin for 36 de novo CNVs. Consistent with previous findings24,25, the majority (n = 22, 61.1%) of them arose on the paternally derived alleles. The paternal bias was mostly due to large deletions and/or CNVs outside rearrangement hotspots (Fig. 2B).

(A) Cumulative proportion of de novo CNVs as a function of physical sizes. More than 90% of all ultra-rare CNVs greater than 1 Mb occur de novo; and large deletions are more likely de novo as compared with duplications of similar sizes. (B) Parental-of-origin for different classes of de novo CNVs. The majority of de novo CNVs arise on the paternal allele. The observed paternal bias was mainly driven by the CNVs larger than 1 Mb and/or outside known hotspots for non-allelic recombination (*p < 0.1, by exact binomial test against equal chance).
Although the parents as a whole did not have significantly increased CNV burden, when stratified by gender, we observed 4.3% of the mothers carried CNVs affecting at least 8 genes, a significant 2.7 fold increase compared to control females (1.6%; OR = 2.82, P = 4.64E-03). Mothers also carried marginally increased number of deletions >500 kb (2.3%) compared with control females (1.1%; OR = 2.07, P = 0.128). We observed similar trend at other thresholds of CNV size/gene content (Supplementary Table S6). The increase in CNV burden in the mothers was not due to underestimation of the CNV burden in controls or platform difference; because we have controlled for the effects of platform difference (Materials and Methods). In addition, the large CNV burden in patients’ fathers cannot be distinguished from control males; and male and female controls had similar CNV burdens. When comparing between the mothers and fathers that were genotyped by the same platform, there was a gradual increase among mothers with gene-affecting CNVs with increasing number of genes (Supplementary Table S11).
Pathogenic CNVs
Using the criteria detailed in Materials and Methods, we identified 40 pathogenic CNVs in 39 patients (26 males, 13 females), including one patient with two such variants (Supplementary Table S10). The number of CNVs fulfilling each pathogenic criterion is shown as a Venn diagram in Fig. 1B.
We found a total of 19 CNVs associated with known genomic disorders (Table 2). Most of them occurred de novo, except three (Xq28, 16p11.2, and 1q21.1) were inherited from mothers. CNVs at 16p11.2, 1q21.1 and 22q11.2 are known to have reduced penetrance and also occur at very low frequencies in controls. Mothers who carried 16p11.2 and 1q21.1 CNVs had reported stillbirth in their previous pregnancies. Fifteen of these CNVs were located in the hotspots of non-allelic homologous recombination (NAHR), which have typical boundaries defined by the flanking segmental duplications. We found three smaller CNVs showing atypical boundaries within NAHR hotspots (Fig. 3), which may help refine the candidate genes. As a proof of principle, we found a smaller 1.4 Mb de novo deletion carried by patient MR_3721 who showed classical sign of Smith-Magenis syndrome. The deletion co-localized with two other deletions and two duplications at the same locus; their smallest overlapping region contained the known disease genes RAI1 and SHMT1(Fig. 3A). The recurrent 1q21.1 microdeletion/duplication have been associated with variable pediatric phenotypes including intellectual disability, autism, congenital heart defects, etc26. We identified a smaller 500 kb duplication in a prenatal case (MR_1194) with congenital absence of abdominal wall. The duplication contained three genes including CHD1L and PRKAB2 (Fig. 3B). A previous study using patient-derived lymphoblast cell lines have documented functional anomalies of CHD1L and PRKAB2 in 1q21.1 deletion syndrome27. A small deletion containing CHD1L has also recently been implicated in autism spectrum disorder28. The left boundary of this duplication was uncertain due to an assembly gap of human genome. Previous studies suggested HYDIN2 gene within the gap be the causative gene26,29. We performed quantitative real-time polymoerase chain reaction (qRT-PCR) experiments and excluded the copy number gain of HYDIN2. However, we cannot exclude the increased copy number of other genes in this gap. The pathogenic effect of this duplication may also be due to functional alternations other than increased gene dosage (see Discussion). We also identified a male patient (MR_1535) carrying a de novo 2.3 Mb duplication located within 16p11.2-p12.1 deletion syndrome30 locus (chr16:22.75–25.02 Mb, hg18). The reciprocal duplication of 16p11.2-p12.1 deletion has only been recently established as a genomic disorder, with only a handful reported cases30,31,32. Our patient was 2 years old at diagnosis, showed speech delay, nystagmus, and mild craniofacial dysmophism (ptosis). The duplicated region contains 20 genes including PLK1 (Fig. 3C), which has been suggested as the most likely dosage sensitive gene at this locus32. Close inspection of array signals (Supplementary Figure S5) revealed two nearby duplications, which most likely reflect a patient-specific inversion at this region.

(A) Five de novo CNVs (two duplications and three deletions) in the Smith-Magenis/Potocki-Lupski syndrome region. The minimal overlapping region encompasses two known haploinsufficient genes RAI1 and SHMT1. (B) An atypical 1q21.1 duplication inherited from mother. The left boundary is uncertain due to gaps and segmental duplications in that region. Previous studies suggested a dosage sensitive gene HYDIN2 in that region be the causative gene26,29. We tested the copy number of this gene using qRT-PCR, but did not find copy number gain of this gene. (C) A de novo 16p12.1 duplication partially overlaps the region of the known 16p11.2-p12.1 deletion/duplication syndrome, including PLK1 gene. The duplication is proximal to 16p12.1 deletion syndrome50. In all cases, green lines above indicate the typical boundaries of genomic disorder-associated CNVs.
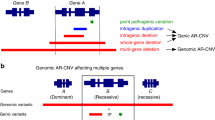
Besides the CNVs associated with genomic disorders, we also identified 15 CNVs (11 deletions, 4 duplications) affecting large number of genes. All of them occurred de novo in patients. Known haploinsufficient (HI) genes were found in 6 deletions. In four cases, patients’ phenotypes well matched to the syndromic features caused by haploinsufficiency of PAX6 (Aniridia, MIM:607108), TCF4 (Pitt-Hopkins syndrome, MIM:610954), FBN1 (skeletal/joint malformations, limb deformity; MIM:154700), and TWIST1 (Saethre-Chotzen syndrome, MIM:101400). Four large duplications overlapped known HI genes including ZEB2 (MIM:605802), MBD5 (MIM:611472), SALL4 (MIM:607343), ENG (MIM:131195); but evidence for the triplosensitivity remains to be established.
In addition to very large CNVs or CNVs associated genomic disorders, we were also able to identify several small deletions (<500 kb and affecting <= 3 genes) (Table 3). These include single-gene exonic deletions in NRXN1, GRM8, CREBBP (Rubinstein-Taybi syndrome, MIM:180849), KDM4B, and DMD (Fig. 4). Three CNVs including duplication of PPP3CC, deletions involving DYNC1I1 and DLGAP2 were considered likely pathogenic, which have been previously reported in a few NDD patients.

(A) GRM8, (B) NRXN1, (C) CREBBP, and (D) KDM4B. The approximate CNV boundaries are shown as parentheses superimposed on gene structures. Deletions of GRM8 and NRXN1 were maternally transmitted; deletions of CREBBP and KDM4B occurred de novo.
Female Protective Model
Interestingly, we noted that all inherited pathogenic CNVs were identified in male patients and maternally inherited (Tables 2 and 3), only two of which were on chromosome X. Among ultra-rare autosomal CNVs identified in patients, inheritance could be determined with high confidence for 143 of them. Overall, the maternal transmission rate was 46.2% (66 out of 143), which was not significantly different from the proportion of mothers in the parents (252 of 510) (OR = 0.83, P = 0.38). However, there was a marginal excess of maternally inherited CNVs that were >300 kb (OR = 2.33, P = 0.047) and CNVs that contains at least one evolutionary conserved gene (OR = 3.16, P = 0.059) (Fig. 5). No such bias was observed in common CNVs (data not shown).

The mosaic plots show the number of autosomal CNVs stratified by inheritance status (maternal or paternal), size (>or <=300 kb), and type (gain or loss). The area of each rectangular partition of the square is proportional to the number of CNVs fall in each classification. (A) CNVs larger than 300 kb tend to be transmitted from mothers as compared with fathers (p < 0.05 by Fisher’s Exact Test (FET)). (B) Maternally transmitted CNVs are also more likely to overlap with the coding exons of evolutionary conserved genes, which are defined by the top 10% of residual variation intolerance score88 (p < 0.1 by FET).
To further explore the maternal transmission bias, we also examined rare inherited CNVs identified in a large clinical cohort of intellectual disability and/or multiple congenital anomalies from a published study19. Data on inheritance were available for 1065 CNVs, including 584 inherited autosomal CNVs in 529 patients. Overall, 330 (56.5%) of them were maternally inherited (P = 1.88E-03). Because parents were not genotyped in that study, rare CNVs might be subject to selection before parental testing. To account for potential ascertainment bias, we compared the inheritance rate at different CNV sizes, gene contents, and HI scores33 (Supplementary Table S12). Maternal inheritance gradually increase with the increase of HI score threshold, and was significantly enriched for CNVs with HI score >12 (25 maternal vs. 5 paternal; 85.3% maternal rate) than for CNVs with HI score <= 0 (56% maternal rate) (OR = 3.83, P = 4.9E-03). The maternally inheritance rate was also marginally higher for larger CNVs (>2.5 Mb) and CNVs affecting more genes (>25 genes). To further predict deleteriousness, we compiled 321 most likely dosage sensitive genes (harboring autosomal dominant disease-causing LoF mutations) implicated in developmental disorders from DDG2P database34. Significantly more maternally inherited CNVs overlap at least one such genes (41 maternal vs. 14 paternal, OR = 2.43, P = 4.28E-03). Therefore, potential pathogenic CNVs were enriched in maternally inherited alleles even after accounting for overall increase of maternal transmission.
We have demonstrated an increased CNV burden in the mothers of MR/DD patients compared to both controls and the fathers of the patients. It also suggests that in addition to transmitted CNVs, un-transmitted CNVs in mothers may also be enriched for pathogenic alleles. So we applied the same criteria to identify potential pathogenic CNVs in parents that are not found in patients. Six (likely) pathogenic CNVs were found (Table 4), including 16p13.11 deletion, deletions of CUL3, BMP4, WHSC2, KANK1, and a duplication of CNTN4-CNTN6. Notably, all of them were carried by the mothers.
The identification of pathogenic CNVs, both transmitted and un-transmitted, in unaffected mothers, can be explained if females are more tolerant than males to deleterious mutations. Under this hypothesis, manifestation of MR/DD for females would require a higher burden of deleterious mutations. Consistent with this, we found that pathogenic CNVs identified in female patients affected a significantly higher number of genes (median: 49) than those in male patients (median: 25; P = 0.022). The increased number of affected genes was not due to duplications affecting more genes than deletions, as only one pathogenic duplication was found in female patients.
Additional Findings
In addition to CNV calling, SNP array data can be further exploited to identify long runs of homozygotes (ROH), constituent uniparental disomy (UPD), and mosaic structural abnormalities35.
By analyzing the Mendel errors in trios, one female patient (MR_1369) was found to have unusually large number (569) of Mendelian transmissions on chromosome 15. Over 99.9% of her genotype on this chromosome exactly matched to her mother without loss of heterozygosity, indicating that it was caused by maternal chromosome 15 heterodisomy. The patient’s phenotype confirmed the diagnosis of Prader-Willi syndrome. We also searched ROH segments, which may harbor recessive disease-causing mutations. By comparing to parental genotypes, the possibility of uniparental isodisomy was excluded for all segments >10 centi-Morgans (cM) in patients of trios. One male patient (MR_2559) was found to have total ROH length over 300 cM. The estimated kinship coefficient of his parents was ~0.13, consistent with first cousin mating. In addition, we found three other patients with total ROH greater than 50 cM, which might result from recent inbreeding. None of them carried pathogenic CNVs; neither did we found homozygous CNVs in their ROH segments. Further sequencing studies will be needed to identify the recessive mutations. A burden analysis showed that the proportion of patients with long ROH length was not significantly higher than that of parents and controls (Supplementary Table S13), which was expected since consanguineous families were not preferentially ascertained in this study. We did not find detectable mosacism in patients, but identified one mosaic segmental UPD from 11p15.3 to p-terminus in a parent with unknown origin (Supplementary Figure S8). Segmental parental UPD for 11p15.5 is a known cause of Beckwith-Wiedemann syndrome in 10–20% of patients36. The clinical significance of the finding was unclear.
Despite the low probe density (~300 k), we were able to identify a number of single gene disruptions in this study, including 3 single gene and 24 exonic deletions in patients (Supplementary Table S5). To opportunistically identify additional gene disrupting CNVs, we implemented a workflow of targeted rare CNV genotyping37 to search for small gene disrupting CNVs that may be missed by the standard calling algorithm (Supplementary Methods). Exon regions spanned by five informative markers in over 2000 candidate genes were interrogated to look for outliers in the signal intensities across the sample. Samples with consecutive probes of low or high intensities are candidate carriers of a rare CNV. This method achieved 85% sensitivity in recovering the ultra-rare CNVs identified in the clinical cohort (positive for 58 out of 68 CNVs that covered targeted regions; Table S8, Table S9). After a series of QC filter, prioritization and validation, we identified a small deletion in a male patient (MR_3590), disrupting three genes including the coding exon 1 of HIRA gene (Supplementary Figure S9) within the 22q11.1 deletion (DiGeorge) syndrome region. HIRA encodes for a histone chaperon, and is considered as a primary candidate for DiGeorge syndrome38. It has a predicted HI probability of 0.9433. No gene-disrupting CNV other than the typical 22q11.2 deletion was observed in controls; and loss-of-function (LoF) variants in this gene were also extremely rare (based on ExAC database). The patient showed mental retardation, speech delay, and hyperactivity. The clinical significance of this deletion remains to be evaluated with new mutations in additional patients.
Discussion
Implications for CNV Interpretation
We adopted three overlapping criteria for evaluating the pathogenicity of CNVs. Those criteria are dynamic since our knowledge about genomic disorders and gene dosage sensitivity are constantly updating. This is especially the case for CNV-disease associations outside NAHR hotspots, because rarity of the events entails the screening on clinical cohorts with very large sample sizes to establish their disease causality.
Among large deletions identified in patients, we found a female patient (MR_3067) with a 3.7 Mb deletion at 4q21 locus encompassing candidate genes PRKG2 and RASGEF1B. The patient showed gross developmental delay since birth, speech impairment, and generalized tonic-clonic seizure since 2 years old. Although patients with overlapping deletions at 4q21 locus have been reported, the locus only gained genome-wide significance in recent studies22,23. Haploinsufficiency of MBD5 gene was known to be responsible for the phenotypes of 2q23 micro-deletion syndrome39. The triplosensitivity of this same gene has only been suggested recently40. We identified a large duplication encompassing 2q23 deletion syndrome region in a female patient (MR_1191) with speech delay and dysmorphic craniofacial features.
The evaluation criteria were based on the presumed functional consequence of CNVs on affected genes. Throughout the study, we consider a CNV affects a gene if it overlaps its CDS region. While it may be obvious that deletion leads to haploinsufficiency, the consequences of duplication are less clear. In addition to increase the dosages of encompassing genes, duplication may also disrupt genes at its insertion site41, or create chimeric genes at breakpoints42. Several duplications that partially overlap the coding sequences known HI genes were noted, including a 190 kb de novo duplication overlapping FBN1 and SLC12A1, and a 750 kb duplication overlapping PDE4D (Supplementary Table S8). The functional consequences cannot be determined until the breakpoints are resolved. Deletions that affect noncoding or regulatory sequences may also causes haploinsufficiency due to the changes in gene transcription or translation, but functional assays would be required for validation.
Additional factors are also considered in practice when evaluating the pathogenicity of CNVs, including inheritance, frequency in natural populations, and recurrence in patients, which are discussed below.
Inheritance
We identified and validated a total of 41 de novo CNVs in 39 patients, 33 of them were judged as pathogenic and 2 likely pathogenic, reinforcing the notion that not all de novo CNVs are disease causing18. The remaining six de novo CNVs in five (1.7%) patients were of unknown clinical significance, four of which did not overlap any coding gene. The carrier rate is consistent with previous reported de novo CNV rates (1~2%) in healthy control trios43,44,45. While most of the pathogenic de novo CNVs were large, we did find two de novo exonic deletions. One notable example is KDM4B, a lysine-specific demethylase. The LoF variants in this gene and several members of the same family (KDM3A, KDM5B, KDM6B) have recently been associated with autism in a large scale genetic investigation46. Other members of the KDM family were also been identified as the disease genes for syndromic mental retardation, including KDM5C (MIM:314690) and KDM6A (MIM: 300128). We also identified a de novo duplication encompassing PPP3CC, which encodes a catalytic subunit of calcineurin, a protein phosphatase involved in the downstream regulation of dopaminergic signal transduction. Similar duplications were observed in at least two other patients with affective and cognitive disorders47,48. And the gene has recently been identified as a putative modulator of antidepressant response49. The de novo occurrences in the above cases add further support for the causal involvement of dosage sensitivity of KDM4B and PPP3CC in NDD.
Our study also confirms the pathogenic roles of inherited CNVs, not restricted to chromosome X, which are transmitted from asymptomatic parents because of incomplete penetrance. The penetrance of some pathogenic CNVs may be mediated by the presence of a second large CNV50,51, and/or due to gender-specific liability thresholds where females are predicted to carry a more penetrant risk variant load to be affected52. We found strong evidence to support for the latter. The findings of our study can be explained a simple transmission model of DD/MR which was previously proposed for autism53. Families with affected children can be classified into at least two types. Patients in low-risk families were caused by de novo mutations that mainly originate from mutations in paternal germline, with high penetrance in male and low penetrance in female offspring. The unaffected females carriers of the causative mutation in turn transmit the mutation in a dominant fashion to their offspring. It also suggests that inheritance of a CNV be incorporated into the review process to aid the evaluation its clinical significance.
Population Frequency
In this study, we focused on ultra-rare CNVs, which had frequency <0.1% in natural population. They are expected to enrich for highly penetrant alleles that are under strong purifying selection. We also demonstrated that CNVs within 0.1~1% frequency range did not contribute to the large CNV burden in patients. However, we may have missed CNVs that only moderately increase the disease risk.
So we checked the population frequencies of CNVs associated with known genomic disorders (Supplementary Table S15). We found all except one (15q11.2 BP1-BP2 deletion) appeared <0.1% in controls, which is also consistent with previous reports22,23,54. Given CNVs outside NAHR hotspots are less frequent, applying population frequency filter of 0.1% would unlikely miss pathogenic CNVs with high penetrance. The 15q11.2 BP1-BP2 deletion is proximal to Prader-Willi/Angleman syndrome deletion separated by a genome assembly gap. The deletion has been associated with schizophrenia55, developmental delay56, congenital heart defects57, and cognitive abilities58 but with low penetrance. The frequency of the deletion in our control databases is 0.35%, similar to previous reports in WTCCC data59, but higher than some other studies22,58,60. We found two cases with the deletion; both also carried the Angelman syndrome deletion. Most likely they represent a single large deletion spanning from BP1 to BP3 (Supplementary Figure S10).
Theoretically, the population frequency of a disease-causing mutation is determined by the inheritance mode, selection coefficient, genetic drift, and locus-specific mutation rate. A relatively high population frequency of 15q11.2 deletion can be explained by a combination of its moderate penetrance22 and a high rate of rearrangement in NAHR hotspots. The association of this deletion with NDD in Chinese remained to be evaluated in future studies.
Recurrence in Patients
Because of the stringent filtering procedure and a relatively small sample size, most of the ultra-rare CNVs identified in patients are private except for a few cases in NAHR hotspots. We also searched for overlapping events outside NAHR hotspots and identified several cases of overlapping CNVs and intersecting genes (Supplementary Figure S11).
Notably, we found an exonic deletion of ARSF at Xp22 shared by two patients: one was carried by a male patient inherited from his mother, the other was carried by female inherited from her father. The deletions found in two patients had exactly the same boundary (Fig. 6A). Both parents were unaffected. The same deletion was not observed in controls or public databases. Since most CNVs outside NAHR hotspots were generated from replication-based mechanisms61 presumably with very low mutation rates, rare CNVs with recurrent boundaries outside hotspot regions are most likely generated by ancestral mutational events (Fig. 6B). Haplotype sharing analysis shows that the two deletions shared a 0.75 cM haplotype identical by descent (Fig. 6C). It suggests that the deletion is likely a local polymorphism, not subject to purifying selection. The absence of this CNV in controls may be caused by the lack of probe coverage at this locus. In an early resequencing study of X-linked mental-retardation pedigrees, the ARSF gene was initially nominated as a candidate but was later rejected because LoF variants were also observed in healthy males62.

(A) The genome browser tracks show the gene structure, log-R ratios of SNP array probes, and CNVs from Database of Genomic Variants at this region. The deletion omitting the exon 8 of ARSF gene was identified in one male patient inherited from mother and in one female patient inherited from father, with identical boundary observed in both cases. The same deletion is absent in other unrelated samples from in-house and public databases. The deletion carriers are unrelated as confirmed by whole-genome SNP genotypes. (B) If the deletion originated from an ancestral mutational event several generations ago, then the mutation carriers are expected to share the same chromosome background identical by descent from the recent most common ancestor (shown in red). Because males are hemizygous on chromosome X, the haplotype background of this deletion can be determined. (C) To quantify the shared ancestry around the deletion locus, we statistically inferred 624 unrelated haplotypes from trios in the cohort. Haplotype sharing with the male patient who carried the deletion was visualized. The extent of haplotype sharing was measured by allele matching starting from the deletion point toward left and right until the first marker showing the mismatched allele. Each line represents an unrelated haplotype, sorted by the shared haplotype length. The second deletion haplotype in the female patient is highlighted in blue. SNPs used to define haplotypes are marked below. Positions were relative to the deletion locus in centiMorgan (cM). The genetic length of shared haplotype background of the deletion is about 0.75 cM.
While recurrent disruption of genes exclusive to patients was commonly used to lend further support on pathogenicity, our case suggests that caution should be exercise to interpret rare CNVs with recurrent boundaries outside NAHR hotspots.
Comparison with Previous Studies
To compare the CNV burdens with previous studies22,23, we considered all low-frequency variants (<1% population frequency). For CNVs >500 kb, we found a total of 64 autosomal CNVs in 61 (21.3%) patients, compared to 592 such events in 571 (9.7%) in controls. For CNVs >750 kb, the carrier rate was 15.0% and 5.5% for cases and controls respectively. The excess of patients was 11.6% with CNVs >500 kb and 9.5% for CNVs >750 kb, which were close to but slightly lower than the previous report (13.5% with >500 kb, and 12.7% with >750 kb). This was probably due to the exclusion of patients with subtelomeric aberrations. Previous studies also report an increased CNV burden in patients with multiple congenital anomalies (MCA)19,22. Among 270 patients with detailed clinical information, 13.7% of them were considered having MCA (showing at least two signs of brain malformations, gross craniofacial dysmorphology, cardiac defects, and neurological deficits). And we observed slightly higher number of patients with MCA carrying CNVs affecting at least 8 genes (15.8%) as compared with patients without MCA (12.1%). But our sample size is limited to reach a significant conclusion (OR = 1.4, P > 0.5).
Including the case of chromosome 15 UPD, we found 41 pathogenic structural variants in 40 (14%) patients. If likely pathogenic CNVs were included, the number increases to 44 variants in 42 (14.6%) patients. We also identified pathogenic CNVs, including 8 transmitted and 4 un-transmitted, in 12 (2.4%) unaffected parents all of whom were patients’ mothers. The reported diagnostic yield range from 10% to 20%63, depending on many factors including sample ascertainment, genotyping platform, analytical methods, and evaluation criteria. The diagnostic yield of our study fall well within this range, and is higher than the observed large CNV burdens due to a number of small pathogenic CNVs (Table 3). Resolution can be further improved by using high-density exon targeted arrays9 or exome sequencing64.
A higher proportion of male MM/DD patients has long been noted in epidemiology studies65, which is also evident in our patient cohort with 46% excess of males patients. The increased proportion of male patients cannot be fully explained by hemizygous causal mutations on chromosome X66. A “female protective model” has been proposed to explain the male excess. While the simple model cannot capture every aspect of gender bias in NDD67, it has gained support from a number of studies, mostly in autism which showed highest male/female ratio (7:1). It has been demonstrated that: (1) female patients are more likely to have highly penetrant mutations than male patients45,52,68 (2) inherited disease-causing mutations are more likely transmitted from mothers68 and (3) mothers of the patients have higher burdens of deleterious mutations69. All of them were also supported by our data. We made a strong case for the maternal transmission bias of pathogenic alleles by showing that pathogenic or likely pathogenic CNVs with strong literature support identified in parents were all carried by mothers, only two of which were on chromosome X. This could also partly due to our ascertainment criteria that included only sporadic cases with healthy parents. Some previous studies also reported inherited pathogenic CNVs transmitted from mildly affected fathers70.
Burdens of inherited CNVs have also previously been investigated in autism by comparing transmitted CNVs between probands and unaffected sibs44,71, but only weak or no evidence of disease association was found. It is possible that most inherited rare variants are likely benign, only a few are disease causing; burden analysis has limited power if signals are buried in large amount of noise. Inherited CNVs of DD/MR in two other large data sets (ISCA, Signature Genomics) were also previously analyzed68. Consistent with our analysis of ref. 19, stronger maternal transmission bias stood out after the putative pathogenic CNVs were being enriched. Some studies also reported an overall maternal transmission bias of small exonic CNVs64, and higher CNV burden in females sampled from general populations69, both of which were not observed in our study. Future studies with increased resolution and expanded populations will be needed to evaluate these findings.
At each individual locus of genomic disorders, most commonly observed CNVs in Europeans were also found in Chinese patients of NDD (Table 2, Supplementary Table S15). But some population-specific locus does exist. A notable example is 17q21.3 micro-deletion syndrome. A local inversion polymorphism at this locus put the segmental duplications in the same orientation, predisposing this locus to NAHR72. As this inversion is almost exclusively found in Europeans, we did not observe similar patients in Chinese and other non-European populations.
Additional population specific loci may exist, because segmental duplications are highly variable between individuals genome-wide73 and thus may also likely differ across populations. We compared the CNV frequencies of our case/control cohorts in NAHR hotspots reported previously22 (Supplementary Table S16). A number of locus showing population differentiations can be noted, although it can also be attributed to platform difference. Further studies will be needed to better characterize population differences in the duplication architecture and understand their consequences on CNVs in disease.
Materials and Methods
Patients Ascertainment
The clinical cohort used in this study was selected from MR/DD trios collected by Department of Pediatrics in Peking University First Hospital. Informed written consent was obtained from the parents. The clinical manifestation of the patients included a wide range of neurodevelopmental phenotypes, typical of clinical diagnostic setting. Patients were ascertained to exclude history of prenatal brain injury, toxication, hypoxia, central nervous system infection, or cranial trauma. They also showed no evidence of recognizable inherited metabolic disorder or specific neurodegenerative disorders by brain imaging and blood/urinary metabolic screening. For patients eligible for intellectual assessment, only moderate to severe cases (IQ < 55, assessed with Gesell Developmental Schedules or Wechsler Intelligence Scale for children) were included. Sanger sequencing on FMR1 gene had been performed to exclude causative mutations for male patients; and clinical features of Rett syndrome were excluded for female patients. All of them had normal karyotype and negative of subtelomeric aberrations screening reported previously74.
Genomic DNA was extracted from peripheral blood for each index patient and his or her parents. DNA samples of 289 patients and 510 parents were genotyped using Illumina CytoSNP12 bead arrays, which had a total 299,671 probes. The research was approved by the ethical committees of Peking University First Hospital and National Research Institute of Family Planning. The methods were carried out in accordance with the approved guidelines.
Control Cohorts
Control samples included in this study were taken from previously published genome-wide association studies (GWAS) collected by two institutions in China (Supplementary Table S1B). The Shanghai Institute of Hypertension (SIH) cohort (n = 902) was recruited for a case-control study of essential hypertension. The GWAS summary statistics were previously included in the GWA meta-analysis of blood pressure by Asian Genetic Epidemiology Network75. The Anhui Medical University (AMU) cohort (n = 5366) was composed of cases of three autoimmune diseases and shared controls. The samples were used in previous GWAS of psoriasis76, systemic lupus erythematosus77, and vitiligo78. All GWAS samples were genotyped using Illumina HumanHap 610 k bead array. Neurodevelopmental disorders were not screened for the GWAS subjects. Although these subjects are not representative of general population, they can be served as controls because no evidence has so far been found that rare and large CNVs are associated with these diseases. But even if large CNVs do increase the risk of some disease, this would only make our comparison with MR/DD more conservative.
Data Processing
We implemented and validated a rigorous computational workflow for CNV calling, quality control, filtering and interpretation (Supplementary Figure S1, details are given in Supplementary Methods). Briefly, we used PLINK79 for genotype management and analysis. After SNP-based QC, genotypes were used to identify duplicated samples, and to verify familial relationships, reported genders, and population ancestry. Genotypes were also used to infer constituent UPD and long ROH. After SNP genotype-based QC, we performed further QC on samples based on array intensities before CNV calling. CNVs were called based on log-R ratio and B-allele frequency signals using PennCNV80. High confidence CNV calls were subject to a series of filters to enrich for causal variants. The steps included the exclusion of known CNPs, CNVs of the same type that occurred at least five times in 5866 population controls, and CNVs detected in at least two other unrelated parents. The resulting ultra-rare CNV calls were subject to iterative QC, manual curation, and experimental validation to generate the final CNV list. The inheritance status of CNVs found in patients was initially determined by comparing to the unfiltered CNV segments in parents, then subject to manual curation. De novo origin in 5 cases where at least one parent was not available for testing was assumed based on the absence in one parent and their well-established causal involvement in severe and fully penetrant phenotypes.
To identify smaller exonic CNVs that might be missed by standard calling algorithm, we also started with a list of known/candidate disease genes, and defined target regions that cover the coding exons and contain enough informative SNP probes. Then, targeted rare CNV genotyping were performed to search for additional small CNVs37. Mosacisms were called using MAD package81.
CNV Validation
Genomic DNA was extracted from peripheral blood leukocytes using standard methods (QIAamp DNA Mini Kit, Qiagen, Hilden, Germany). The exon regions of identified genic CNVs were targeted for primer design. Primer sequences of candidate genes were designed based on sequence data obtained from the NCBI database using Primer Express software 3.0 (Applied Biosystems).
The targeted regions were amplified in patients, parents (when available), and 12 unrelated healthy control individuals with matched ancestry by qRT-PCR with a set of gene-specific primers (available upon request). The gDNA was used as template in qRT-PCR reactions with SYBR® Green PCR Master Mix (Takara Biotechnology (DaLian) Co. LTD) and performed on 7000 real-time PCR system (Applied Biosystems). The quantification of the target was normalized to an assay from chromosome 21, C2, and the relative copy number (RCN) was determined on the basis of the comparative ΔΔCt method with a normal control DNA as the calibrator82. The experiments were repeated three times. A ~0.5-fold RCN and a ~1.5-fold RCN were used for deletion and duplication, respectively.
CNV Burden Analysis
To analyze the burden of ultra-rare and large CNVs in patients and parents, we need an accurate estimate of the baseline rate in control populations. CNVs in controls were first subject to the identical filtering process as done for the clinical cohort. Namely, we excluded CNVs whose 50% of the region overlapped CNP regions, or were covered by CNVs of the same type found in at least 5 unrelated controls or 2 parents. Due to platform differences, CNV calls in controls were further required to cover at least 10 markers on CytoSNP12 array. We also checked that all ultra-rare large CNVs found in the clinical cohort also had enough probe coverage (>=10 probes) on HumanHap 610 k array. CNVs >=500 kb or overlapping the coding exons of >=8 refSeq coding genes (over 90% of them >=250 kb) were subject to manual curation by the analyst with proven accuracy validated by experiments (Supplementary Methods). The manual curation were applied uniformly to the CNVs retained after automated filtering to correct for erroneous boundaries, and to reject false positives caused by abnormally behaving probes, CNVs artifacts in regions of segmental duplications or genome assembly gaps, etc. All comparisons between cases and controls were made on the curated CNV set, for which we believe should be least influenced by the platform differences.
We also analyze the burden of low frequency (0.1~1%) large CNVs. The low-frequency set was derived by using different filtering criteria; namely, CNVs were excluded if 50% its region overlap CNVs of the same type in at least 59 unrelated controls and 6 unrelated parents. We then manually reviewed signal plots of all low frequency CNV calls that were not in the ultra-rare CNV set.
Parental Origin (POO) of De novo CNVs
To determine the POO of de novo CNVs, we first generated CNV specific genotype calls for the offspring using PennCNV (infer_snp_allele utility). SNP markers informative of POO were then identified by comparing with the genotypes of parents. For example, when a father and mother’s genotypes at a SNP marker are AA and AB, respectively, then if offspring carry a duplication whose genotype call is ABB, then we can infer this de novo duplication happened on the maternal chromosome. Similarly, if the offspring carry a deletion whose genotype call at that marker in B, then we can infer the deletion happened on the paternal chromosome. For complete trios, we inferred the POO from all such informative SNPs with the CNV segment; a binomial p-value was assigned to test the predominance of paternal or maternal origin (against the 50% equal chance). When p < 0.01, POO of the de novo CNV was determined by the majority vote. The POO of small de novo CNVs without enough informative SNPs could not be determined. In four rare cases, only genotypes of one parent were available, but the de novo origin was presumed based on the well-established causal involvement in severe and fully penetrant phenotypes. The CNV-specific genotype was compared to the genotypes of the available parent, to check whether the CNV is consistent with the mutation on that parent. In case of large number of consistencies, CNV was presumed to originate from mutation in the other parent. Because all four CNVs had >100 informative SNPs, the POO determined by this heuristic approach should be accurate.
CNV Interpretation
To judge the pathogenicity of CNVs, we adopted the following criteria in spirit of ACMG recommendation83 and an evidence-based review procedure20. A CNV is judged causal if it overlaps at least 50% of the critical region of known genomic disorders matched for the copy numbers; or deletes the CDS regions of known haplo-insufficient genes or duplicates known triplo-sensitive genes84,85. Typical phenotypes of well known syndromes were checked with clinical records to confirm the diagnosis. If no such genes was found, we subjectively judge a CNV as pathogenic if it overlaps the CDS regions of a large number of genes: >=15 genes for deletions and >=20 genes for duplications. The gene number threshold were determined empirically such that >99% of CNVs detected from population-based controls should fall below these levels (Supplementary Figure S7). We also considered a CNV to be probable pathogenic, if it overlaps critical regions or dosage sensitive genes recently emerged from large-scale genetic studies of NDD with strong statistical significance or with convincing experimental evidence. CNVs were judged as likely pathogenic, if it were also reported in patients with similar phenotypes. The remaining was considered as unknown clinical significance. CNVs judged as pathogenic or probable pathogenic are considered as pathogenic variants throughout the paper.
Haplotype Analysis
To shed light on the evolutionary origin of the recurrent ARFS exonic deletion at Xp22 (Supplementary Figure S11G), we analyzed its SNP haplotype background. Genotypes from 218 complete trios centered around the deletion (+/−1 cM) were extracted (physical position: chrX:2720840-3628132, hg18 assembly). We only included common SNPs with MAF >0.05. Male heterozygotes and Mendel errors were set to missing. If a sample had more than 4% missing genotypes, then the complete trios was excluded from analysis. In trios consisting of father-mother-son, hemizygous genotypes of the son were used for phasing the genotypes of mother; and father’s haplotype was assumed known. This resulted in 143 phased mothers and 143 phase known fathers, or 429 unrelated haplotypes in total. In trios consisting of father-mother-daughter, father’s genotypes were treated as diploid homozygotes. We then used 429 resolved haplotypes to “guide” the phasing of genotypes of 65 father-mother-daughter trios using Beagle version 3.3.286,87. A total of 624 unrelated haplotypes were inferred. The shared haplotype background was determined by allele matching extending from the deletion locus toward left and right sides until the first marker with mismatched allele was encountered.
Additional Information
How to cite this article: Wang, B. et al. CNV analysis in Chinese children of mental retardation highlights a sex differentiation in parental contribution to de novo and inherited mutational burdens. Sci. Rep. 6, 25954; doi: 10.1038/srep25954 (2016).
References
Cook, E. H., Jr. & Scherer, S. W. Copy-number variations associated with neuropsychiatric conditions. Nature 455, 919–923, 10.1038/nature07458 (2008).
Merikangas, A. K., Corvin, A. P. & Gallagher, L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet 25, 536–544, 10.1016/j.tig.2009.10.006 (2009).
Stankiewicz, P. & Lupski, J. R. Structural variation in the human genome and its role in disease. Annu Rev Med 61, 437–455, 10.1146/annurev-med-100708-204735 (2010).
Girirajan, S. et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLos Genet 7, e1002334, 10.1371/journal.pgen.1002334 (2011).
Miller, D. T. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86, 749–764, 10.1016/j.ajhg.2010.04.006 (2010).
Stankiewicz, P. & Beaudet, A. L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev 17, 182–192, 10.1016/j.gde.2007.04.009 (2007).
Boone, P. M. et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 31, 1326–1342, 10.1002/humu.21360 (2010).
Bruno, D. L. et al. Extending the scope of diagnostic chromosome analysis: detection of single gene defects using high-resolution SNP microarrays. Hum Mutat 32, 1500–1506, 10.1002/humu.21581 (2011).
Tucker, T. et al. Single exon-resolution targeted chromosomal microarray analysis of known and candidate intellectual disability genes. Eur J Hum Genet 22, 792–800, 10.1038/ejhg.2013.248 (2014).
Riggs, E. R. et al. Chromosomal microarray impacts clinical management. Clin Genet 85, 147–153, 10.1111/cge.12107 (2014).
Henderson, L. B. et al. The impact of chromosomal microarray on clinical management: a retrospective analysis. Genet Med 16, 657–664, 10.1038/gim.2014.18 (2014).
Coe, B. P. et al. Resolving the resolution of array CGH. Genomics 89, 647–653, 10.1016/j.ygeno.2006.12.012 (2007).
Hehir-Kwa, J. Y. et al. Genome-wide copy number profiling on high-density bacterial artificial chromosomes, single-nucleotide polymorphisms, and oligonucleotide microarrays: a platform comparison based on statistical power analysis. DNA Res 14, 1–11, 10.1093/dnares/dsm002 (2007).
Halper-Stromberg, E. et al. Performance assessment of copy number microarray platforms using a spike-in experiment. Bioinformatics 27, 1052–1060, 10.1093/bioinformatics/btr106 (2011).
Pinto, D. et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 29, 512–520, 10.1038/nbt.1852 (2011).
Tsuchiya, K. D. et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med 11, 866–873, 10.1097/GIM.0b013e3181c0c3b0 (2009).
Hehir-Kwa, J. Y., Pfundt, R., Veltman, J. A. & de Leeuw, N. Pathogenic or not? Assessing the clinical relevance of copy number variants. Clin Genet 84, 415–421, 10.1111/cge.12242 (2013).
Vermeesch, J. R., Balikova, I., Schrander-Stumpel, C., Fryns, J. P. & Devriendt, K. The causality of de novo copy number variants is overestimated. Eur J Hum Genet 19, 1112–1113, 10.1038/ejhg.2011.83 (2011).
Vulto-van Silfhout, A. T. et al. Clinical significance of de novo and inherited copy-number variation. Hum Mutat 34, 1679–1687, 10.1002/humu.22442 (2013).
Riggs, E. R. et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet 81, 403–412, 10.1111/j.1399-0004.2011.01818.x (2012).
Hoischen, A., Krumm, N. & Eichler, E. E. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci 17, 764–772, 10.1038/nn.3703 (2014).
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846, 10.1038/ng.909 (2011).
Coe, B. P. et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–1071, 10.1038/ng.3092 (2014).
Hehir-Kwa, J. Y. et al. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J Med Genet 48, 776–778, 10.1136/jmedgenet-2011-100147 (2011).
Sibbons, C., Morris, J. K., Crolla, J. A., Jacobs, P. A. & Thomas, N. S. De novo deletions and duplications detected by array CGH: a study of parental origin in relation to mechanisms of formation and size of imbalance. Eur J Hum Genet 20, 155–160, 10.1038/ejhg.2011.182 (2012).
Mefford, H. C. et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 359, 1685–1699, 10.1056/NEJMoa0805384 (2008).
Harvard, C. et al. Understanding the impact of 1q21.1 copy number variant. Orphanet J Rare Dis 6, 54, 10.1186/1750-1172-6-54 (2011).
Girirajan, S. et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet 92, 221–237, 10.1016/j.ajhg.2012.12.016 (2013).
Doggett, N. A. et al. A 360-kb interchromosomal duplication of the human HYDIN locus. Genomics 88, 762–771, 10.1016/j.ygeno.2006.07.012 (2006).
Ballif, B. C. et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat Genet 39, 1071–1073, 10.1038/ng2107 (2007).
Behjati, F. et al. M-banding characterization of a 16p11.2p13.1 tandem duplication in a child with autism, neurodevelopmental delay and dysmorphism. Eur J Med Genet 51, 608–614, 10.1016/j.ejmg.2008.06.007 (2008).
Barber, J. C. et al. 16p11.2-p12.2 duplication syndrome; a genomic condition differentiated from euchromatic variation of 16p11.2. Eur J Hum Genet 21, 182–189, 10.1038/ejhg.2012.144 (2013).
Huang, N., Lee, I., Marcotte, E. M. & Hurles, M. E. Characterising and predicting haploinsufficiency in the human genome. PLos Genet 6, e1001154, 10.1371/journal.pgen.1001154 (2010).
Wright, C. F. et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314, 10.1016/S0140-6736(14)61705-0 (2015).
Bruno, D. L. et al. Pathogenic aberrations revealed exclusively by single nucleotide polymorphism (SNP) genotyping data in 5000 samples tested by molecular karyotyping. J Med Genet 48, 831–839, 10.1136/jmedgenet-2011-100372 (2011).
Romanelli, V. et al. Beckwith-Wiedemann syndrome and uniparental disomy 11p: fine mapping of the recombination breakpoints and evaluation of several techniques. Eur J Hum Genet 19, 416–421, 10.1038/ejhg.2010.236 (2011).
Mefford, H. C. et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res 19, 1579–1585, 10.1101/gr.094987.109 (2009).
Farrell, M. J. et al. HIRA, a DiGeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ Res 84, 127–135 (1999).
Talkowski, M. E. et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 89, 551–563, 10.1016/j.ajhg.2011.09.011 (2011).
Mullegama, S. V. et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet 22, 57–63, 10.1038/ejhg.2013.67 (2014).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347, 10.1038/nature13394 (2014).
Newman, S., Hermetz, K. E., Weckselblatt, B. & Rudd, M. K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet 96, 208–220, 10.1016/j.ajhg.2014.12.017 (2015).
Itsara, A. et al. De novo rates and selection of large copy number variation. Genome Res 20, 1469–1481, 10.1101/gr.107680.110 (2010).
Sanders, S. J. et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885, 10.1016/j.neuron.2011.05.002 (2011).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94, 677–694, 10.1016/j.ajhg.2014.03.018 (2014).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215, 10.1038/nature13772 (2014).
Saus, E. et al. Comprehensive copy number variant (CNV) analysis of neuronal pathways genes in psychiatric disorders identifies rare variants within patients. J Psychiatr Res 44, 971–978, 10.1016/j.jpsychires.2010.03.007 (2010).
Costain, G. et al. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum Mol Genet 22, 4485–4501, 10.1093/hmg/ddt297 (2013).
Fabbri, C. et al. PPP3CC gene: a putative modulator of antidepressant response through the B-cell receptor signaling pathway. Pharmacogenomics J 14, 463–472, 10.1038/tpj.2014.15 (2014).
Girirajan, S. et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 42, 203–209, 10.1038/ng.534 (2010).
Girirajan, S. et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 367, 1321–1331, 10.1056/NEJMoa1200395 (2012).
Gilman, S. R. et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907, 10.1016/j.neuron.2011.05.021 (2011).
Zhao, X. et al. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci USA 104, 12831–12836, 10.1073/pnas.0705803104 (2007).
Kaminsky, E. B. et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13, 777–784, 10.1097/GIM.0b013e31822c79f9 (2011).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236, 10.1038/nature07229 (2008).
Burnside, R. D. et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet 130, 517–528, 10.1007/s00439-011-0970-4 (2011).
Soemedi, R. et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 91, 489–501, 10.1016/j.ajhg.2012.08.003 (2012).
Stefansson, H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366, 10.1038/nature12818 (2014).
Grozeva, D. et al. Independent estimation of the frequency of rare CNVs in the UK population confirms their role in schizophrenia. Schizophr Res 135, 1–7, 10.1016/j.schres.2011.11.004 (2012).
Kirov, G. et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry 75, 378–385, 10.1016/j.biopsych.2013.07.022 (2014).
Hastings, P. J., Lupski, J. R., Rosenberg, S. M. & Ira, G. Mechanisms of change in gene copy number. Nat Rev Genet 10, 551–564, 10.1038/nrg2593 (2009).
Tarpey, P. S. et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 41, 535–543, 10.1038/ng.367 (2009).
Hochstenbach, R., Buizer-Voskamp, J. E., Vorstman, J. A. & Ophoff, R. A. Genome arrays for the detection of copy number variations in idiopathic mental retardation, idiopathic generalized epilepsy and neuropsychiatric disorders: lessons for diagnostic workflow and research. Cytogenet Genome Res 135, 174–202, 10.1159/000332928 (2011).
Krumm, N. et al. Transmission disequilibrium of small CNVs in simplex autism. Am J Hum Genet 93, 595–606, 10.1016/j.ajhg.2013.07.024 (2013).
Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr Res 65, 591–598, 10.1203/PDR.0b013e31819e7203 (2009).
Mandel, J. L. & Chelly, J. Monogenic X-linked mental retardation: is it as frequent as currently estimated? The paradox of the ARX (Aristaless X) mutations. Eur J Hum Genet 12, 689–693, 10.1038/sj.ejhg.5201247 (2004).
Polyak, A., Rosenfeld, J. A. & Girirajan, S. An assessment of sex bias in neurodevelopmental disorders. Genome Med 7, 94, 10.1186/s13073-015-0216-5 (2015).
Jacquemont, S. et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet 94, 415–425, 10.1016/j.ajhg.2014.02.001 (2014).
Desachy, G. et al. Increased female autosomal burden of rare copy number variants in human populations and in autism families. Mol Psychiatry 20, 170–175, 10.1038/mp.2014.179 (2015).
Asadollahi, R. et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J Med Genet 51, 677–688, 10.1136/jmedgenet-2014-102588 (2014).
Levy, D. et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897, 10.1016/j.neuron.2011.05.015 (2011).
Koolen, D. A. et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38, 999–1001, 10.1038/ng1853 (2006).
Alkan, C. et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41, 1061–1067, 10.1038/ng.437 (2009).
Wu, Y. et al. Submicroscopic subtelomeric aberrations in Chinese patients with unexplained developmental delay/mental retardation. BMC Med Genet 11, 72, 10.1186/1471-2350-11-72 (2010).
Kato, N. et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 43, 531–538, 10.1038/ng.834 (2011).
Zhang, X. J. et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet 41, 205–210, 10.1038/ng.310 (2009).
Han, J. W. et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41, 1234–1237, 10.1038/ng.472 (2009).
Quan, C. et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat Genet 42, 614–618, 10.1038/ng.603 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, 10.1086/519795 (2007).
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17, 1665–1674, 10.1101/gr.6861907 (2007).
Gonzalez, J. R. et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics 12, 166, 10.1186/1471-2105-12-166 (2011).
Sun, M. et al. Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long range, limb-specific SHH enhancer. J Med Genet 45, 589–595, 10.1136/jmg.2008.057646 (2008).
Kearney, H. M. et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13, 680–685, 10.1097/GIM.0b013e3182217a3a (2011).
Seidman, J. G. & Seidman, C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest 109, 451–455, 10.1172/JCI15043 (2002).
Dang, V. T., Kassahn, K. S., Marcos, A. E. & Ragan, M. A. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 16, 1350–1357, 10.1038/ejhg.2008.111 (2008).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81, 1084–1097, 10.1086/521987 (2007).
Browning, B. L. & Browning, S. R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84, 210–223, 10.1016/j.ajhg.2009.01.005 (2009).
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S. & Goldstein, D. B. Genic intolerance to functional variation and the interpretation of personal genomes. PLos Genet 9, e1003709, 10.1371/journal.pgen.1003709 (2013).
Lionel, A. C. et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet 23, 2752–2768, 10.1093/hmg/ddt669 (2014).
Ching, M. S. et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet 153B, 937–947, 10.1002/ajmg.b.31063 (2010).
Elia, J. et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 44, 78–84, 10.1038/ng.1013 (2012).
Prasad, A. et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2, 1665–1685, 10.1534/g3.112.004689 (2012).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372, 10.1038/nature09146 (2010).
Ullmann, R. et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat 28, 674–682, 10.1002/humu.20546 (2007).
Hannes, F. D. et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet 46, 223–232, 10.1136/jmg.2007.055202 (2009).
Lumaka, A. et al. Variability in expression of a familial 2.79 Mb microdeletion in chromosome 14q22.1-22.2. Am J Med Genet A 158A, 1381–1387, 10.1002/ajmg.a.35353 (2012).
Reis, L. M. et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet 130, 495–504, 10.1007/s00439-011-0968-y (2011).
Bakrania, P. et al. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 82, 304–319, 10.1016/j.ajhg.2007.09.023 (2008).
Zaidi, S. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223, 10.1038/nature12141 (2013).
Kerzendorfer, C. et al. Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum Mol Genet 21, 2181–2193, 10.1093/hmg/dds033 (2012).
Hu, J. et al. CNTN6 copy number variations in 14 patients: a possible candidate gene for neurodevelopmental and neuropsychiatric disorders. J Neurodev Disord 7, 26, 10.1186/s11689-015-9122-9 (2015).
Kashevarova, A. A. et al. Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol Cytogenet 7, 97, 10.1186/s13039-014-0097-0 (2014).
Lerer, I. et al. Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum Mol Genet 14, 3911–3920, 10.1093/hmg/ddi415 (2005).
Vanzo, R. J., Martin, M. M., Sdano, M. R. & South, S. T. Familial KANK1 deletion that does not follow expected imprinting pattern. Eur J Med Genet 56, 256–259, 10.1016/j.ejmg.2013.02.006 (2013).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81300131) to J.W.; Beijing Municipal Natural Science Key Project (7151010), National Key Research Project “973” (2012CB944602), Special Project of Ministry of Health of China (201002006) to Y.J.; the National Basic Research Program of China (2012CB316504) to X-G.Z.; and National Science and Technology Basic Work Project (2014FY130100) to B.W.
Author information
Authors and Affiliations
Contributions
Y.J., X.M., B.W. and X.Y.Z. designed the study; T.J., J.M.W. and X.W. collected samples and analyzed the phenotypes; B.W. coordinated the research; J.W. performed genotyping and validation experiments; D.Z. and X.J.Z. contributed control data; X.Y.Z. analyzed the data and drafted manuscript with the input from all other authors; X.G.Z. and P.C.S. revised paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, B., Ji, T., Zhou, X. et al. CNV analysis in Chinese children of mental retardation highlights a sex differentiation in parental contribution to de novo and inherited mutational burdens. Sci Rep 6, 25954 (2016). https://doi.org/10.1038/srep25954
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25954
This article is cited by
-
The diagnostic yield of intellectual disability: combined whole genome low-coverage sequencing and medical exome sequencing
BMC Medical Genomics (2020)
-
Copy number variations in candidate genomic regions confirm genetic heterogeneity and parental bias in Hirschsprung disease
Orphanet Journal of Rare Diseases (2019)
-
Clinical Application of Chromosome Microarray Analysis in Han Chinese Children with Neurodevelopmental Disorders
Neuroscience Bulletin (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
