
Abstract
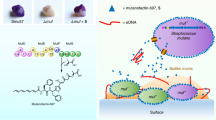
Biofilm formation on biotic or abiotic surfaces has unwanted consequences in medical, clinical and industrial settings. Treatments with antibiotics or biocides are often ineffective in eradicating biofilms. Promising alternatives to conventional agents are biofilm-inhibiting compounds regulating biofilm development without toxicity to growth. Here, we screened a biofilm inhibitor, raffinose, derived from ginger. Raffinose, a galactotrisaccharide, showed efficient biofilm inhibition of Pseudomonas aeruginosa without impairing its growth. Raffinose also affected various phenotypes such as colony morphology, matrix formation and swarming motility. Binding of raffinose to a carbohydrate-binding protein called LecA was the cause of biofilm inhibition and altered phenotypes. Furthermore, raffinose reduced the concentration of the second messenger, cyclic diguanylate (c-di-GMP), by increased activity of a c-di-GMP specific phosphodiesterase. The ability of raffinose to inhibit P. aeruginosa biofilm formation and its molecular mechanism opens new possibilities for pharmacological and industrial applications.
Similar content being viewed by others
Introduction
Most bacteria are able to live in two different states: planktonic and sessile. A biofilm is an assemblage of a single or multiple species that are encapsulated in self-produced extracellular polymeric substances (EPS). Notably, bacterial infections are frequently associated with biofilms1. For example, cystic fibrosis patients infected with Pseudomonas aeruginosa show a progressive loss of pulmonary function due to its biofilm formation in the lung2. Moreover, biofilms tend to foul medical devices, contact lens and artificial implants, which eventually lead to critical medicinal problems3,4. Beyond these, biofilms in the industrial settings (e.g. ship hulls, water pipes and membrane filters) cause loss of performance and increased cost for maintenance and quality control5. Nevertheless, biofilms formed on biotic and abiotic surfaces are notoriously difficult to eradicate, mainly because biofilm cells are insensitive to antimicrobial agents or biocides6. Biofilm cells are known to be 10–1,000 fold more resistant to antimicrobial agents than planktonic cells3.
In contrast to traditional bactericidal or bacteriostatic approaches to inhibit biofilms, a recently developed control method is targeted at interference with biofilm development without affecting bacterial growth7. For example, bacterial cell-to-cell communication called quorum sensing (QS) is mediated by chemical signal molecules8 and QS is known to be associated with biofilm maturation9. These findings are used to develop strategies to disrupt biofilm maturation using structural analogues of QS signal molecules (e.g. furanone, azithromycin and 4-nitro-pyridine-N-oxide) or enzymes degrading QS molecules (e.g. acylase and lactonase). Some molecules have been shown to enhance cell dispersion during biofilm development. Enzyme dispersion B showing EPS degradation has been applied for this purpose10. Nitric oxide and cis-2-decenoic acid, an unsaturated fatty acid, are also reported to induce biofilm dispersion7.
Various natural products are effective in regulating biofilm development. The advantages of using natural products in biofilm inhibition are their lower toxicity and higher specificity compared to synthetic compounds11. Bean sprouts, chamomile, carrots, propolis, yellow peppers, water lilies, harbanero, garlic, fruit of a south-east Asian tree (Lagerstroemia speciosa), peas, medicinal plants from southern Florida (e.g. Conocarpus erectus), grapefruit, etc. have been shown to possess anti-QS activity12,13,14,15 and a few (e.g. garlic extract) show antibiofilm performance. We previously demonstrated that ginger extract had a capability to reduce biofilm formation for various bacteria including P. aeruginosa by reducing cellular cyclic diguanylate (c-di-GMP)16, but the active compound(s) associated with the finding has not been identified.
The goal of this study was to screen novel compound(s) of ginger extract that can inhibit P. aeruginosa biofilm formation and explore the biofilm inhibition mechanism. We identified raffinose, a galactotrisaccharide, which was effective in reducing biofilm formation by attaching to a carbohydrate-binding protein and by controlling c-di-GMP that affects the initial and probably, the dispersal stages of biofilm development.
Results
Screening of biofilm inhibitors in ginger
Five putative ingredients frequently detected in ginger extract (6-gingerol, farnesol, L-ascorbic acid, myricetin and raffinose, Fig. 1a) were tested for their potential to inhibit biofilm formation. Those ingredients were selected because they were identified in the ginger extract showing antibiofilm effect (data not shown). The degree of inhibition of biofilm formation by these candidates was evaluated using a static biofilm assay based on P. aeruginosa PA14 as a model bacterium in 96-well microtiter plates. Furanone C-30, a well reported biofilm inhibitor showing significant antibiofilm effect at 10 μM via QS inhibition17, was used as the positive control in this experiment. Figure 1b shows that 6-gingerol and raffinose reduced biofilm formation by 39% and 43%, respectively, while farnesol, L-ascorbic acid and myricetin did not significantly reduce biofilm formation. The biofilm inhibiting effects of 6-gingerol and raffinose were comparable to furanone C-30 which showed 46% inhibition. Since the effect of 6-gingerol on biofilm inhibition has been previously discussed by us18, in this study, we report on the ability of raffinose to inhibit biofilm formation and the associated inhibitory mechanisms.

Screening of compounds from ginger inhibiting biofilm formation.
(a) Putative ginger compounds used in this study. (b) Quantification of P. aeruginosa biofilm formation with the putative ginger compounds and furanone C-30 (a positive control) for 24 h in the wells of microtiter plates. The concentration of each compound was 10 μM. At least two independent experiments were conducted using 8 wells of microtiter plates, error bars indicate one standard deviations. **P < 0.0005 versus negative control.
Effects of raffinose on growth and biofilm formation
The growth of P. aeruginosa was measured for batch cultures with 0–100 μM raffinose. The growth curves for different raffinose concentrations were not significantly different in lag, exponential and stationary phases during 14 h of incubation (Fig. 2a). The results suggested that the growth of P. aeruginosa was unaffected by the addition of raffinose up to 100 μM.

Effects of raffinose on P. aeruginosa phenotypes.
(a) Growth at different concentrations of raffinose (0, 1, 10 and 100 μM) for 14 h incubation in flasks. Error bars indicate the standard deviations of 3 independent cultures. (b) 3-dimensional analyses of CLSM images of P. aeruginosa biofilm untreated (left) and with 10 μM raffinose treatment (right), on the glass slides. The biofilms were then stained using three fluorophores specific for nucleic acids (DAPI; blue), carbohydrates (ConA; green) and proteins (Ruby; red), before being subject to confocal laser scanning microscopy (CLSM) imaging. Arrows indicate hollow structure in the biofilm. (c) Colony morphology of P. aeruginosa without (left) and with 10 μM raffinose treatment (right) on Congo red agar plates. (d) Top images show swarming motility of P. aeruginosa cultured with different concentrations of raffinose (0, 1, 10, 100 and 1,000 μM) for 24 h on 0.5% BM-2 plate. Scale bars indicate 1 cm. Bottom graph shows the length of dendrites generated by swarming motility for each plate. **P < 0.0005 versus the control.
A static biofilm assay was performed with different concentrations of raffinose (0–1,000 μM). Biofilm formation was reduced gradually (17–58%) with increasing raffinose concentration (Fig. S1), demonstrating that raffinose inhibited biofilm formation in a concentration-dependent manner. The biofilm inhibition capacity of the raffinose’s hydrolyzed products of α-galactosidase (D-galactose and sucrose) was evaluated. α-galactosidase is widely distributed in microorganisms, plants and animals19. D-galactose and sucrose did not reduce or increase the biofilm formation up to 1,000 μM concentration (Fig. S1). There were no statistical differences in biofilm formation between control and D-galactose or sucrose treatment (P > 0.05).
The effect of raffinose on biofilm formation was also tested in a flow reactor where growth medium with or without 10 μM raffinose was continuously dripped on glass slides for 24 hours. Figure 2b shows typical biofilms. The biofilm formed with raffinose treatment showed relatively lower thickness (8.4 μm) and smaller volume (4.8 μm3/μm2) than that without raffinose treatment (35.2 μm and 10.5 μm3/μm2, respectively). Although the biofilm formed without raffinose treatment demonstrated a typical P. aeruginosa mushroom morphology, the biofilm formed with 10 μM raffinose treatment was relatively flat. Interestingly, the biofilm with 10 μM raffinose treatment showed hollow structures (1,210 ± 170 per mm2) (see arrows in Fig. 2b) which were not evident in the biofilm without raffinose treatment.
Effect of raffinose on the other phenotypes
Congo-red containing plates were used to observe P. aeruginosa colony morphology and to evaluate the effect of raffinose on exopolysaccharide production. Congo red is a pigment that binds to the exopolysaccharide matrix of bacterial biofilms and leads to rugose colony morphology in P. aeruginosa20. As shown in Fig. 2c, a typical pink rugose morphology was evident for the colony without raffinose treatment, while the colony with raffinose treatment (10 μM) showed pink smooth morphology. This result is an indication of a reduction of exopolysaccharide production by raffinose treatment in P. aeruginosa.
The effect of raffinose on exopolysaccharide production was further evaluated by directly measuring total carbohydrate content in EPS from both planktonic and biofilm cells grown with 0–1,000 μM raffinose in growth medium. Exoprotein (a component of the biofilm matrix) production was also analyzed. Both planktonic and biofilm cells treated with raffinose significantly reduced the total carbohydrate and protein content of the EPS (Fig. S2). Total carbohydrate reduced by 21–53% and 22–48% (planktonic and biofilm cells, respectively), while total protein was reduced by 1–41% and 9–42% (planktonic and biofilm cells, respectively), in a dose-dependent manner, when treated with raffinose.
Biofilm formation in P. aeruginosa is reported to be inversely regulated by swarming motility21. Effect of raffinose (0–1,000 μM) on swarming motility was evaluated by growing P. aeruginosa cells on 0.5% soft agar plates and measuring the length of dendrites from the center (Fig. 2d). The dendrite lengths of the cells grown with raffinose were 1.6–2.4 fold longer than those grown without raffinose treatment and the length increased with increasing raffinose concentration. This suggests that raffinose promoted swarming motility on the soft agar plates. However, there were no changes in swimming or twitching motility by raffinose treatment (Fig. S3).
Binding of raffinose to LecA
P. aeruginosa produces two carbohydrate-binding proteins (lectins): LecA (PA-IL) and LecB (PA-IIL), which show a specificity for D-galactose and L-fucose, respectively22,23. Because LecA demonstrated high affinity to D-galactose and its derivatives22,24 as well as involvement in P. aeruginosa biofilm development25, it is reasonable to hypothesize that raffinose, a D-galactose derivative, may bind to LecA and thereby to suggest a mechanism for affecting P. aeruginosa biofilm development. To test this hypothesis, we initially evaluated the binding of raffinose to LecA based on 1D relaxation-edited nuclear magnetic resonance (NMR)-binding experiments26,27 using raffinose and LecA (Fig. 3a). NMR signals of 0.1 mM raffinose were monitored in the absence and presence of 50 μM LecA in buffer solution. In addition, 1 mM galactose was added to monitor the change of raffinose signals by competition at the same binding site. In the absence of LecA protein (third row spectra, Fig. 3a), indicative peaks for H1 and H2 signals of raffinose at 5.33 and 4.13 ppm were seen. These broadened and weakened in the presence of LecA protein (second row spectra, Fig. 3a) and the signal intensities recovered with the addition of a 10-fold excess of D-galactose (first row spectra, Fig. 3a). The results indicated that raffinose binds to LecA and its binding site overlaps with the galactose-binding site. Moreover, the signals for the protons at C-1 and C-6 of the galactose at 5.17 and 3.40 ppm in 50 mM LecA solution were also recovered by the addition of 1 mM raffinose (Fig. S4). Conversely, the galactose (0.1 mM) signals in LecA solution were also enhanced with treatment of a 10-fold excess of raffinose (Fig. S4). These results suggest that raffinose and galactose compete for the same site of the LecA protein.

Interactions of raffinose and P. aeruginosa LecA protein.
(a) Binding of raffinose to LecA monitored by CPMG NMR spectroscopy. First row: NMR spectrum of raffinose (0.1 mM) and LecA (50 μM) with the addition of galactose (1 mM) Second row: NMR spectrum of raffinose (0.1 mM) in the presence of LecA (50 μM). Third row: NMR spectrum of raffinose (0.1 mM) in the absence of LecA. Fourth row: NMR spectrum of LecA only. (b) In silico analysis of raffinose and LecA. Hydrogen-bonding interactions between LecA and the best-docked pose of raffinose (left). The docked raffinose on the surface of LecA (right). (c) Biofilm formation at different concentrations of raffinose (0, 1, 10 and 100 μM) in ΔlecA mutant for 24 h in microtiter plates. (d) Biofilm formation at different concentrations of raffinose (0, 1, 10, 100 and 1,000 μM) in ΔlecA-pUCP18-LecA for 24 h in microtiter plates. At least two independent experiments were conducted using 10 wells of microtiter plates, error bars indicate one standard deviations. **P < 0.0005 versus the control. *P < 0.005 versus the control.
To confirm the direct binding of raffinose to LecA, the binding affinities of raffinose, D-galactose and sucrose to LecA were measured by isothermal titration calorimetry. Raffinose showed a 1.5-fold increase in binding (Kd = 32 μM) to LecA as compared with D-galactose (Kd = 47 μM), while sucrose which lacks the galactose moiety did not bind to LecA (Fig. S5), indicating that the galactose moiety of raffinose is a determinant of LecA binding. Binding enthalpies (ΔH), entropies (ΔS) and free energies (ΔG) of raffinose and D-galactose are summarized in Table S1.
To better understand the binding of raffinose to LecA, in silico docking studies of raffinose were performed using the X-ray structure of LecA in complex with melibiose (PDB: 4AL9). Most of the docked raffinose poses showed common binding patterns in which the galactose moiety was projected toward the calcium ion of the LecA active site, while the glucose and fructose moieties were oriented toward the surface and the loop region of LecA. The best-docked pose of raffinose is shown in Fig. 3b. The galactose moiety of the raffinose interacted strongly with Tyr 36, His 50, Pro 51, Asp 100, Val 101 and Thr 104. The glucose and fructose regions also interacted by hydrogen-bonding with Gln 53, Gly 37 and Asn 107, respectively.
Binding of raffinose to LecA and its relevance to biofilm inhibition were further verified using P. aeruginosa strains, one that cannot produce LecA and one that overproduces LecA. As shown in Fig. 3c, the lecA knockout mutant (a strain not producing LecA) demonstrated reduced biofilm formation and, furthermore, the biofilm formation did not decrease with increases in the raffinose concentration (P > 0.05). On the contrary, as shown in Fig. 3d, the lecA knockout mutant carrying pUCP18-LecA (a LecA overproducing strain) formed a biofilm similar to the wild-type strain carrying an empty vector (pUCP18). Additionally, the lecA knockout mutant carrying pUCP18-LecA showed a reduction in biofilm formation in the presence of raffinose, in a concentration-dependent manner (0–1,000 μM) (Fig. 3d). These results confirm that LecA is essential to form a biofilm and suggested that raffinose binding to LecA is required for biofilm inhibition.
Decrease in cellular cyclic diguanylate (c-di-GMP) production
Cellular c-di-GMP plays important roles in the molecular determination between planktonic and sessile lifestyles, the regulation of motility and the induction of virulence factor production28. In our previous study, we reported that ginger extract reduced the cellular c-di-GMP levels in both planktonic and biofilm cells of P. aeruginosa16. Accordingly, we evaluated the effect of raffinose on cellular c-di-GMP in both planktonic and biofilm cells. In general, biofilm cells exhibited higher c-di-GMP levels than planktonic cells (Fig. 4a), in agreement with other studies29. Furthermore, treatment with 10 μM raffinose reduced c-di-GMP levels in both planktonic (47%) and biofilm cells (59%).

Effect of raffinose on cellular c-di-GMP levels.
(a) Cellular c-di-GMP levels in P. aeruginosa planktonic and biofilm cells cultured without and with 10 μM raffinose for 24 h. Error bars indicate the standard deviations of 3 independent measurements. **P < 0.005 versus the control. *P < 0.05 versus the control. (b) Quantification of biofilm formation in P. aeruginosa wspF mutant with different concentrations (0, 1, 10 and 100 μM) of raffinose for 24 h in the wells of microtiter plates. At least two independent experiments were conducted using 8 wells of microtiter plates, error bars indicate one standard deviations. **P < 0.00005 versus the control. *P < 0.005 versus the control. (c) Swarming motility of wspF mutant cultured without (left) and with 10 μM raffinose (right) for 24 h on 0.5% BM-2 plate. (d) Degradation of the PDE-specific substrate bis-pNPP by P. aeruginosa cells cultured with raffinose (0, 1, 10 and 100 μM). **P < 0.000005 versus the control. *P < 0.00005 versus the control.
In addition, to verify the relevance of raffinose treatment to cellular c-di-GMP regulation, a P. aeruginosa mutant which can overproduce c-di-GMP (ΔwspF) was used. The wspF gene in P. aeruginosa is reported to encode a methylesterase30. The methylesterase is presumed to control the activity of WspR protein which contains a CheY-like receiver and a diguanylate cyclase (GGDEF) domain30. Because ΔwspF shows constitutive activation of the WspR protein, ΔwspF leads to elevation of cellular c-di-GMP level and EPS production31. As shown in Fig. 4bΔwspF induced around a 2-fold increase in biofilm formation compared to the wild-type strain. However, biofilm formation of ΔwspF was reduced 12–26% by raffinose treatment (0–100 μM), although the reduction of biofilm formation did not reach that of the wild type strain. This might be due to the fact that raffinose treatment was not sufficient to reduce cellular c-di-GMP level of ΔwspF mutant to the level of the wild type strain (Fig. S6). Interestingly, raffinose enhanced the swarming motility of ΔwspF. As shown in Fig. 4cΔwspF showed limited swarming motility on a 0.5% soft agar plate, but ΔwspF grown with 10 μM raffinose clearly showed lengthened dendrites.
The above result appears to be related to a reduction of cellular c-di-GMP levels. One of the routes in reducing cellular c-di-GMP level is enhanced c-di-GMP specific phosphodiesterase (PDE) activity of P. aeruginosa. This possibility was tested by measuring the PDE activity, by analyzing the degradation of a PDE-specific substrate, bis-p-nitrophenol phosphate (bis-pNPP), in P. aeruginosa grown with raffinose. Figure 4d shows that P. aeruginosa grown with raffinose increased the activity of bis-pNPP degradation 1.1–2.3 fold, as compared with the same strain grown without raffinose. This result demonstrates that raffinose may reduce the P. aeruginosa cellular c-di-GMP levels via induction of PDE activity.
Discussion
Besides biofilm reduction, raffinose altered other phenotypes of P. aeruginosa, such as inhibition of rugose colony formation in Congo-red plate (Fig. 2c), reduction of EPS production (Fig. S2) and increasing swarming motility (Fig. 2d). These altered phenotypes are typically observed when cellular c-di-GMP levels decrease 16,32. Therefore, it is reasonable to infer that raffinose triggered various phenotypic alternations by reducing cellular c-di-GMP. This speculation is confirmed by the experiments using a P. aeruginosa mutant that overproduces cellular c-di-GMP (ΔwspF): raffinose reduced biofilm formation (Fig. 4b) concomitant with recovery of swarming motility (Fig. 2d).
C-di-GMP levels are regulated by two enzymes acting oppositely. C-di-GMP is produced from two GTPs by diguanylate cyclases (DGCs) and is degraded by phosphodiesterases (PDEs) into two GTPs via pGpG33. Two routes are possible for decreasing cellular c-di-GMP levels: decreasing the activity of DGCs with GGDEF domain or increasing the activity of PDEs with EAL and/or HD-GYP domains. The experimental results show that increasing concentrations of raffinose increase the PDE activity of P. aeruginosa (Fig. 4d), suggesting that the latter route was the mechanism decreasing c-di-GMP levels by raffinose. This hypothesis is supported by the study of Chung et al.31 who increased PDE activity by cloning pvrR, a PDE gene, in a plasmid vector using the P. aeruginosa ΔwspF mutant. The transformed ΔwspF mutant showed increased PDE activity along with decreased c-di-GMP levels and enhanced swarming motility. In explaining the decrease in c-di-GMP level by raffinose treatment, the possibility of a decrease in the activity of DGCs could not be excluded because of the lack of supporting evidence leaving the mechanism open.
On the other hand, a reduced level of cellular c-di-GMP is reported to be associated with the motile mode of growth and dispersal as well as reduced biofilm formation32. Biofilm dispersal is initiated by localized cell death and lysis, to provide nutrients for escaping cells 7. This entails hollowing of biofilm microcolonies34. In this study, hollow structures in the biofilm treated with raffinose (Fig. 2b) suggest that raffinose triggers biofilm dispersion as well as reduction in biofilm formation by reducing cellular c-di-GMP level. This speculation was further evaluated by measuring released planktonic cells from P. aeruginosa biofilms formed on glass slides. The number of released cells increased by 1.8–3.6 fold when treated with 100 μM raffinose, as compared with untreated cells (Fig. S7). Taken together, our data demonstrating the regulation of cellular c-di-GMP levels by raffinose have enhanced our understanding about galactosides binding to LecA and their inhibition of P. aeruginosa biofilm formation (i.e., LecA-ligand competitive inhibition was not the sole mechanism for biofilm inhibition). Nevertheless, it is not clear whether LecA-raffinose binding caused the decrease in c-di-GMP or raffinose decreased c-di-GMP independent from LecA-raffinose binding, which will be investigated in the near future.
It is also important to note that LecA production is related to biofilm formation. Diggle et al.25 observed lecA gene expression in a P. aeruginosa biofilm and showed enhanced biofilm formation by a LecA-overproducing strain and reduction by a lecA mutant. We investigated a possible interference of lecA gene expression by raffinose treatment using RT-qPCR analyses (Fig. S8). The expression of the lecA gene was reduced by 50% in the wild type P. aeruginosa by raffinose treatment. Interestingly, the P. aeruginosa mutant overproducing cellular c-di-GMP (ΔwspF) demonstrated a 1.5-fold increase in lecA gene expression over the wild-type strain, with a 30% decrease in lecA gene expression after raffinose treatment. Furthermore, the P. aeruginosa lecA knockout mutant (ΔlecA) showed a decrease in cellular c-di-GMP levels and a 1.5-fold increase in the activity of c-di-GMP specific PDE as compared with the wild-type strain. These results imply that binding of raffinose to LecA reduced lecA gene expression and in turn, decreased cellular c-di-GMP levels, which resulted in biofilm inhibition. Nevertheless, detailed molecular mechanisms should be investigated for a better understanding about LecA-mediated biofilm formation.
Raffinose is a common galactooligosaccharide found in various vegetables (e.g. beans, cabbage and broccoli) and plant seeds35, as well as in ginger. It is not clear why some plants produce bacterial biofilm inhibitors such as raffinose. It would be interesting to study whether such biofilm inhibitors are produced accidentally or via evolutionary processes. Whatever the reason, the production of biofilm inhibitors appears to provide survival benefits for the plants. For example, Austrian red algae are reported to produce halogenated furanones effective in inhibiting biofilm formation17, which avoids colonization by microorganisms that, in turn, block light to the plant. Biofilm inhibitors could be potentially applied in various medical and industrial settings. Increasing numbers of antibiotic-resistant pathogenic bacteria cause difficulties in antibiotic therapy. For example, more than 95% P. aeruginosa strains are reported to be resistant to gentamicin, carbenicillin, co-trimoxazol, ceftizoxime and tetracycline 3. Biofilm inhibitors that do not affect bacterial growth are an emerging alternative to antibiotics in treating infectious diseases involving biofilm formation. They can also reduce biofouling problems, replacing biocides used in membrane filters for water treatment, water distribution pipes, ship hulls, laundry machines, humidifiers, dishwashers, etc. We believe that the findings from this study will be helpful to design more potent LecA-mediated biofilm inhibitors for combating unwanted biofilms.
Experimental Procedures
Bacterial strains and chemicals
P. aeruginosa PA14 was used as a model biofilm-forming bacterium. wspF or lecA knockout mutant of P. aeruginosa36 was used to test whether raffinose was also effective for bacteria that overproduce cellular c-di-GMP or could not produce LecA. Ginger’s ingredients, such as 6-gingerol, farnesol, L-ascorbic acid, myricetin and D-(+)-raffinose pentahydrate, were purchased from Sigma Aldrich (St. Louis, MO, USA) and were dissolved in dimethyl sulfoxide (Carl Roth, Karlsruhe, Germany).
Growth inhibitory test
Overnight culture of P. aeruginosa was grown in AB medium (300 mM NaCl, 50 mM MgSO4, 0.2% vitamin-free casamino acids, 10 mM potassium phosphate, 1 mM L-arginine and 1% glucose, pH 7.5)37 with varying concentrations of raffinose (0, 1, 10, or 100 μM) using a shaking incubator maintained at 37 °C, 250 rpm, for 14 h. Bacterial growth was evaluated by measuring optical density (OD) at 595 nm using a UVmini-1240 spectrophotometer (Shimadzu, Kyoto, Japan) in triplicate samples collected hourly.
Static biofilm formation assay
Overnight cultures of test bacteria (OD at 595 nm = 1.5) were diluted in fresh AB medium (1:20) and aliquoted into wells of a TPP® 96-well polystyrene microtiter plates (Sigma Aldrich). After forming biofilms in the wells at 37 °C without agitation for 24 h, OD of suspended cultures was measured at 595 nm using an iMark microplate absorbance reader (BioRad, Richmond, CA, USA), after which the suspended cultures were discarded. The plate was washed with phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4, pH 7.2) to remove remaining suspended cells. The biofilms were stained with 1.0% crystal violet for 30 min and then washed with autoclaved deionized water to remove remaining unbound dye. The crystal violet bound to biofilms was eluted by 100% ethanol and OD of the eluent was measured at 545 nm. The biofilms were quantified as the amount of crystal violet bound to biofilms (OD at 545 nm), normalized by the amount of suspended cells (OD at 595 nm).
Biofilm formation assay using a drip-flow reactor
One ml of overnight culture of P. aeruginosa (OD at 595 nm = 1.5) was diluted in 19 ml fresh AB medium (1:20) in a petri dish. A glass slide was dipped into the petri dish containing the dilution and incubated at 37 °C for 24 h to form a biofilm on the glass slide. The glass slide covered with biofilm cells was inserted into the DFR-110 drip-flow biofilm reactor (BioSurface Technologies, Bozeman, MT, USA). Fresh AB medium without or with raffinose (10 μM) was continuously fed into the reactor using a peristaltic pump (Masterflex C/L tubing pumps, Cole-Parmer, Vernon Hills, IL, USA) at 20 ml∙h−1 and operated in an incubator at 37 °C for 24 h. Then, the glass slide was carefully washed with PBS (pH 7.2). The attached biofilm cells were stained with DAPI (Carl Roth), FITC-labeled ConA (Sigma Aldrich) and SYPRO Ruby (Invitrogen, Carlsbad, CA, USA) sequentially for 20 min each. The stained biofilm cells were washed twice with PBS (pH 7.2) after each staining step. DAPI, ConA and Ruby solutions were targeted to stain DNA, carbohydrate and protein, respectively. The biofilm cells were then observed using CLSM (Carl Zeiss LSM700, Jena, Germany). Confocal images were taken with a 40× objective lens (C-Apochromat 40×/1.20 W Korr M27, Carl Zeiss) and the images of blue (DAPI), green (ConA) and red (RUBY) fluorescence were observed simultaneously with the Z-stack mode using the Zen 2011 program (Carl Zeiss). Detailed methods about the fluorescent filters are described in a previous study16. Bio-volume (μm3/μm2), biofilm average thickness (μm) and roughness coefficient were measured by Comstat2 in the ImageJ program38.
Construction of a pUCP-LecA expression plasmid
LecA protein was ectopically expressed using a multicopy plasmid, pUCP1839. The sequence of the lecA gene region was amplified by PCR using Pfu polymerase (PfuUltra II Fusion HS DNA Polymerase, Agilent Technologies, Santa Clara, CA, USA) with the following oligonucleotides: 5′- ATGGCCGGATCCCGTTGCTGTGCTTTGCTG (BamHI restriction site is underlined) and 5′- CAGAAGCTTCGGCCACACGGGCAACTTCAC (HindIII restriction site is underlined). The amplified 540-bp fragment and plasmid pUCP18 were digested with both BamHI and HindIII endonucleases, gel purified and ligated using T4 ligase (Promega, Madison, WI, USA). The ligated plasmid was transformed into Escherichia coli (XL10-Gold Ultracompetent cells, Agilent Technologies), as per the manufacturer’s protocol.
Transformation of P. aeruginosa with pUCP-LecA expression plasmids
The preparation of competent cells for P. aeruginosa wild type and ΔlecA mutant and the transformation of P. aeruginosa using an electroporation method, were based on a previous study40. 1.5 mL of overnight culture of P. aeruginosa wild type and ΔlecA cells grown in LB medium, were harvested by centrifugation at room temperature for 3 min at 16,000× g. The cell pellet was washed twice using 1 mL of 300 mM sucrose and then the cell pellet was resuspended in 100 μL of 300 mM sucrose. For electroporation, purified plasmid DNA (pUCP18 and pUCP-LecA) using the Qiagen plasmid prep kit (Qiagen, Chatsworth, CA, USA) was mixed with 100 μL of P. aeruginosa competent cells. The purified plasmid DNA pUCP18 (empty vector) was mixed with wild type and ΔlecA competent cells, while the purified pUCP-LecA was mixed with ΔlecA competent cell. The mixture was transferred to a 0.2 cm gap width electroporation cuvette (BIO-RAD, Hercules, CA, USA) and pulsed (25 μF, 200 Ω, 2.5 kV) using the Gene Pulser XcellTM electroporation system (BIO-RAD). The electroporated cells (100 μL) were transferred to a 15-ml conical tube, 1 mL of fresh LB medium was added and the mixture was shaken at 250 rpm at 37 °C for 1 h. The cells were spread on an LB plate with 100 μg/mL carbenicillin and incubated at 37 °C for 14 h. Colonies were picked and grown in LB broth. Plasmid DNA was isolated, treated with BamHI and HindIII endonucleases and separated using gel electrophoresis, to confirm insertion of the lecA gene into the plasmid.
Colony morphology assay
Overnight culture of P. aeruginosa was diluted in fresh T-broth medium (10 g/L tryptone) with raffinose (0 or 10 μM) and the dilution was incubated using a shaking incubator at 37 °C, 250 rpm for 14 h (OD at 595 nm = 1.0). 2 μL of the culture was spotted on a Congo-red plate16 and incubated at room temperature for 3–7 days.
EPS analysis
EPS of planktonic and biofilm cells was analyzed using a modified sonication method16. Planktonic cells were prepared by diluting an overnight culture of P. aeruginosa in fresh AB medium (1:20) with 0–1,000 μM raffinose and incubated on a shaking incubator at 37 °C, 250 rpm, overnight. Biofilm cells were prepared using borosilicate bottles by diluting overnight cultures of P. aeruginosa in fresh AB medium (1:20) with raffinose (0–1,000 μM) and by incubating the dilution at 37 °C for 24 h without agitation. OD of the suspended cells from both planktonic and biofilm cultures were measured at 595 nm using a spectrophotometer. The biofilm cells were resuspended in 3 mL 0.01 M KCl by vortexing. Planktonic and biofilm cultures were centrifuged at 8,000× g and the harvested cells were resuspended in 10 mL and 3 mL 0.01 M KCl, respectively. The resuspended cells were disrupted using a sonicator (VCX 750, SONICS, Newtown, CT, USA) for 4 cycles of 5 s of operation and 5 s of pause at a power level of 3.5 Hz. The sonicated cells were centrifuged at 4,000× g and the supernatants were filtered through 0.22-μm Millex filter (Car Roth). Filtered supernatants were used for the quantification of protein and carbohydrate. Protein analysis was performed by the Lowry method in 96-well microtiter plate: 40 μL filtrate and 200 μL Lowry reagent (L3540, Sigma Aldrich) were mixed in a microtiter plate and the mixture was incubated for 10 min at room temperature, after which 20 μL Folin-Ciocalteu reagent (F9252, Sigma Aldrich) was added to the mixture and the mixture was incubated for 30 min. Protein quantity was measured by measuring OD at 750 nm using the iMark microplate reader. Carbohydrate was analyzed by phenol-sulfuric acid method in a 96-well microtiter plate: 15 μL filtrate and 150 μL sulfuric acid were mixed in a microtiter plate and the mixture was incubated for 30 min at room temperature: 30 μL 5% phenol was added to the mixture and the mixture was incubated at 90 °C in a water bath. Carbohydrate quantity in the mixture was measured by measuring OD at 490 nm using the iMark microplate reader. Protein (OD at 750 nm) and carbohydrate (OD at 490 nm) quantity were normalized by suspended cells (OD at 595 nm).
Swarming motility assay
Swarming motility was assayed based on a previous method16. 10 μL overnight cultures of P. aeruginosa with raffinose (0–1,000 μM) was spotted on a BM-2 plate (62 mM potassium phosphate [pH 7.0], 2 mM MgSO4, 10 mM FeSO4, 1% casamino acid, 0.4% glucose and 0.5% Bacto agar) and incubated at 37 °C for 24 h. The degree of swarming motility was evaluated by measuring average length of dendrites.
RT-qPCR analysis
RT-qPCR was performed to quantify and compare the levels of P. aeruginosa lecA gene expression. The lecA and housekeeping gene proC primer sets were designed using Primer 3 version 0.4.0 (http://frodo.wi.mit.edu/) (Table S2). Total RNA extraction was performed from P. aeruginosa biofilm cells using TRI REAGENT (Molecular Research Center, OH, USA), following the manufacturer’s instruction. RT-qPCR was performed using the Bio-Rad CFX-96 real time system (Bio-Rad, Hercules, CA, USA). The reaction mixture consisted of the following reagents: 10 μL SYBR Premix Ex TaqTM (Takara, Shiga, Japan), 0.8 μL each of the forward and reverse primers (10 μM), 0.4 μL of 50 X ROXTM Reference Dye I, 2 μL extracted total RNA (100 ng/ml) and RNase free water, to generate a 20 μL final volume. Detailed protocols about RNA extraction and RT-qPCR methods are described in our previous study18.
Biofilm dispersion assay
An overnight culture of P. aeruginosa was diluted in fresh AB medium (OD at 595 nm = 0.05). A polystyrene petri dish was filled with 20 mL of the dilution. A sterilized glass slide was dipped into the petri dish and incubated at 37 °C for 24 h to develop biofilms on the glass slide. The slide was then gently rinsed using PBS (pH 7.2) and dipped into the petri dish containing 20 mL of AB medium with or without 100 μM raffinose. The petri dish was incubated for 2 h and 4 h at 37 °C, respectively. 100 μL of the culture was serially diluted to 10−5 in PBS (pH 7.2). For counting colony forming units, 100 μL of the final dilution was spread on LB agar plates and incubated overnight at 37 °C.
Statistical analysis
P-values for testing statistical differences between measurements were estimated by a student’s t-test (Excel software, Microsoft, Redmond, WA, USA).
NMR analysis
All spectra were measured at 298 K on a Bruker Avance 600 MHz spectrometer (Billerica, MI, USA). The 1D relaxation-edited NMR experiments utilized CPMG pulse sequences with excitation sculpting for water suppression. The pre-acquisition delay was 1.2 s and total spin lock time was 400 ms. The delay for spin echo between successive 180 pulses in CPMG experiment was 2 ms. The data were collected with a sweep width of 16 ppm (9,600 Hz) and 128 scans. Data processing was performed with Topspin 3.0 (Bruker) software with a sine square window function over 16384 complex data points before Fourier transformation. Samples contained 50 μM LecA protein (L9895, Sigma Aldrich) and 0.1 mM of raffinose or galactose in a 10% D2O buffered solution. For the displacement experiment, 1 mM of competitor (raffinose vs. galactose) was added to each sample.
In silico docking studies
Ligand preparation and optimization: Raffinose was generated as 2D and 3D structures by ChemBioDraw (ver. 11.0.1) and Chem3D Pro (ver. 11.0.1), respectively, being saved in .mol file format. The process of ligand preparation and optimization was performed by the ‘Sanitize’ preparation protocol in SYBYL-X 2.1.1 (Tripos Inc., St Louis, MI, USA).
Protein preparation: The protein structures of lectin PA-IL from P. aeruginosa in PDB format were downloaded from the RCSB protein data bank (PDB ID: 4AL9). A structure preparation tool in SYBYL-X 2.1.1 was employed for protein preparation. The crystal ligand melibiose, water molecules, Ca2+ ion and other chains except chain F were removed from the protein-ligand complexes for docking. Conflicted side-chains of amino acid residues were fixed. Hydrogen atoms were added under the application of TRIPOS Force Field as a default setting. A minimization process was performed by the POWELL method, the initial optimization option was changed to None from a default setting SIMPLEX, and the termination gradient and max iteration were set to 0.5 kcal/(mol*Å) and 1000 times, respectively. After protein minimization, processed LecA protein structure was saved as .mol2 file format.
Docking and scoring function studies: The docking study of raffinose was performed by Surflex-Dock GeomX module in SYBYL-X 2.1.1. Docking was guided by the Surflex-Dock protomol. Protomol, containing information on the binding site of Lectin PA-IL for Surflex-Dock, was defined according to the location information of the original crystal ligand melibiose. Two factors related with the generation of Protomol are Bloat(Å) and Threshold which were set to 0.5 and 0, respectively. The maximum number of generated poses per each ligand and the minimum RMSD between final poses were set to 20 and 0.05, respectively. Other parameters were applied with its default settings in all runs.
Cellular c-di-GMP concentration analysis
For the analysis of c-di-GMP in planktonic cells, 4.9 mL of 37% formaldehyde (Carl Roth) was added to one liter of overnight cultures of P. aeruginosa (OD at 595 nm = 1.5) grown without or with raffinose (10 μM). The cultures were centrifuged at 8,000× g for 10 min and the harvested cells were resuspended in 40 mL PBS (pH 7.2) containing 0.18% formaldehyde. For the analysis of c-di-GMP in biofilm cells, initially, an overnight culture of P. aeruginosa was diluted in fresh AB medium (1:20). The dilution was aliquoted into borosilicate bottles without or with raffinose (10 μM) and incubated at 37 °C for 24 h without agitation. After decanting the suspended cells, the biofilm cells were washed twice with PBS (pH 7.2). Biofilm cells were resuspended by vortexing in 3 mL PBS containing 0.18% formaldehyde. Planktonic and biofilm cells were centrifuged at 8,000× g for 10 min and the harvested cells were resuspended in 5 mL deionized water. The resuspensions were boiled for 10 min and cooled in ice for 10 min. The cooled resuspensions were mixed with 100% ethanol (9.3 mL) for extraction of nucleotides and then the mixtures were centrifuged at 8,000× g for 10 min at 4 °C. The supernatants were filtered through a 0.22-μm filter (Car Roth, Karlsruhe, Germany) for analyzing cellular c-di-GMP. The harvested cells were used to analyze total protein using the Lowry method described above. The filtrated supernatants were lyophilized using Savant SpeedVac (SC201A, Thermo Scientific, San Jose, CA, USA) and then the lyophilized cells were dissolved in 200 μL buffer A (100 mM KH2PO4, 4 mM tetrabutyl ammonium hydrogen sulfate, pH 5.9). The dissolved solutions were filtered through 0.45-μm filter (Car Roth). Filtered solutions (20 μL) were analyzed using high-performance liquid chromatography (HPLC) (Hewlett Packard 1100 Series, Agilent, Santa Clara, CA, USA) with an ultraviolet detector (254 nm) and a Kinetex C-18 column (4.6 × 250 mm, 5 u, Phenomenex, Torrance, CA, USA) with mixing buffer (75% of buffer A and 25% of pure methanol) as the mobile phase at the Korea Basic Science Institute (Seoul, Korea). C-di-GMP separation was achieved using a gradient HPLC method as follow: 0–4 min; 50:50, 5–20 min; 0:100 and 21–30 min; 50:50 (operational time (min); ratio of buffer A and methanol) at a flow rate of 0.8 mL∙min-1.
In vitro PDE activity assay
In vitro PDE activity was assayed based on a previous study29. Overnight cultures of P. aeruginosa were diluted in fresh AB medium (1:100), treated with varying raffinose concentrations (0–1,000 μM) and incubated in a shaking incubator at 37 °C, 250 rpm for 14 h. The suspended cultures were centrifuged at 6,000× g for 10 min at 4 °C and the harvested cells were resuspended in 1 ml of PDE activity assay buffer (50 mM NaCl, 50 mM Tris base [pH 8.1], 1 mM MnCl2 and 5 mM bis-pNPP). Bis-pNPP (N3002, Sigma Aldrich) used as a synthetic substrate of c-di-GMP specific PDE. The resuspended cells were lysed using CelLytic Express (C5491, Sigma Aldrich). For the PDE reaction, the mixture was incubated at 37 °C for 2 h in a shaking incubator at 60 rpm. The OD of the mixture was then measured at 410 nm using an iMark microplate absorbance reader for analyzing bis-pNPP degradation into p-nitrophenol. PDE activity (OD at 410 nm) was normalized by total proteins measured by the Lowry method, as described above.
Additional Information
How to cite this article: Kim, H.-S. et al. Raffinose, a plant galactoside, inhibits Pseudomonas aeruginosa biofilm formation via binding to LecA and decreasing cellular cyclic diguanylate levels. Sci. Rep. 6, 25318; doi: 10.1038/srep25318 (2016).
References
Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS 121, 1–58 (2013).
Sriramulu, D. D., Lünsdorf, H., Lam, J. S. & Römling, U. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 54, 667–676 (2005).
Davies, D. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov 2, 114–122 (2003).
Passerini, L., Lam, K., Costerton, J. W. & King, E. G. Biofilms on indwelling vascular catheters. Crit Care Med 20, 665–673 (1992).
Baker, J. S. & Dudley, L. Y. Biofouling in membrane systems–A review. Desalination 118, 81–89 (1998).
Lewis, K. Riddle of biofilm resistance. Antimicrob Agents Chemother 45, 999–1007 (2001).
McDougald, D., Rice, S. A., Barraud, N., Steinberg, P. D. & Kjelleberg, S. Should we stay or should we go: mechanisms and ecological consequences for biofilm dispersal. Nat Rev Microbiol 10, 39–50 (2012).
Waters, C. M. & Bassler, B. L. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Bi 21, 319–346 (2005).
Davies, D. G. et al. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280, 295–298 (1998).
Landini, P., Antoniani, D., Burgess, J. G. & Nijland, R. Molecular mechanisms of compounds affecting bacterial biofilm formation and dispersal. Appl Microbiol Biot 86, 813–823 (2010).
Koo, H. & Jeon, J. Naturally occurring molecules as alternative therapeutic agents against cariogenic biofilms. Adv Dent Res 21, 63–68 (2009).
Teplitski, M., Robinson, J. B. & Bauer, W. D. Plants secrete substances that mimic bacterial N-acyl homoserine lactone signal activities and affect population density-dependent behaviors in associated bacteria. Mol Plant Microbe In 13, 637–648 (2000).
Singh, B. N. et al. Lagerstroemia speciosa fruit extract modulates quorum sensing-controlled virulence factor production and biofilm formation in Pseudomonas aeruginosa. Microbiol-Sgm 158, 529–538 (2012).
Rasmussen, T. B. et al. Screening for quorum-sensing inhibitors (QSI) by use of a novel genetic system, the QSI selector. J Bacteriolo 187, 1799–1814 (2005).
Girennavar, B. et al. Grapefruit juice and its furocoumarins inhibits autoinducer signaling and biofilm formation in bacteria. Int J Food Microbiol 125, 204–208 (2008).
Kim, H.-S. & Park, H.-D. Ginger extract inhibits biofilm formation by Pseudomonas aeruginosa PA14. PloS one 8, e76106 (2013).
Hentzer, M. et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J 22, 3803–3815 (2003).
Kim, H.-S., Lee, S.-H., Byun, Y. & Park, H.-D. 6-Gingerol reduces Pseudomonas aeruginosa biofilm formation and virulence via quorum sensing inhibition. Sci Rep 5, 8656 (2015).
Dey, P. M. & Pridham, J. B. Biochemistry of -galactosidases. Adv Enzymol Ramb 36, 91–130 (1972).
Friedman, L. & Kolter, R. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51, 675–690 (2004).
Caiazza, N. C., Merritt, J. H., Brothers, K. M. & O’Toole, G. A. Inverse regulation of biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol 189, 3603–3612 (2007).
Garber, N., Guempel, U., Belz, A., Gilboa-Garber, N. & Doyle, R. J. On the specificity of the D-galactose-binding lectin (PA-I) of Pseudomonas aeruginosa and its strong binding to hydrophobic derivatives of D-galactose and thiogalactose. BBA-Gen Subjects 1116, 331–333 (1992).
Glick, J. & Garber, N. The intracellular localization of Pseudomonas aeruginosa lectins. J Gen Microbiol 129, 3085–3090 (1983).
Kadam, R. U. et al. A glycopeptide dendrimer inhibitor of the galactose‐specific lectin LecA and of Pseudomonas aeruginosa biofilms. Angew Chem Int Edit 50, 10631–10635 (2011).
Diggle, S. P. et al. The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ Microbiol 8, 1095–1104 (2006).
Hajduk, P. J., Olejniczak, E. T. & Fesik, S. W. One-dimensional relaxation-and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J Am Chem Soc 119, 12257–12261 (1997).
Palmer, A. G., Kroenke, C. D. & Loria, J. P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol 339, 204–238 (2001).
Romling, U., Gomelsky, M. & Galperin, M. Y. C-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol 57, 629–639 (2005).
Barraud, N. et al. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels and enhanced dispersal. J Bacteriol 191, 7333–7342 (2009).
Hickman, J. W., Tifrea, D. F. & Harwood, C. S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. P Natl Acad Sci USA 102, 14422–14427 (2005).
Chung, I. Y., Choi, K. B., Heo, Y.-J. & Cho, Y.-H. Effect of PEL exopolysaccharide on the wspF mutant phenotypes in Pseudomonas aeruginosa PA14. J Microbiol Biotechn 18, 1227–1234 (2008).
Ueda, A. & Wood, T. K. Connecting quorum sensing, c-di-GMP, pel polysaccharide and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog 5, e1000483 (2009).
Schirmer, T. & Jenal, U. Structural and mechanistic determinants of c-di-GMP signalling. Nat Rev Microbiol 7, 724–735 (2009).
Barraud, N. et al. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 188, 7344–7353 (2006).
Kuo, T. M., Vanmiddlesworth, J. F. & Wolf, W. J. Content of raffinose oligosaccharides and sucrose in various plant seeds. J Agr Food Chem 36, 32–36 (1988).
Liberati, N. T. et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. P Natl Acad Sci USA 103, 2833–2838 (2006).
Greenberger, J. S., Donahue, D. & Sakakeeny, M. Induction of ecotropic endogenous murine leukemia virus in long-term bone marrow cultures from mouse strains of varying natural leukemia incidence. J Reticuloendothel Soc 26, 839–853 (1979).
Heydorn, A. et al. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiol-Uk 146, 2395–2407 (2000).
Schweizer, H. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97, 109–112 (1991).
Choi, K.-H., Kumar, A. & Schweizer, H. P. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Meth 64, 391–397 (2006).
Acknowledgements
We would like to thank Professor You-Hee Cho in Cha University for kindly providing PA14 mutants (ΔwspF and ΔlecA) and Joo Hee Chung for performing HPLC analysis and Dr. Eunha Hwang for performing ITC experiments at the Korea Basic Science Institute. In addition, we sincerely thank Professor Arnold Demain in Drew University for critical reading of the manuscript and his advice. This research was funded by National Research Foundation of Korea grants (2015R1D1A1A09057657 and 2014R1A4A1007304).
Author information
Authors and Affiliations
Contributions
H.-S.K., Y.B. and H.-D.P. designed the experiments. H.-S.K., E.C., Y.H.K. and Y.H.J. performed experiments. H.-S.K., Y.H.J., Y.B., B.H.O. and H.-D.P. analyzed the experimental data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, HS., Cha, E., Kim, Y. et al. Raffinose, a plant galactoside, inhibits Pseudomonas aeruginosa biofilm formation via binding to LecA and decreasing cellular cyclic diguanylate levels. Sci Rep 6, 25318 (2016). https://doi.org/10.1038/srep25318
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25318
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
