
Abstract
L-edge soft X-ray spectroscopy has been proven to be a powerful tool to unravel the peculiarities of electronic structure of transition metal compounds in solution. However, the X-ray absorption spectrum is often probed in the total or partial fluorescence yield modes, what leads to inherent distortions with respect to the true transmission spectrum. In the present work, we combine photon- and electron-yield experimental techniques with multi-reference first principles calculations. Exemplified for the prototypical FeCl2 aqueous solution we demonstrate that the partial yield arising from the Fe3s → 2p relaxation is a more reliable probe of the absorption spectrum than the Fe3d → 2p one. For the bonding-relevant 3d → 2p channel we further provide the basis for the joint analysis of resonant photoelectron and inelastic X-ray scattering spectra. Establishing the common energy reference allows to assign both spectra using the complementary information provided through electron-out and photon-out events.
Similar content being viewed by others
Introduction
X-ray spectroscopic methods are indispensable for probing the structure of matter. The recent developments in the vacuum liquid-microjet technique have broadened the applicability of especially soft X-ray spectroscopy to highly volatile liquid solutions1,2. The microjet in conjunction with X-rays provides in situ access to the electronic structure of atomic and molecular solutes. Hence, solute–solvent interactions as well as short-lived chemically reactive intermediates produced upon X-ray excitation or ionization can be studied in great detail1,2,3,4. Among the prominent applications is the investigation of aqueous transition-metal (TM) ions, in particular iron-water complexes, which play a key role in biological and physical-chemical processes5,6,7,8,9,10,11.
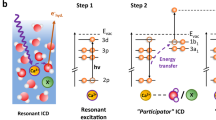
In L-edge soft X-ray spectroscopy of TM ions, the excitation and ionization of 2p core electrons gives insight into chemical bonding and functionality due to the dipole-allowed probing of frontier d-orbitals. Upon absorption of a soft X-ray photon, a 2p core hole is created at the metal centre; this photon energy is large enough to ionize an electron from the oxygen 1s orbital of the water solvent. Subsequent radiative decay channels of the core-excited TM ion comprise 3d → 2p and 3s → 2p pathways which represent about 1% of the total decay probability for the first-row TM ions12. The majority channel is due to Auger-electron decay; other autoionization processes although being possible13,14,15 will not be considered here.
In pioneering studies16,17,18,19,20,21, L-edge X-ray absorption (XA) spectra from aqueous TM ions have been reported, based, however, solely on the detection of the total fluorescence yield (TFY). The TFY detection mode corresponds to an action spectroscopy. It enjoys popularity since the direct measurement of XA in a transmission experiment is challenging for a liquid-jet with a typical diameter of 10–20 μm1,22,23,24,25. In a TFY measurement, a photodiode is used that integrates the signal from all possible radiative decay channels including the solvent background. However, individual relaxation channels have not been explored separately, with the 3d → 2p one being an exception19,26. In general, the assumption that the detected TFY signal intensity is proportional to the number of initially absorbed X-ray photons at a given excitation photon energy is not valid27,28. There are X-ray optical effects responsible for the deviation between TFY and true XA signals such as concentration-dependent self-absorption and saturation as well as signal/background ratio variation29,30. On a more fundamental level, in order to resemble the first-order XA spectrum the corresponding second-order fluorescence must follow an incoherent two-step model31, and the resulting signal is a product of absorption and emission spectra. This holds true only when the interference effects between different radiative decay channels and non-trivial dependence on the polarization of incoming and detected photons can be neglected. This assumption was found to be violated in several studies27,28. Moreover, the state-dependent electron delocalization into the neighbouring ligands after core-excitation could influence the fluorescence intensity19,20,32. Such charge migration processes may occur on a timescale comparable to the few femtosecond lifetime of the core hole33. These effects together, and our lack of understanding of the role of various decay channels, have caused a fair amount of speculations about the electronic structure which is often calculated by semi-empirical theoretical models34.
Disentangling individual channels requires to resolve the energy of the emitted second-order electrons and photons. This is possible through resonant photoelectron (RPE) spectroscopy for the electron channels, and via resonant inelastic X-ray scattering (RIXS) spectroscopy for photon-decay channels. In the present study, RIXS and RPE experimental techniques are applied together with the first principles restricted active space self-consistent field/second order perturbation theory (RASSCF/RASPT2) calculations in order to explore the complementary character of photon- and electron-emission spectra for the exemplary case of an FeCl2 aqueous solution. The focus is put on the interplay of the decay channels, which is addressed by detecting state-dependent pathways involving 3d valence orbitals versus stronger-bound 3p and 3s iron orbitals. The joint analysis of experimental and theoretical spectra provides two key results. First, the question is answered under which conditions partial-yield photon and electron detection can give an accurate measurement of the true X-ray absorption from TM aqueous solutions. As mentioned before, a transmission spectrum cannot be measured with the present cylindrical microjet, but this has no effect on the analysis of the multiple relaxation channels which is the focus of the present work. Partial fluorescence yield (PFY) and partial electron yield (PEY) spectra are obtained by integration of a signal within photon and electron channel-dependent energy ranges. Second, the basis for a back-to-back analysis of RPE and RIXS measurements from the same FeCl2 aqueous solution is established, providing the assignment of the decay channels and energies of the respective electronic states. It is shown that the joint interpretation of RIXS and RPE spectra for the 3d → 2p channel can considerably contribute to our understanding of solute–solvent interactions.
Results and Discussion
The XA spectra from the iron aqueous solutions obtained in different partial-yield modes all exhibit L3 (706–715 eV) and L2 (719–726 eV) edge signals arising from the spin-orbit coupling of the 2p core-hole created by X-ray excitation. Figure 1 summarizes the PFY and PEY spectra, obtained for the various decay channels as indicated in the insets. The assignment of the different Auger-electron channels is presented in Figure S1 in the Supplementary Information (SI). The specific relaxation channels differ with respect to the refill of the core-hole which can be either from valence- or core-shells. For photon detection, the signal from Fe 3d → 2p and 3s → 2p relaxation channels was integrated over the emitted photon energy within the channel-dependent energy ranges 660–720 eV and 605–640 eV, respectively. Analogously, photoelectron spectra from 2p3d3d and 2p3p3p Auger decay channels were integrated within 672–715 eV and 560–600 eV kinetic energy ranges, resulting in the respective PEY-XA spectra.

Comparison of (a) experimental valence and core PFY versus theoretical XAS and PFY spectra; (b) valence and core PEY; (c) valence spectra involving 3d core-hole refill; (d) core spectra due to 3p (PcEY) and 3s (PcFY) relaxations. The corresponding relaxation channels are depicted as insets. All spectra are normalized to the intensity of the highest peak at 708.4 eV to ease comparison.
The different decay channels can be classified according to the nature of the involved orbitals. In PFY (Fe 3d → 2p) and PEY (2p3d3d) the electron, which refills the core hole comes from a Fe 3d valence orbital (spectra are denoted as PvFY and PvEY). These 3d orbitals carry information on the mixing with ligand-centred orbitals. In contrast, in PFY (Fe 3s → 2p) and PEY (2p3p3p) the core-hole is refilled by electrons from deeper (3s/3p) Fe orbitals which do not appreciably interact with the ligands (spectra are denoted as PcFY and PcEY).
The calculated PvFY, PcFY spectra, and the transmission XA spectrum shown in Fig. 1a will serve as reference for the following discussion. The two PFY spectra exhibit distinct differences, i.e. the pre-edge intensity near 707 eV excitation photon energy is lower, while the post-edge at 709–714 eV and the L2 edge are more intense for PvFY than for PcFY. The calculated XA and PcFY spectra are essentially indistinguishable from each other. Further, both computed PvFY and PcFY spectra are in very good agreement with the experimental results that are also shown in Fig. 1a. Notice, however, that the calculations did not take into account the X-ray optical effects mentioned above.
The difference in the L3/L2 edge intensity ratio observed in Fig. 1 can be attributed to several effects. These include self-absorption as well as Coster-Kronig transitions, both enhancing the photon emission at the L2 edge4. The fact that these effects are not included into the calculation points to yet another reason for the difference in the spectra, i.e., the spin-orbit coupling. For the two radiative relaxation channels (3s → 2p and 3d → 2p) core-excited intermediate states are mixtures of the wave functions with different multiplicities due to the strong 2p core-hole spin-orbit coupling, whereas valence initial and final states almost do not mix, and represent pure quintet and triplet states. Thus, the second-order fluorescence process, initial → intermediate → final state, can be described as quintet (ground) → mixed → quintet and triplet states, for details see ref. 35. This leads to the appearance of new formally spin-forbidden radiative channels for L3 post-edge and L2 bands, where spin-orbit coupling is stronger than for the lower energy wing of the L3-edge20,26,35. Since spin-orbit coupling has no effect for 3s orbitals and is notable for 3d orbitals, it leads to distortions of PvFY as compared to the XA spectrum, whereas PcFY is not affected. In the following discussion we will focus on the L3 edge only, including its post-edge region.
Compared to the fluorescence yield mode, the PvEY and PcEY spectra (Fig. 1b) show similar although less pronounced differences; see also panels c and d of Fig. 1. The analysis of spin-orbit coupling is more involved in case of PEY, due to transitions between mixed intermediate states of the Fe2+ ion and final valence spin manifolds of the ionized Fe3+ system. Hence, these effects are not considered here.
Another source of differences between PEY/PFY spectra with respect to the transmission XA spectrum is the electronic interaction of the TM ion with the environment, i.e., solvation-shell water molecules. The excited electron can be further involved in other relaxation processes such as electron delocalization and energy transfer20,36. The delocalized excited electron has a lower probability to refill the 2p hole within its lifetime (sub-10 fs), and hence the indirect PEY/PFY probe should be state-dependent according to the extent of delocalization and should not track the true X-ray absorption probability20. The observed behaviour in Fig. 1 thus correlates with the orbital extensions: core 3s and 3p orbitals are strongly localized, and are not or barely involved in iron–ligand orbital mixing. Thus core orbitals are subject to less distortion since delocalization and also spin-orbit channels are of minor importance, while both channels are essential for the 3d shell, since 3d and water lone-pair orbitals are considerably mixed. This behaviour seems to be generally valid, for another example see refs 19,20.
The preceding discussion has pointed to the different sensitivity of valence- and core-level relaxation channels to electron delocalization and spin-orbit coupling. Hence, it is not surprising that PcEY and PcFY detection (Fig. 1c) leads to spectra which are more closely reflecting the true X-ray absorption. Here we argue that the computed XA spectrum accurately captures the true (transmission) XA spectrum; this is yet to be confirmed experimentally though.
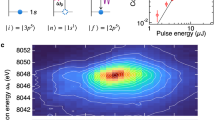
In the following we focus on the particular electron- or photon-energy resolved channels involving frontier Fe 3d orbitals. The RPE and RIXS spectra measured at the Fe 2p → 3d resonance are shown in Fig. 2a,b, respectively, in a two-dimensional (2D) representation depending on the incoming photon energy. Panels c and d of Fig. 2 display four cuts through the 2D spectra in the L3-edge region at those excitation energies which correspond to the main absorption features (compare Fig. 1). The theoretical direct photoionization signal from the iron centre contributing to total RPE is shown in Figure S2 of the SI, and the calculated RIXS spectrum is very similar to that discussed in ref. 26.

(a) 2D RPE as a function of electron kinetic energies; (b) 2D RIXS; (c) 1D RPE cuts for the selected excitation energies, the electron binding energy is used to facilitate comparison with the RIXS spectra; (d) 1D RIXS cuts for the selected excitation energies.
The RPE spectrum is assigned with the help of the well-understood off-resonant photoelectron spectrum, obtained here at 705 eV excitation energy (see Figs 2c and 3a). It essentially represents the neat-water PE spectrum37,38 with the features at 11.2, 13.5, 17.3, and 31.0 eV binding energies (BE) being due to ionization from the 1b1, 3a1, 1b2, and 2a1 water valence orbitals (blue labels in Fig. 3a), respectively. A very weak signal from Fe 3d at 7.09 eV39 corresponding to the lowest ionization energy of the solution can be also seen.

(a) Aligned RPE and RIXS spectra. Red, blue, and green labels correspond to Fe, H2O, and Cl− transitions, respectively; (b) Sketch of Fe 3d orbital symmetries for weakly Jahn-Teller distorted system; (c) Scheme connecting the corresponding transitions in RIXS (elastic, inelastic) and RPE (both direct and Auger ionization channels).
Since the energetic structure of the valence levels does not depend on the excitation energy, we can discuss the principle features of the RPE spectra for an excitation energy for which signal intensities are strongest. This is the case at maximum absorption, i.e., at 708.4 eV, shown in Fig. 3a. The large Auger-electron signal in the ~8–20 eV BE range, occurring for the Fe L3-edge excitation, mainly arises from the 2p3d3d Auger transition. The Fe 3d orbitals of weakly the tetragonally distorted Jahn-Teller d6 system with approximate D4h point group symmetry split into four components as shown in Fig. 3b. According to the theoretical photoelectron spectrum (Figure S2 of the SI), the Fe transitions (red labels in Fig. 3a) group into three bands: a distinct feature due to ionization of a spin-down (β) electron from the lowest b2g(β) orbital at 7.09 eV BE, a broad band comprising numerous main and combination transitions of spin-up (α) electrons from a1g(α), b1g(α), and eg(α) orbitals overlapping in energy and being centred at 9.5 eV BE, and from b2g(α) at 12.24 eV BE. The b2g(β) electron occupying the deepest 3d orbital has the lowest ionization energy because the orbital binding energy is compensated by pairing (exchange) energy. The chloride 3p valence ionization (green labels in Fig. 3a) leads to a small contribution around 9 eV BE40 coinciding with the Fe a1g(α)/b1g(α)/eg(α) feature as well as with the low-binding energy side of the water 1b1 orbital signal. The feature at 15.3 eV is most probably due to first-solvation shell water 3a1 orbitals, which form σ-bonding combinations with iron a1g and b1g ( and ) orbitals. Note that the 1b2 water signal is also shifted to higher binding energies upon such interactions.
For the resonant excitation at 708.4 eV, the overall RPE signal intensity dramatically increases in comparison to the off-resonant region. Moreover, a signal increase occurs at the energies of the water peaks 1b1, 3a1, and 1b2, and of the Fe 3d peaks. Such a strong signal enhancement results from the interference of the outgoing electron waves from two simultaneous electron emission processes sharing the same final state (see Fig. 3c), and leading to identical kinetic energies of the emitted electrons41,42. One process is the direct ionization of the valence orbitals excited by the 708.4 eV energy photon. The other process is the 2p3d3d Auger-decay releasing a 3d electron from the valence level with the same kinetic energy. This interference effect is a qualitative unequivocal evidence of the mixing between iron and water valence orbitals, and represents a central experimental finding of this study. Note that if there was no mixing, only the Fe 3d signal would be enhanced.
A quantitative comparison of RPE and RIXS spectra in terms of the energies of the occurring peaks requires a proper alignment of both spectra on the energy axis. As a consequence of strong correlation effects and pronounced spin-mixing this requires switching from the one-electron orbital picture to the many-electron states picture as illustrated in Fig. 3c. The lowest-energy peak in the RPE spectrum is due to the transition between ground states of initial Fe2+(aq) and ionized Fe3+(aq) systems for direct ionization or between the X-ray core-excited state of Fe2+(aq) and the ground state of Fe3+(aq) as shown by blue arrows in Fig. 3c. This corresponds to elastic scattering in RIXS when the same core-excited state relaxes back to the ground state of Fe2+(aq) (green arrow in Fig. 3c).
The alignment of the lowest-binding energy RPE feature with the elastic RIXS peak, as is done in Fig. 3a, establishes a common energy reference which allows connecting all other features of the two spectra. Let us consider a particular valence excited state. In RPE, it corresponds to the transition to a final valence excited state of Fe3+(aq) with the excitation energy ΔW (cf. grey arrows in Fig. 3c). Analogously, the inelastic RIXS transition (red arrow) results in a valence excited state of Fe2+(aq) with excitation energy ΔE. If electronic relaxation upon ionization Fe2+(aq) → Fe3+(aq) + e− is small and ΔE ≈ ΔW, the present alignment allows the back-to-back comparison and assignment of RPE and RIXS.
According to the suggested alignment, the feature at 2.13 eV loss energy in RIXS is connected with the broad band at 9.5 eV BE in RPE, and thus can be assigned to predominant radiative relaxation from a1g, b1g, and eg orbitals. The RIXS spectral shoulder near 5.13 eV which is in accordance with the 12.24 eV shoulder in RPE can be attributed to the relaxation from the b2g orbital, and a weak band near 9.0 eV loss energy is assigned to weak charge-transfer transitions from first-solvation shell water orbitals. The charge-transfer transitions again evidence the orbital mixing between metal and ligand. Remarkably, the 0.9 eV RIXS feature arises from the relaxation of the same b2g(β) electron as in the first RPE peak. The energy shift of 0.9 eV can be explained taking into account that in RPE the b2g(β) electron is completely removed from the system, and its corresponding Coulomb and exchange energies contribute to the ionization energy. In contrast, in RIXS the energy of the final state is determined by the change of the corresponding terms due to excitation of this electron from b2g to other orbitals. The assignment based on our spectra alignment is supported by the theoretical analysis of the direct photoionization and the RIXS spectra.
Since RPE and RIXS transitions have different selection rules, having a common reference in energy does not imply that the intensities are the same. This explains the absence of prominent RIXS transitions involving water orbitals. Thus, the intensity differences provide information on the rate of different radiative and non-radiative transitions following the core-excitation.
An assignment of the higher-energy excitations, a particularly near 712 eV, where the PEY and PFY spectra exhibit substantially different intensities (Fig. 1), cannot be made based on our spectra. According to the theoretical analysis, this region corresponds to strong mixing between states of different multiplicities, and thus is difficult to interpret taking into account the different influence of spin-orbit coupling onto electron- and photon-out events.
Conclusions
Complementary electron and photon emission spectra are necessary for a full understanding of electronic-structure interactions between metal solute and water solvent. Previously reported X-ray spectroscopy studies relied on the photon detection and did not consider the electronic structure information from the electron-emission channels. The parallel consideration of PFY and PEY spectra, originating from various channels, reveals large deviations from the true XA spectrum in the case of the 3d → 2p relaxation. In contrast, the “core” PFY and PEY spectra due to the 3s → 2p relaxation are accurate measures of the transmission XA spectrum. In fact our measurements suggest that the latter channel (investigated here for fluorescence) is the most accurate probe. Looking at the 3d → 2p relaxation, with the discussed energy alignment, one can find experimentally find the correspondence between spectral signatures in the RPE and RIXS spectra. Moreover, comparing RPE and RIXS intensities one may obtain information on the competition between the radiative and non-radiative decay channels. Our analysis suggests a new protocol for investigation of X-ray spectra of transition metal solute–solvent electronic interactions. The provided insight shall promote understanding structure and rational design of catalytic and other functional materials.
Methods
Experimental
FeCl2 hexahydrate salt, purchased from Sigma Aldrich with >99% purity, was dissolved in deionised water to make a 1 M aqueous solution for both photon and electron measurements. The colour of the solution is pale-lime and the molar ratio of [Fe(H2O)5Cl]+ and [Fe(H2O)6]2+ species is ~77/23 43,44. The microjet technique was employed to introduce the liquid sample into the vacuum chamber through a 20 μm diameter glass nozzle. The electron and photon signals were taken from the laminar area of the jet which is approximately 2–3 mm in length starting from the nozzle head.
The experiments were carried out at the U41-PGM undulator beamline at the synchrotron facility BESSY II, Berlin. Two experimental stations, LiXEdrom and LiquidPES, were used for photon and electron detection, respectively. The U41-PGM beamline provides horizontally linear polarized light with high photon flux and a micro-focus spot, which is crucial to micro-jet measurements. In LiXEdrom, photons emitted from the jet were collected along the polarization direction of the incident photon beam (to suppress the elastic peak) and subsequently dispersed by a spherical grating with 1200 lines/mm and 7.5 m radius. The dispersed photons were then detected by a microchannel plate (MCP)/fluorescence screen/CCD assembly. The sample, grating, and photon detector are arranged on a Rowland circle geometry for accurate focusing. For the LiquidPES experiment, electrons were collected perpendicular to the polarization of the incident photon beam by a hemispherical energy analyzer. Note that the 2p3s3s Auger-decay channel is not considered because of a too low signal. The details of the two experimental stations have been described elsewhere1,2,3,4.
Computational
Theoretical calculations were performed for [Fe(H2O)6]2+ and [Fe(H2O)5Cl]+ complexes, where geometries were optimized at the BLYP/cc-pVTZ level with Gaussian 09 package45. The valence- and core-excited electronic states were further studied using the restricted active space self-consistent field (RASSCF) method46 with the relativistic ANO-RCC basis sets47,48 of triple-zeta quality. The second order perturbation theory correction (RASPT2) with an imaginary level shift of 0.1 Hartree was applied on top of RASSCF to account for dynamic correlation. The active space for XAS, RIXS, and PFY calculations included 2p in the RAS1 space (1 hole allowed) and 3s together with 3d orbitals in RAS2 space (full CI); all occupied inactive orbitals were kept frozen. The resulting configuration space includes dipole-allowed 2p → 3d excitations and 3d → 2p and 3s → 2p radiative relaxation transitions. Spin-orbit coupling was treated in the LS-coupling limit49 taking into account 75 quintet (S = 2) and 375 triplet (S = 1) directly interacting states. Scalar relativistic effects were included within the Douglas-Kroll-Hess approach50,51. RIXS and PFY spectra were evaluated using the Kramers-Heisenberg expression properly including coherence effects and polarization dependence (for details, see ref. 35). Valence photoelectron spectra were obtained using the Dyson orbital approach as described in ref. 52 with the active space including only 3d orbitals in RAS2 space. A single quintet initial state for [Fe(H2O)6]2+ and 1 sextet (S = 5/2), 24 quartet (S = 3/2), and 75 doublet (S = 1/2) final states coupled via spin-orbit coupling were included for [Fe(H2O)6]3+. All calculations were performed with a locally modified MOLCAS 8.0 code53.
Additional Information
How to cite this article: Golnak, R. et al. Joint Analysis of Radiative and Non-Radiative Electronic Relaxation Upon X-ray Irradiation of Transition Metal Aqueous Solutions. Sci. Rep. 6, 24659; doi: 10.1038/srep24659 (2016).
References
Winter, B. & Faubel, M. Photoemission from liquid aqueous solutions. Chem. Rev. 106, 1176–1211 (2006).
Winter, B. Liquid microjet for photoelectron spectroscopy. Nucl. Instrum. Methods Phys. Res. Sect. Accel. Spectrometers Detect. Assoc. Equip. 601, 139–150 (2009).
Lange, K. M. et al. High resolution X-ray emission spectroscopy of water and aqueous ions using the micro-jet technique. Chem. Phys. 377, 1–5 (2010).
Gotz, M. D. et al. Probing Coster-Kronig transitions in aqueous Fe2+ solution using inverse partial and partial fluorescence yield at the L-edge. J. Phys. Chem. Lett. 3, 1619–1623 (2012).
Casci, J. L., Lok, C. M. & Shannon, M. D. Fischer-Tropsch catalysis: The basis for an emerging industry with origins in the early 20th century. Catal. Today 145, 38–44 (2009).
Gärtner, F. et al. Light-driven hydrogen generation: Efficient iron-based water reduction catalysts. Angew. Chem. Int. Ed. 48, 9962- 9965 (2009).
Thomson, A. J. & Gray, H. B. Bio-inorganic chemistry. Curr. Opin. Chem. Biol. 2, 155–158 (1998).
Marcus, Y. Effect of ions on the structure of water: Structure making and breaking. Chem. Rev. 109, 1346–1370 (2009).
Wasserscheid, P. & Keim, W. Ionic liquids—new “solutions” for transition metal catalysis. Angew. Chem. Int. Ed. 39, 3772–3789 (2000).
Ashcroft, A. T. et al. Selective oxidation of methane to synthesis gas using transition metal catalysts. Nature 344, 319–321 (1990).
Ikariya, T. & Blacker, A. J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 40, 1300–1308 (2007).
Kotani, A. & Shin, S. Resonant inelastic X-ray scattering spectra for electrons in solids. Rev. Mod. Phys. 73, 203–246 (2001).
Öhrwall, G. et al. Charge dependence of solvent-mediated intermolecular Coster-Kronig decay dynamics of aqueous ions. J. Phys. Chem. B 114, 17057–17061 (2010).
Pokapanich, W. et al. Ionic-charge dependence of the intermolecular coulombic decay time scale for aqueous ions probed by the core-hole clock. J. Am. Chem. Soc. 133, 13430–13436 (2011).
Ottosson, N., Öhrwall, G. & Björneholm, O. Ultrafast charge delocalization dynamics in aqueous electrolytes: New insights from Auger electron spectroscopy. Chem. Phys. Lett. 543, 1–11 (2012).
Aziz, E. F. The solvation of ions and molecules probed via soft X-ray spectroscopies. J. Electron Spectrosc. Relat. Phenom. 177, 168–180 (2010).
Freiwald, M., Cramm, S., Eberhardt, W. & Eisebitt, S. Soft X-ray absorption spectroscopy in liquid environments. J. Electron Spectrosc. Relat. Phenom. 137–140, 413–416 (2004).
Fuchs, O. et al. A liquid flow cell to study the electronic structure of liquids with soft X-rays. Nucl. Instrum. Methods Phys. Res. Sect. Accel. Spectrometers Detect. Assoc. Equip. 585, 172–177 (2008).
Seidel, R. et al. Origin of dark-channel X-ray fluorescence from transition-metal ions in water. J. Am. Chem. Soc. 134, 1600–1605 (2012).
Bokarev, S. I., Dantz, M., Suljoti, E., Kühn, O. & Aziz, E. F. State-dependent electron delocalization dynamics at the solute-solvent interface: Soft X-ray absorption spectroscopy and ab initio calculations. Phys. Rev. Lett. 111, 083002 (2013).
Nagasaka, M., Hatsui, T., Horigome, T., Hamamura, Y. & Kosugi, N. Development of a liquid flow cell to measure soft X-ray absorption in transmission mode: A test for liquid water. J. Electron Spectrosc. Relat. Phenom. 177, 130–134 (2010).
Siegbahn, H. & Siegbahn, K. ESCA applied to liquids. J. Electron Spectrosc. Relat. Phenom. 2, 319–325 (1973).
Faubel, M., Steiner, B. & Toennies, J. P. Photoelectron spectroscopy of liquid water, some alcohols, and pure nonane in free micro jets. J. Chem. Phys. 106, 9013 (1997).
Lange, K. M., Kothe, A. & Aziz, E. F. Chemistry in solution: Recent techniques and applications using soft X-ray spectroscopy. Phys. Chem. Chem. Phys. 14, 5331–5338 (2012).
Wilson, K. R. et al. X-ray spectroscopy of liquid water microjets. J. Phys. Chem. B 105, 3346–3349 (2001).
Atak, K. et al. The nature of the chemical bond of aqueous Fe2+ probed by soft X-ray spectroscopies and ab initio calculations. J. Phys. Chem. B 117, 12613–12618 (2013).
de Groot, F. M. F., Arrio, M. A., Sainctavit, P., Cartier, C. & Chen, C. T. Fluorescence yield detection: Why it does not measure the X-ray absorption cross section. Solid State Commun. 92, 991–995 (1994).
Kurian, R. et al. Intrinsic deviations in fluorescence yield detected X-ray absorption spectroscopy: The case of the transition metal L2,3 edges. J. Phys.: Condens. Matter 24, 452201 (2012).
de Groot, F. M. F. Dips and peaks in fluorescence yield X-ray absorption are due to state-dependent decay. Nat. Chem. 4, 766–767 (2012).
Regier, T. Z., Achkar, A. J., Peak, D. & Tse, J. S. & Hawthorn, D. G. Dark channel fluorescence observations result from concentration effects rather than solvent-solute charge transfer. Nat. Chem. 4, 765–766 (2012).
Luo, Y., Ågren, H. & Gel’mukhanov, F. Polarization anisotropy in resonant X-ray emission from molecules. Phys. Rev. A 53, 1340–1348 (1996).
Aziz, E. F., Rittmann-Frank, M. H., Lange, K. M., Bonhommeau, S. & Chergui, M. Charge transfer to solvent identified using dark channel fluorescence-yield L-edge spectroscopy. Nat. Chem. 2, 853–857 (2010).
Kuleff, A. I. & Cederbaum, L. S. Radiation generated by the ultrafast migration of a positive charge following the ionization of a molecular system. Phys. Rev. Lett. 106, 053001 (2011).
de Groot, F. & Kotani, A. Core level spectroscopy of solids. (CRC Press, Boca Raton, 2008).
Bokarev, S. I. et al. Unraveling the electronic structure of photocatalytic manganese complexes by L-edge X-ray spectroscopy. J. Phys. Chem. C 119, 19192–19200 (2015).
Thürmer, S. et al. On the nature and origin of dicationic, charge-separated species formed in liquid water on X-ray irradiation. Nat Chem 5, 590–596 (2013).
Winter, B. et al. Full valence band photoemission from liquid water using EUV synchrotron radiation. J. Phys. Chem. A 108, 2625–2632 (2004).
Kurahashi, N. et al. Photoelectron spectroscopy of aqueous solutions: Streaming potentials of NaX (X = Cl, Br, and I) solutions and electron binding energies of liquid water and X− . J. Chem. Phys. 140, 174506 (2014).
Seidel, R. et al. Valence photoemission spectra of aqueous Fe2+/3+ and [Fe(CN)6]4−/3− and their interpretation by DFT calculations. J. Phys. Chem. B 115, 11671–11677 (2011).
Winter, B. et al. Electron binding energies of aqueous alkali and halide ions: EUV photoelectron spectroscopy of liquid solutions and combined ab initio and molecular dynamics calculations. J. Am. Chem. Soc. 127, 7203–7214 (2005).
Thürmer, S., Seidel, R., Eberhardt, W., Bradforth, S. E. & Winter, B. Ultrafast hybridization screening in Fe3+ aqueous solution. J. Am. Chem. Soc. 133, 12528–12535 (2011).
Hüfner, S. Photoelectron spectroscopy: Principles and applications, 3 ed. (Springer-Verlag, Berlin Heidelberg, 2003).
Heinrich, C. A. & Seward, T. M. A spectrophotometric study of aqueous iron (ii) chloride complexing from 25 to 200 °C. Geochim. Cosmochim. Acta 54, 2207–2221 (1990).
Apted, M. J., Waychunas, G. A. & Brown, G. E. Structure and specification of iron complexes in aqueous solutions determined by X-ray absorption spectroscopy. Geochim. Cosmochim. Acta 49, 2081–2089 (1985).
Frisch, M. J. et al. Gaussian 09, revision C.1 (Gaussian, Inc., Wallingford CT, 2009).
Malmqvist, P. A., Rendell, A. & Roos, B. O. The restricted active space self-consistent-field method, implemented with a split graph unitary group approach. J. Phys. Chem. 94, 5477–5482 (1990).
Roos, B. O., Lindh, R., Malmqvist, P.-Å., Veryazov, V. & Widmark, P.-O. Main group atoms and dimers studied with a new relativistic ANO basis set. J. Phys. Chem. A 108, 2851–2858 (2004).
Roos, B. O., Lindh, R., Malmqvist, P.-Å., Veryazov, V. & Widmark, P.-O. New relativistic ANO basis sets for transition metal atoms. J. Phys. Chem. A 109, 6575–6579 (2005).
Malmqvist, P. Å., Roos, B. O. & Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin-orbit coupling. Chem. Phys. Lett. 357, 230–240 (2002).
Douglas, M. & Kroll, N. M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 82, 89–155 (1974).
Hess, B. A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 33, 3742–3748 (1986).
Grell, G. et al. Multi-reference approach to the calculation of photoelectron spectra including spin-orbit coupling. J. Chem. Phys. 143, 074104 (2015).
Karlström, G. et al. Molcas: A program package for computational chemistry. Comput. Mater. Sci. 28, 222–239 (2003).
Acknowledgements
S.I.B, G.G., S.G.A. and O.K. acknowledge financial support by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, (grant No. D-003-435) and the Deutsche Forschungsgemeinschaft (KU 952/10-1). K.A. acknowledges financial support of the Einstein Foundation Berlin for a postdoctoral scholarship. B.W. and R.S. gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (Collaborative Research Centre 1109), and B.W. also gratefully acknowledges support from the Deutsche Forschungsgemeinschaft Research Unit FOR 1789. The authors thank the HZB staff for assisting in the operation of the beamline.
Author information
Authors and Affiliations
Contributions
B.W. and E.F.A. conceived and designed experiments. R.G., R.S., J.X., K.A., I.U. and S.T. conducted experiments and analyzed the data. S.I.B. and G.G. performed theoretical calculations. E.F.A., S.G.A. and O.K. coordinated the project. S.I.B., R.S., J.X., O.K. and B.W. co-wrote the paper; all authors were involved in the discussion and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Golnak, R., Bokarev, S., Seidel, R. et al. Joint Analysis of Radiative and Non-Radiative Electronic Relaxation Upon X-ray Irradiation of Transition Metal Aqueous Solutions. Sci Rep 6, 24659 (2016). https://doi.org/10.1038/srep24659
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24659
This article is cited by
-
Water at charged interfaces
Nature Reviews Chemistry (2021)
-
Chemical bonding in aqueous hexacyano cobaltate from photon- and electron-detection perspectives
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
