
Abstract
High sensitivity of the Kβ fluorescence spectrum to electronic state is widely used to investigate spin and oxidation state of first-row transition-metal compounds. However, the complex electronic structure results in overlapping spectral features, and the interpretation may be hampered by ambiguity in resolving the spectrum into components representing different electronic states. Here, we tackle this difficulty with a nonlinear resonant inelastic X-ray scattering (RIXS) scheme, where we leverage sequential two-photon absorption to realize an inverse process of the Kβ emission, and measure the successive Kα emission. The nonlinear RIXS reveals two-dimensional (2D) Kβ-Kα fluorescence spectrum of copper metal, leading to better understanding of the spectral feature. We isolate 3d-related satellite peaks in the 2D spectrum, and find good agreement with our multiplet ligand field calculation. Our work not only advances the fluorescence spectroscopy, but opens the door to extend RIXS into the nonlinear regime.
Similar content being viewed by others

Introduction
X-ray fluorescence spectroscopy has been a standard technique for non-destructive element analysis due to the one-to-one relationship between elements and emission photon energies. Recent investigation1,2,3 on the spectral shape has revealed a close correlation of the Kβ fluorescence spectrum to the electronic states, such as the spin and oxidation state of first-row transition-metal compounds. Now, X-ray fluorescence spectroscopy becomes a powerful tool for investigating even biological samples, e.g., the oxidation state of Mn in the oxygen-evolving complex of photosystem II4,5,6,7, in addition to the various field of physics and chemistry1,2,3. The Kβ fluorescence is a radiative transition from a 1s core-hole state (1s1) to a 3p core-hole state (3p5) and does not explicitly involve a 3d state. It is an exchange interaction between 3d and 3p electrons that reflects the 3d electronic state in the Kβ fluorescence8. At the same time, the exchange and other interactions split the electronic states and result in heavily overlapping spectral features as we will see later. This could make it difficult to extract the key spectral components related to the electronic states of interest and hamper the in-depth discussion.
A good playground for the fluorescence spectrum is copper metal, which is a simple system but has been investigated for decades both experimentally and theoretically9,10,11,12,13,14,15. There are two approaches to analyzing the fluorescence spectrum. As for the Kβ spectrum, it can be analytically decomposed into five Lorentzians10. This phenomenological analysis may have a problem that each component is not necessarily connected to a particular electronic state. The Kβ spectrum can also be well reproduced as a sum of theoretical spectra calculated for possible electronic states11,12. However, different theoretical models can fit the spectrum, and thus the theoretical fitting may not discriminate subtle changes or differences in the electronic state. In order to perform more deterministic analysis, we need to get more spectroscopic information from the fluorescence process.
In this article, we discuss the expansion of the one-dimensional fluorescence spectrum to two dimensions. We propose a fusion of nonlinear absorption and resonant inelastic X-ray scattering (RIXS) and demonstrate our idea on copper metal. Our analysis of the 2D fluorescence spectrum resolves 3d-related satellite peaks, which are hidden by a strong main peak in the conventional Kβ fluorescence spectrum. We also present multiplet ligand calculations and interpret the experimentally resolved satellites based on the theoretical prediction.
Results
Nonlinear RIXS scheme
Our idea to extract more information from the Kβ fluorescence process is to expand the one-dimensional spectrum to two dimensional by adding a second axis. The spectral components, which overlap in the Kβ fluorescence spectrum, may separate along the second axis direction. Suppose we could use an absorption process from 3p5 to 1s1. This is an inverse process of the Kβ fluorescence and should be governed by the same transition matrix elements. After this absorption process, the resultant 1s1 state might decay by emitting the Kα fluorescence, i.e., a 1s1 → 2p5 radiative transition. Consequently, we could have two tuneable parameters, the photon energies for the absorption and emission processes. This absorption-emission process is to be treated in a similar manner to RIXS16,17,18 because the absorption is resonant. If we had measured the Kβ emission, the process should be described as resonance fluorescence19.
The biggest hurdle to realize our idea is the fact that all core levels are occupied in the ground state, and consequently, the 3p5 → 1s1 absorption process is strictly forbidden by the Pauli principle. The key is sequential two-photon absorption (Fig. 1a). In this nonlinear process, the first photon creates a 3p core hole, and opens a new resonant absorption channel from 3p5 to 1s1 within a very short period of the 3p core-hole lifetime. The second photon can excite a 1s core electron to the 3p core hole if the photon energy is carefully tuned to the resonance. We note that the sequential absorption on the core-to-core resonance was already reported in the soft-X-ray region20,21, however, has not been combined with the X-ray emission spectroscopy.

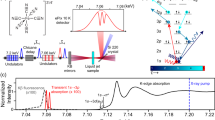
a Schematic nonlinear RIXS process. The system is excited by sequential two-photon absorption including the inverse process of the Kβ fluorescence, and then decays by the Kα emission. Uninvolved orbitals and electrons are omitted for simplicity. b The pulse-energy dependence of the 8048 eV emission count rate at a pump photon energy of 8905 eV (circles). Vertical bars indicate the standard error of the mean. Solid line represents the best fit with a quadratic function without a linear or constant term. The coefficient of the quadratic term gives \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\). c Image plot of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\). The pump photon energy covers the whole Kβ spectral range, while the emission photon energy does the main parts around the Kα1 and Kα2 peaks.
As we use the nonlinear process and RIXS, we refer to the above process as 3p5 → 1s1 → 2p5 nonlinear RIXS. An inverse process, i.e., 2p5 → 1s1 → 3p5 nonlinear RIXS, is also possible but may be less efficient due to the shorter lifetime for the 2p5 state than 3p5, and an order of magnitude lower Kβ yield than Kα. We label the excited state after the first absorption as i, the intermediate state after the second absorption as n, the final state after the emission as f, and the ground state as g (Fig. 1a). The excited state i may include multiple hole state due to a shake-off process and cascade of the Auger decay. The shake-off process can create two-hole states having one 3p hole and an additional hole in 3s, 3p, or 3d subshell22. The pump photon energy higher than the L1 absorption edge may create a 2s, 2p, or 3s hole, and initiate the Auger cascade, during which multi-hole state with at least one 3p hole may appear. Among various multi-hole states possible, however, a 3p53d9 state is found important to interpret the experimental result, as we will see later.
First, we formulate the nonlinear RIXS process. We exclude the Auger cascade from the formulation for simplicity. The absorption of the first photon can be treated as an independent process from the subsequence because a promoted high-energy photo-electron hardly interacts with the rest system. Therefore, this part can be described by rate equations23. Below, we will omit the electrons in the high-energy continuum from the configuration expression, e.g., |i〉 = |3p5〉 instead of |i〉 = |3p5ε〉, unless we need to indicate it explicitly. On the other hand, the absorption of the second photon is resonant, and thus the i → n absorption and the n → f emission should be described as a coherent second-order process like the conventional RIXS. Combining all processes, the differential cross-section for the nonlinear RIXS may be expressed as:
where ωp is the pump angular frequency, ωe is the angular frequency at which the emission is measured, dΩ is an element of solid angle into which the photon specified by the subscript is emitted, σgi is the g → i absorption cross-section, and τi is the lifetime of i. We introduced a normalized dimensionless instrumental function, Φ, which represents the finite photon-energy resolution of the detecting system. The dimension of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) is (length)4(time), which is the same as the cross-section for two-photon absorption. The double differential scattering cross-section of the RIXS part may be written as24:
by keeping only a resonant term in the Kramers–Heisenberg formula and applying the dipole approximation. Here, ω1 and ω2 are the angular frequencies of the incident and emitted photons, respectively, m is the electron mass, r0 is the classical radius of electron, Γn is the lifetime broadening of n, ωab=(Ea−Eb)/ħ with Ex being the energy of the state x, and \(\hat{r}\) is the position operator of the electron. Summation over all electrons is omitted for simplicity. We point out here that the initial state of the RIXS part includes many excited states with different energies, whereas the conventional RIXS starts from the ground state.
Measurement of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\)
Since the lifetime of the 3p core-hole state, τi, is on a sub-femtosecond timescale, nonlinear RIXS requires intense femtosecond X-rays. We used an X-ray free-electron laser, SACLA25, which delivered an X-ray beam with the central photon energy at 8905 eV, corresponding to the Kβ photon energy of copper, a full-width-at-half-maximum (FWHM) bandwidth of 43 eV, and an FWHM pulse duration as short as 8 fs (ref. 26). We used a Si (111) double-crystal monochromator to select a 1.0 eV monochromatic beam, and to scan the pump-photon energy over the Kβ range, and then, focused the beam down to 0.72(H) × 0.92(V) μm2 (FWHM) by a Kirkpatrick–Baez (KB) mirror27 (see Methods and Supplementary Fig. 2 for details). The average pulse energy in front of the KB mirror was 8.0 μJ at 8905 eV, from which we estimate the peak intensity and the peak photon-flux density on the focus to be 1.5 × 1017 W/cm2 and 11 photons/Å2/fs, respectively. We measured the Kα emission spectrum from a 10-μm-thick copper foil set on the focus using a single-shot polychromator.
Figure 1b shows a typical pump pulse-energy dependence of the emission count rate measured at ωe = 8048 eV, i.e., the peak of the Kα1 fluorescence spectrum. The pump photon energy was tuned to the Kβ fluorescence peak at ωp = 8905 eV (Supplementary Fig. 3). The pulse-energy dependence is well reproduced by a quadratic term only, as expected for sequential two-photon absorption. This indicates that non-resonant fluorescence by higher harmonic radiation and multi-photon processes of more than two are negligible under the present condition. The coefficient of the quadratic term is proportional to \({\sigma }_{{{{{{\rm{RIX}}}}}}S}^{\left(2\right)}\). Analyzing the pulse-energy dependence of the Kα emission counts at each pump and emission photon energies, we map out \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) on the ωp−ωe plane as shown in Fig. 1c.
2D fluorescence spectrum
The \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) plane shows two prominent peaks at (ωp, ωe)=(8905 eV, 8027 eV) and (8905 eV, 8048 eV), which correspond to the peak photon energies for the Kβ and Kα1,2 fluorescence spectra. These two peaks are not simple Lorentzian but are accompanied by additional weak peaks. To make clear the weak structures, three characteristic sections along constant ωp lines are shown in Fig. 2. We also plot the Kα fluorescence spectrum measured at an excitation photon energy of 9100 eV well above the K edge at 8980 eV for comparison. Among the three emission spectra, only that at ωp = 8905 eV, i.e., the Kβ peak, is found similar to the Kα fluorescence spectrum.

Circles show sections of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) at three characteristic ωp. The error of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) due to the fitting with a quadratic function (Fig. 1b) is smaller than the marker size. Solid lines in the middle panel represent the Kα fluorescence spectrum measured by one-photon excitation at 9100 eV well above the K edge. Dashed lines represent the fitting with Lorentzian(s), and each component is plotted by a dotted line.
Here, we employ the phenomenological approach to analyze the emission spectra. The Kα emission spectrum is much simpler than Kβ because the exchange interaction between the 3d and 2p electrons is weaker than that between 3d and 3p. We may associate analytically decomposed Lorentzians with particular electronic transitions. The Kα1 emission peak around ωe = 8048 eV can be decomposed into two Lorentzians labeled F and G. The spectral shape of the main peak G coincides with the Kα1 fluorescence spectrum. Component G is considered to originate predominantly from the 1s1 → 2p5 diagram transition. Comparing with the previous analysis on the Kα fluorescence spectrum10,13, we assign the weaker peak F to a satellite due to the 1s13d9 → 2p53d9 transition. However, we consider that component F may contain just a part of the 1s13d9 → 2p53d9 transition, and the rest may be included in the main component G. The Kα2 peak also consists of two Lorentzians D and E, which are considered to have the same origins as F and G, respectively. The branching ratio (Kα2/Kα1) of the nonlinear RIXS is found slightly higher than the Kα fluorescence, which may be due to the resonant excitation at a single photon energy of 8905 eV.
When ωp was detuned from the Kβ peak, the Kα emission spectrum changed shape as expected for RIXS. We need at least three Lorentzians, G´, B, and C, to reproduce the Kα1 emission spectrum at ωp = 8897 eV. The broad peak labeled G´ lies near the constant energy transfer line, ωp−ωe = 857 eV, which passes through the peak G on the two-dimensional spectrum. Therefore, we consider G´ is the tail of G. This is a characteristic of the coherent process, where the conservation of energy must be satisfied between the states i and f, but not for the intermediate state n in Eq. (2). On the other hand, the Kα1 emission spectrum for ωp = 8912 eV has no sign of the tail of G, and can be reproduced by two Lorentzians, I and J. The three peaks, C, G, and J, have similar central photon energies, indicating that they have the same origin, while larger energy difference among the satellite peaks, B, F, and I, implies that they might come from different electronic states. Unlike Kα1, the Kα2 emission spectra can be reproduced with one or two components.
Analysis of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\)
Now, we apply the same procedure as Fig. 2 to decompose all emission spectra into Lorentzian components in order to understand the overall structure of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\). We independently fitted the emission spectrum at each ωp with a minimum number of Lorentzians and no constant background (Supplementary Fig. 4). One 2D spectral feature may be sliced into several one-dimensional Lorentzians. We did not impose any constraint on the fitting parameters, such as the peak photon energy, to connect a Lorentzian to its neighbors along the ωp direction. Figure 3a and b show the decomposition result in the Kα1 and Kα2 regions, respectively. The components in the Kα1 region can be classified into several clusters, while those for Kα2 are not dissolved well as is pointed out above.

a Decomposition of the Kα1 emission spectra. Each symbol and its size represent the peak center and area of the Lorentzian, respectively. Components denoted by the same symbol are considered to have the same origin. The cluster ID for closed diamonds (B), squares (F), and hexagons (I) is taken from the corresponding peak in Fig. 2. Open circles and open diamonds are considered to be the main peak and the Kα1 fluorescence from relaxed state (see text for details). Triangles are assigned to the tail of main peak. Dashed line indicates the constant energy transfer line, ωp−ωe = 857 eV, which pass through the main peak. b Decomposition of the Kα2 emission spectra. Circles indicate the center of the Lorentzian with the peak area represented by the size. c, d Theoretical \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) calculated for the 3p53d9 satellite peaks. The calculated spectrum is shifted to align with the experimental data. e Schematic energy-level diagram of copper metal for |i〉 = |3p5 3d9〉, |n〉 = |1s1 3d9〉, and |f〉 = |2p5 3d9〉. The energy difference is taken from the theoretical calculation.
To decipher the spectral feature, we compare theoretical calculation based on Eq. (1). We used the standard multiplet ligand field theory16,17,28 with octahedral (Oh) crystal fields of a face-centered cubic structure of copper metal (see the “Methods” section for details). In the rest of this article, we focus on the 3p53d9 satellites, i.e., transitions involving |i〉 = |3p5 3d9〉. We ignored the i-dependence of τi in Eq. (1), and calculated the scattering cross-section of the RIXS part as 3p53d9ε → 1s13d9ε → 2p53d9ε using Eq. (2), where we considered a Cu2+ ion instead of explicitly taking into account the shake-off process10.
Figure 3c and d show \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) calculated for the 3p53d9 satellites. Our simulation predicted three peaks for both the Kα1 and Kα2 emission. Since the 3p53d9 configuration has two holes, the possible LS terms are 1,3P, 1,3D, and 1,3F, corresponding to the orbital angular momentum of L = 1, 2, and 3, respectively17. The superscript indicates the spin multiplicity, e.g., 2 S + 1 = 1 for singlet and 3 for triplet, where S is the spin angular momentum. These L terms split due to the direct Coulomb interaction (Fig. 3e). Each L term further splits by the 3p−3d exchange interaction. The 3p and weaker 3d spin–orbit interactions remove the degeneracy of the triplet, and mixes up all the LS terms. The spin–orbit interaction is weaker than the exchange interaction, and the effect would be experimentally indistinguishable. We retain the LS notation for the sake of clarity. In addition to the above interactions within an atom, the crystal field also splits the 3d level into eg and t2g.
Roughly speaking, the 3p53d9 configuration can be categorized energetically into three groups of two terms each, i.e., 1F and 1P, 3D and 3P, or 1D and 3F, which are separated by several electron volts. The two terms in each group happen to have close energies. The intermediate state, |n〉 = |1s1 3d9〉, is characterized by 1s1t2g6eg3 and 1s1t2g5eg4, because the separation (crystal-field splitting), which was set to 1 eV in the calculation, is larger than a calculated energy splitting of 0.21 eV between 1s13d9 1D and 1s13d9 3D by the exchange interaction. The energy split in |n〉 = |1s1 3d9〉 is smaller than those in |i〉 = |3p5 3d9〉. Thus, three satellite peaks in Fig. 3c and 3d correspond to the three groups in |i〉 = |3p5 3d9〉. This also applies to the Kβ fluorescence and is the origin of a three-peak structure, which has been theoretically predicted but experimentally unresolved9,10,11,12. The final state, f, is dominated by the spin–orbit interaction for the 2p orbital. We note that the 1s1t2g6eg3 and 1s1t2g5eg4 levels form bands in the real system, and they may be experimentally inseparable in the emission spectrum.
Comparing Fig. 3a with Fig. 3c, we interpret the experimentally resolved clusters as follows. A cluster of strong components (open circles) around (8905 eV, 8048 eV) is the main peak by the 3p5 → 1s1 → 2p5 RIXS process. The main peak may be doublet, because the 3p5 level splits into 3p1/2 and 3p3/2 by the spin–orbit interaction. The separation is reported at about 2.5 eV (ref. 10), and hence, the 3p5 (3p1/2) related peak may appear around ωp = 8903 eV, where we found another cluster (open diamonds). The diagonal streak (open triangles and a part of open diamonds) extending from the main peak towards the lower left have similar energy transfers, ωp−ωe = 857 eV. We assign the streak to the tail of the main peak. However, no counterpart was found in the higher photon-energy region. We consider that the components lying along ωe = 8048 eV apart from the main peak are unseparated satellites and the Kα1 fluorescence from relaxed states.
Three clusters labeled B, F, and I (see also Supplementary Fig. 5) are considered to be the satellites involving the 3p53d9 1F/1P, 3D/3P, and 1D/3F terms, respectively. It is such separation of the 3p53d9 satellites from the main peak that we expected for the 2D fluorescence spectrum, and has not been experimentally resolved for the one-dimensional Kβ fluorescence spectrum9,10,11,12. The theoretical calculation predicted almost the same emission photon energy for these three components, while we find the energies are slightly different. The gravities of the clusters B, F, and I are ωe = 8045.4 ± 0.06, 8045.6 ± 0.11, and 8046.8 ± 0.04 eV, respectively. This implies that transition probabilities to each state in |n〉 = |1s1 3d9〉 may differ among the 3p53d9 1F/1P, 3D/3P, and 1D/3F terms in the real system.
The 3p53d9 1F/1P, 3D/3P, and 1D/3F components less disperse along ωe, therefore, we discuss the ωp dependence by integrating \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) over ωe within the Kα1 region (Fig. 4). The theoretical calculation well reproduces the experimentally deduced spectrum. The agreement is satisfactory, considering that we did not make any empirical adjustments during the calculation. Especially, the energy separations among the three peaks determined mainly by the direct and exchange interactions are in good agreement. However, the relative peak height among the three peaks differs between theory and experiment. This discrepancy may be due to partial extraction of the satellite contribution in our phenomenological analysis of the Kα1 emission spectra, and ignored contributions from multi-hole states other than 3p53d9.

Symbols represent the peak area of the fitted Lorentzian. Vertical bar indicates the error of the Lorentzian fitting. Diamonds, squares, and circles represent the same components as shown in Fig. 3a. Solid line represents the ωe-integrated value over the Kα1 region of the calculated \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) shown in Fig. 3c.
Discussion
We successfully realized our nonlinear RIXS scheme on copper metal and measured the 2D fluorescence spectrum as \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\). Although the main part of the spectrum is simple due to the complete 3d subshell in copper metal, we showed that we could isolate the weaker satellite peaks and identify them arising from the two-hole 3p53d9 configuration with the help of our multiplet ligand field calculation. Because of the Auger cascade, the initial state of the RIXS part, i, is not exactly the same as the final state of the Kβ fluorescence. Nevertheless, \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\) retained the three-peak satellite structure expected for the Kβ fluorescence spectrum. A possible reason is that solid-state effects could suppress multi-hole states other than the 3p53d9 configuration (see Supplementary Note 1).
In the case of compounds including transition metals with incomplete 3d subshell, the exchange interaction splits the main peak into the Kβ´ and Kβ1,3 peaks, which are known as a useful indicators of the spin and oxidation state1,2,3. For example, the Kβ´ peak of Mn(II) compounds is a transition to a 5P final state with the 3p spin antiparallel to the five 3d spins, while the Kβ1,3 peak is to a parallel-spin 7P final state. However, the Kβ1,3 peak is also shaped by the 5P term with a different 3d spin configuration29. The spectrum becomes more complicated for Mn(III) and Mn(IV), which may relate to the oxygen-evolving complex4,5,6,7. The nonlinear RIXS could serve for a detailed understanding of the spin and oxidation state by resolving such overlapping terms. Our analysis still relies on the phenomenological decomposition of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\), which worked relatively well due to the simpler Kα1 spectrum for copper metal. However, we anticipate that further theoretical and experimental investigation would lead to sophisticated analysis free from phenomenological modeling.
In the present study, we incorporate the sequential two-photon absorption into RIXS to realize core-to-core absorption. It would be possible to use absorption channels from core to valence30. Apart from realizing absorption to the occupied state, other nonlinear processes are also interesting candidates for nonlinear RIXS. Inclusion of direct two-photon absorption is a straightforward application of the present study. Combination of direct two-photon absorption31,32,33 and RIXS might be useful to investigate directly the 3d state because the 1s → 3d transition is allowed and the 1s → 4p is forbidden by the selection rule. Stimulated emission34,35,36, which can enhance the fluorescence yield, and select a particular radiative transition of interest, is another interesting nonlinear process to be combined.
Methods
Experimental setup
SACLA was operated in self-amplified spontaneous emission (SASE) mode and at a repetition rate of 60 Hz. The half of X-ray pulses were delivered to experimental station 4 of BL3, where we conducted our experiment. The broad-band SASE was tuned to cover the whole Kβ spectral range so that the nonlinear RIXS plane could be measured simply by scanning the monochromator without changing the accelerator and the undulator parameters. The focus size was measured by knife-edge scans with 200-μmϕ gold wires. The pulse energy at the focal position was determined shot by shot using a calibrated beam monitor37. The shot-by-shot fluctuation was used to reveal the pulse-energy dependence of the emission intensity (Supplementary Fig. 6). The copper foil was continuously shifted to expose the undamaged fresh surface. The foil was tilted by 30° from the normal incidence to measure the X-ray emission at right angles to the incoming beam in the polarization plane (Supplementary Fig. 2). This geometry suppresses stray X-rays due to elastic and Compton scattering.
The single-shot polychromator consisted of four cylindrically bent Si (444) crystals set in the von Hamos geometry2 and a multi-port charge-coupled device38 (MPCCD). The crystal dimension was 100 mm along the circumferential direction and 25 mm along the dispersion direction, and the curvature radius was 500 mm. The MPCCD was able to detect a single photon and was suitable to measure the weak nonlinear signal. The nominal photon-energy resolution of the polychromator is calculated to be 0.075 eV/pixel. The pixel number of MPCCD was 1024 along the dispersed direction. The corresponding photon-energy range of the polychromator was about 77 eV, which was enough to cover both the Kα1 and Kα2 spectral ranges. The absolute photon energy of the polychromator was calibrated to the Kα1 peak in literature10. The photon energy of the monochromator was calibrated to the absorption edge of a copper foil beforehand.
Theoretical calculation
Our aim in this study is to calculate the 3d9 components of \({\sigma }_{{{{{{\rm{RIXS}}}}}}}^{\left(2\right)}\), i.e., the 3d9 → 3p53d9ε and 3p53d9ε → 1s13d9ε→2p53d9ε RIXS. We used the standard multiplet ligand field theory16,17,28 with octahedral (Oh) crystal field and did not make any empirical adjustments to the term averaged values of the Slater integrals for the 3d9, 3p53d9, 2p53d9, and 1s13d9 configurations. In the spirit of the early phase of the theoretical study, this is a sensible approach and has a more general interest beyond the specific experiment discussed here. The intra-atomic correlation not included in this scheme was treated approximately by reducing the values of the Slater integrals. The Slater integrals and the spin-orbit coupling constants were calculated by the Hartree–Fock code39, and then the Slater integrals were scaled down to 80% as usual17. The crystal field describes the breaking of the 3d orbital degeneracy in copper metal due to the effect of the electrical field of neighboring copper atoms. The crystal-field splitting parameter was set to 10Dq = 1.0 eV. We assumed that the shake-up process is negligible12, and the shake-off probability does not strongly depend on the details of the multiplet. We calculated σgi in Eq. (1) as a 3d9 → 3p53d9ε transition process instead of 3d10 → 3p53d9ε2 (ref. 10). Because of a similar reason, we set τi = 0.15 eV, however, it may strongly depend on the spin state in other 3d configurations28.
Data availability
The datasets that support the results of this study can be found in this published article. Source data are provided with this paper.
Code availability
The codes of the computer simulations are available from M.T. upon request.
Change history
10 August 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41467-023-40664-5
References
van Bokhoven, J. A. & Lamberti, C. (eds) X-ray Absorption and X-ray Emission Spectroscopy (Wiley, West Sussex, 2016).
Bergmann, U. & Glatzel, P. X-ray emission spectroscopy. Photosynth. Res. 102, 255–266 (2009).
Glatzel, P. & Bergmann, U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev. 249, 65–95 (2005).
Davis, K. M. et al. Rapid evolution of Photosystem II electronic structure during Water Splitting. Phys. Rev. X 8, 041014 (2018).
Zaharieva, I. et al. Room-temperature energy-sampling Kβ X‐ray emission spectroscopy of the Mn4Ca complex of photosynthesis reveals three manganese-centered oxidation steps and suggests a coordination change prior to O2 formation. Biochemistry 65, 4197–4211 (2016).
Visser, H. et al. Mn K-edge XANES and Kβ XES studies of two Mn-Oxo binuclear complexes: investigation of three different oxidation states relevant to the oxygen-evolving complex of Photosystem II. J. Am. Chem. Soc. 123, 7031–7039 (2001).
Messinger, J. et al. Absence of Mn-centered oxidation in the S2→S3 transition: implications for the mechanism of photosynthetic water oxidation. J. Am. Chem. Soc. 123, 7804–7820 (2001).
Tsutsumi, K., Nakamori, H. & Ichikawa, K. X-ray Mn Kβ emission spectra of manganese oxides and manganates. Phys. Rev. B 13, 929–933 (1976).
LaVilla, R. E. Double-vacancy transition in the copper Kβ1,3 emission spectrum. Phys. Rev. A 19, 717–720 (1979).
Deutsch, M. et al. Kα and Kβ x-ray emission spectra of copper. Phys. Rev. A 51, 283–296 (1995).
Enkisch, H., Sternemann, C., Paulus, M., Volmer, M. & Schülke, W. 3d spectator hole satellites of the Cu Kβ1,3 and Kβ2,5 emission spectrum. Phys. Rev. A 70, 022508 (2004).
Pham, T., Nguyen, T., Lowe, J., Grant, I. & Chantler, C. Characterization of the copper Kβ x-ray emission profile: an ab initio multi-configuration Dirac–Hartree–Fock approach with Bayesian constraints. J. Phys. B: Mol. Opt. Phys. 49, 035601 (2016).
Galambosi, S. et al. Near-threshold multielectronic effects in the Cu Kα1,2 x-ray spectrum. Phys. Rev. A 67, 022510 (2003).
Chantler, C. T., Hayward, A. C. L. & Grant, I. P. Theoretical determination of characteristic X-ray lines and the copper Kα spectrum. Phys. Rev. Lett. 103, 123002 (2009).
Diamant, R., Huotari, S., Hämäläinen, K., Kao, C. C. & Deutsch, M. Cu Khα1,2 hypersatellites: suprathreshold evolution of a hollow-atom x-ray spectrum. Phys. Rev. A 62, 052519 (2000).
Kotani, A. & Shin, S. Resonant inelastic x-ray scattering spectra for electron in solids. Rev. Mod. Phys. 73, 203–246 (2001).
de Groot, F. & Kotani, A. Core Level Spectroscopy of Solids (CRC Press, Boca Raton, 2008).
Gel’mukhanov, F., Odelius, M., Polyutov, S. P., Föhlisch, A. & Kimberg, V. Dynamics of resonant x-ray and Auger scattering. Rev. Mod. Phys. 93, 035001 (2021).
Scully, M. O. & Zubairy M. S. Quantum Optics (Cambridge University Press, Cambridge, 1997).
Kanter, E. P. et al. Unveiling and driving hidden resonances with high-fluence, high-intensity X-ray pulses. Phys. Rev. Lett. 107, 233001 (2011).
Rudek, B. et al. Ultra-efficient ionization of heavy atoms by intense X-ray free-electron pulses. Nat. Photon 6, 858–865 (2012).
Mukoyama, T. & Taniguchi, K. Atomic excitation as the result of inner-shell vacancy production. Phys. Rev. A 36, 639–698 (1987).
Rohringer, N. & Robin, S. X-ray nonlinear optical processes using a self-amplified spontaneous emission free-electron laser. Phys. Rev. A 76, 033416 (2007).
Tulkki, J. & Åberg, T. Behaviour of Raman resonance scattering across the K x-ray absorption edge. J. Phys. B: Mol. Phys. 15, L435–L440 (1982).
Ishikawa, T. et al. A compact X-ray free-electron laser emitting in the sub-ångström region. Nat. Photon. 6, 540–544 (2012).
Inubushi, Y. et al. Measurement of the X-ray spectrum of a free electron laser with a wide-range high-resolution single-shot spectrometer. Appl. Sci. 7, 584 (2017).
Yumoto, H. et al. Focusing of X-ray free-electron laser pulses with reflective optics. Nat. Photon. 7, 43–47 (2013).
Taguchi, M., Uozumi, T. & Kotani, A. Theory of X-ray photoemission and X-ray emission spectra in Mn compounds. J. Phys. Soc. Jpn. 66, 247–256 (1997).
Peng, G. et al. High-resolution manganese X-ray fluorescence spectroscopy. oxidation-state and spin-state sensitivity. J. Am. Chem. Soc. 116, 2914–2920 (1994).
Lee, N., Petrenko, T., Bergmann, U., Neese, F. & DeBeer, S. Probing valence orbital composition with iron Kβ X-ray emission spectroscopy. J. Am. Chem. Soc. 132, 9715–9727 (2010).
Tamasaku, K. et al. X-ray two-photon absorption competing against single and sequential multiphoton processes. Nat. Photon. 8, 313–316 (2014).
Ghimire, S. et al. Nonsequential two-photon absorption from the K shell in solid zirconium. Phys. Rev. A 94, 043418 (2016).
Tamasaku, K. et al. Nonlinear spectroscopy with X-ray two-photon absorption in metallic copper. Phys. Rev. Lett. 121, 083901 (2018).
Beye, M. et al. Stimulated X-ray emission for materials science. Nature 501, 191–194 (2013).
Kroll, T. et al. Stimulated X-ray emission spectroscopy in transition metal complexes. Phys. Rev. Lett. 120, 133203 (2018).
Kroll, T. et al. Observation of seeded Mn Kβ stimulated X-ray emission using two-color X-ray free-electron laser pulses. Phys. Rev. Lett. 125, 037404 (2020).
Tono, K. et al. Beamline, experimental stations and photon beam diagnostics for the hard x-ray free electron laser of SACLA. New. J. Phys. 15, 083035 (2013).
Kameshima, T. et al. Development of an X-ray pixel detector with multi-port charge-coupled device for X-ray free-electron laser experiments. Rev. Sci. Instrum. 85, 033110 (2014).
Cowan, R. D. The Theory of Atomic Structure and Spectra (University of California Press, Berkeley, 1981).
Acknowledgements
This work was supported by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan under grant number 21H03746 (K.T.). The experiments were performed with the approval of JASRI under proposal numbers 2017B8034, 2018A8046, 2018B8057, 2019A8049, 2020A8016, and 2021A8018 (K.T.).
Author information
Authors and Affiliations
Contributions
K.T. conceived the study. T.O., I.I., Y.I., and K.T. acquired the experimental data. M.T. performed multiplet ligand field calculation. K.T. analyzed data. M.T. and K.T. wrote the manuscript. T.I. and M.Y. supervised the work. All authors contributed to discussion and manuscript revisions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Frank de Groot, Victor Kimberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tamasaku, K., Taguchi, M., Inoue, I. et al. Two-dimensional Kβ-Kα fluorescence spectrum by nonlinear resonant inelastic X-ray scattering. Nat Commun 14, 4262 (2023). https://doi.org/10.1038/s41467-023-39967-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39967-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
