
Abstract
Femtosecond pump-probe spectroscopy using ultrafast optical and infrared pulses has become an essential tool to discover and understand complex electronic and structural dynamics in solvated molecular, biological, and material systems. Here we report the experimental realization of an ultrafast two-color X-ray pump X-ray probe transient absorption experiment performed in solution. A 10 fs X-ray pump pulse creates a localized excitation by removing a 1s electron from an Fe atom in solvated ferro- and ferricyanide complexes. Following the ensuing Auger–Meitner cascade, the second X-ray pulse probes the Fe 1s → 3p transitions in resultant novel core-excited electronic states. Careful comparison of the experimental spectra with theory, extracts +2 eV shifts in transition energies per valence hole, providing insight into correlated interactions of valence 3d with 3p and deeper-lying electrons. Such information is essential for accurate modeling and predictive synthesis of transition metal complexes relevant for applications ranging from catalysis to information storage technology. This study demonstrates the experimental realization of the scientific opportunities possible with the continued development of multicolor multi-pulse X-ray spectroscopy to study electronic correlations in complex condensed phase systems.
Similar content being viewed by others

Introduction
Femtosecond (fs) pump–probe spectroscopy is now used extensively in the optical and infrared (IR) regimes to understand complex chemical phenomena in the condensed phase following the development of high intensity, commercially available, and tunable ultrafast laser systems. In these widely used experiments, the pump pulse initiates a non-equilibrium process in the excited-state (or ground-state) of the system of interest, and the delayed probe pulse monitors the time evolution of the system. For example, optical transient spectroscopy experiments measure signatures of evolving electronic states following photoexcitation in complex systems1,2. On the other hand, transient IR experiments measure structural dynamics and non-equilibrium vibrational relaxation in photo-excited systems3,4. Femtosecond pump–probe spectroscopy in the condensed phase has resulted in new discoveries in the fields of biology, chemistry, and material science. The pump–probe experiments have served as precursors to coherent nonlinear multidimensional techniques that are now used extensively to map charge and energy transfer pathways in complex systems5,6. An important limitation, however, is that fs optical and IR spectra are only indirectly sensitive to valence charge distributions across specific atomic sites.
The advent of third generation synchrotrons and the development of table-top X-ray sources has resulted in the development of transient X-ray techniques for measuring the X-ray absorption and emission spectra of core electrons in solvated complexes following photoexcitation with an optical pump pulse7,8,9,10,11. X-ray spectroscopy is an element-specific probe of electronic and atomic structure and fs X-ray pulses from X-ray free electron lasers (XFELs) are allowing researchers to routinely measure X-ray absorption and emission spectra of ultrafast photochemical processes in solution12,13,14,15,16. Optical pump X-ray probe experiments have also enabled researchers to measure coherently coupled electronic and atomic motions and ultrafast electron delocalization in complex photochemical phenomena in solution17,18,19,20.
The generation of tunable, high intensity, time-delayed, femtosecond X-ray pulse pairs has been recently demonstrated at various XFELs around the world21,22,23,24,25. These technological developments have enabled X-ray pump X-ray probe experiments studying nuclear and electronic dynamics at different atomic sites in small molecules in the gas phase through various electron ionization detection schemes26,27,28. Two-pulse X-ray photon correlation techniques have measured non-equilibrium structure correlations on short length scales in solutions and solids29,30,31,32,33,34,35. Additionally, nonlinear light matter interactions with intense X-ray pulses have resulted in seeded stimulated emission signals at the Mn K-edge in concentrated solutions36.
Recently, we proposed and theoretically modeled a novel approach to measure core-valence electronic correlations in solvated chemical systems via ultrafast X-ray pump X-ray probe (XPXP) transient absorption spectroscopy, using the transition metal complex, K4FeII(CN)6, as a model system37. X-ray pump pulses were used to create a localized (element-specific) excitation consisting of a 1s core hole in the Fe atom. We used a combination of atomic electron cascade calculations and excited-state time-dependent density functional theory (TDDFT) calculations to predict changes in the X-ray probe transmission near the Fe K-edge following the X-ray pump interaction. Our work found several spectral features below the Fe K-edge absorption edge that reported on the ligand-field splitting and chemically relevant 3p and 3d electron interactions.
This paper is the experimental realization of our previous theoretical work described above37, presenting an XPXP transient absorption experiment of molecules in solution. The measured XPXP transient absorption spectra combined with simulations directly measure the oxidation state dependent electronic cascade pathways and the chemically relevant 3p–3d valence interaction strengths in FeII and FeIII hexacyanoferrates dissolved in water. This study also serves as an experimental demonstration of the feasibility of two-color XPXP transient absorption, paving the way for multi-pulse nonlinear multidimensional X-ray spectroscopy in solution.
Results
Implementation of the XPXP experiment
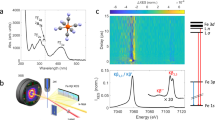
Figure 1a depicts the generation of an energy tunable ~10 fs X-ray pulse pair, separated by a time delay (τ) coincident on a thin liquid jet (250 µm) containing an aqueous solution of either K4FeII(CN)6 or K3FeIII(CN)638. The X-ray pump pulse (7.2 keV, blue dashed line in Fig. 1c) ionizes the sample by removing a 1s electron from the Fe atom. The energy of the X-ray pump pulse is chosen to ensure that fluctuations in its photon energy or spectral shape do not influence the character of the initially prepared ionized state in the Fe complex. Figure 1b provides an example of an Auger-Meitner electron cascade and we stress that this is only one of a multitude of possible pathways explored by the system following the interaction with the X-ray pump pulse. The representative cascade pathway in Fig. 1b proceeds in experimental time as follows: (1) a pump photon ejects a 1s electron to the continuum, (2) a Kα fluorescence event fills the 1s hole and emits a photon, (3) an Auger–Meitner decay fills the new 2p hole while removing a 3p electron, (4) Coster–Kronig decay fills the 3s hole, ejecting a 3dt2g electron, resulting in an electron configuration of [Ne]3s23px=53dt2gy=4. Following the Auger–Meitner electronic cascade (5), the second X-ray pulse centered at 7.06 keV probes the ensemble of resultant core-electronic excited states containing 3p holes. The transmitted X-ray probe pulse is spectrally resolved to measure the transient X-ray absorption spectrum (see Fig. 1a, c).

a Experimental layout. Both the 7.20 keV X-ray pump (blue) and the 7.060 keV X-ray probe (red) are generated collinearly by separate undulators from the same electron bunch (black circle). Insertion of a chicane delay allows for the pump–probe timing (τ) to be controlled. Both X-ray pulses are focused onto the thin liquid jet by a pair of Kirkpatrick–Baez (KB) mirrors to a spot size of 100 nm. The transmitted X-ray spectra are measured using an analyzer crystal and an X-ray detector (ePix 10 K). b Example of an Auger–Meitner cascade in the iron complexes following removal of an Fe 1s electron. The example shows that the 1s hole is quickly filled by various processes (labelled 2–4) including fluorescence, Auger–Meitner decay, and Coster–Kronig decay events. The resultant electronic states have an electronic configuration of [Ne]3s23px3dt2gy. After delay time, τ, the X-ray probe pulse, resonant with the 1s → 3p transitions interrogates the core electronic excited states formed following the X-ray pump interaction. c Equilibrium ground state Fe K-edge X-ray absorption spectrum and Kβ X-ray emission spectrum of a 500 mM aqueous solution of K4FeII(CN)6. The X-ray absorption reference spectrum (solid black, normalized to the post-edge) displays weak pre-edge features of 1s → 3deg and 1s → π* transitions characteristic of an Fe(II) complex as described in ref. 38. Transient 3p absorption features measured by the X-ray probe pulse are shown in red. See Supplementary Note 4 for further details on the treatment of the X-ray absorption, emission spectra, and transient signals.
Prediction of transient core-excited electronic states probed in the XPXP experiment
To predict the transient core-excited electronic states created in the X-ray probe’s spectral window (7060 ± 10 eV) following the removal of a 1s core electron in the Fe atom by the X-ray pump pulse, we performed an electron cascade Markov-chain Monte Carlo (MC-MC) calculation, as described previously37. These calculations serve as a starting point for understanding the spectral features observed in this XPXP experiment. Figure 2 shows the time evolution of [Ne]3s23p53dt2gy electronic configurations in the FeII (solid lines) and FeIII (dashed lines) atomic systems produced by the MC-MC simulations in the first 100 fs after ionization of a 1s electron. These electronic states are the most probable (~20%) among all states populated at the timescale probed in our experiment. Identically colored lines represent states that have lost an equivalent number of electrons from the valence through Auger–Meitner events (NAuger) as indicated in the legend of Figure 2. For example, the solid and dashed blue lines (NAuger=0) in Fig. 2 correspond to the initial 3d6 and 3d5 configurations for FeII and FeIII atoms, respectively. Interestingly, we note that the probability of observing the [Ne]3s23p53dt2g6 state of the FeII system decays to zero within 1 fs. Given the experimental X-ray pulse durations of ~10 fs, we expect the largest contributions from 3p5 core-excited states with 0, 1, and 3 Auger–Meitner events (NAuger = 0, 1, and 3) depending on the starting oxidation state of the Fe atom. The relative contributions of each of the core-excited states to the measured XPXP transient absorption spectrum depend on their calculated probabilities plotted in Fig. 2. The probabilities in Fig. 2 are on the same order of magnitude as those calculated from experimentally determined fluorescence yields of the Fe atom39, and discussed in Supplementary Note 4. The MC-MC simulations also predict the average oxidation state during the first 10 fs of the electronic cascade to be 3d4.2 and 3d3.8 for the FeII and FeIII systems, respectively (see Supplementary Note 3). Combining all the information gleaned from the electron cascade calculations, we would expect to observe the following in the transient XPXP signal: (i) two or three spectral features representing 1s → 3p transitions to distinct electronic states for the FeII and FeIII samples, and (ii) a lack of the 1s → [Ne]3s23p53dt2g6 transition in the FeII data.

Monte Carlo electron cascade calculations of atomic FeII (solid lines) and FeIII (dashed lines) are shown above where the color corresponds to an equivalent number of electrons lost from the valence through Auger–Meitner events (NAuger) in the two Fe species. The probabilities for each state are given with respect to all electronic configurations (including non [Ne]3s23p5 states). At long times (≥1 ps), [Ne]3s23p53dt2gy states comprise 18 and 21% of all possible electronic states for FeII and FeIII atoms, respectively. The instrument response function of the present experiment (~14 fs) is denoted by the red diamond on the time axis. Source data are provided as a Source data file.
Femtosecond XPXP signal measures core-valence interactions
Figure 3 displays the XPXP signal as a change in transmission of the X-ray probe spectrum following 1s excitation of the Fe atom in aqueous solutions of K4FeII(CN)6 or K3FeIII(CN)6 complexes by the X-ray pump pulse. The data are measured at a nominal delay time of 0 fs between the two 10 fs X-ray pulses. Both plots display the presence of transient 1s → 3p absorption features as two negative spectral features, blue shifted from the molecular Fe Kβ energy (7058 eV and 7059 eV for FeII and FeIII molecular complexes respectively, vertical dashed lines in Fig. 3). These transitions are a result of new core-excited electronic states with 3p holes formed following the initial steps of an Auger–Meitner cascade (see Fig. 1b for an example of electron cascade). The XPXP data were processed by separately averaging and subtracting the X-ray pumped and unpumped probe spectra to produce the change in transmission (ΔT/T) data and error bounds in Fig. 3. Individual X-ray shots exhibiting abnormal characteristics in intensity or spectral distribution were excluded from the analysis. Details on the processing of the raw data are provided in Supplementary Note 2. The XPXP signal is dependent on the X-ray pump and probe pulse fluence and the XPXP transient absorption signal is absent in pure water (see Supplementary Figs. 3 and 6).

XPXP signal plotted as change in transmission of the X-ray probe (ΔT/T) following X-ray pumping of the aqueous solutions of (a) K4FeII(CN)6 and (b) K3FeIII(CN)6 measured at τ = \(0\) fs. Negative peaks centered at ~7.060 and 7.062 keV correspond to additional 1s → 3p absorption due to the presence of 3p holes resulting from the Auger–Meitner cascade, reducing the intensity of the X-ray probe pulse through the sample. The transient data is fit (red line) using a sum of two Gaussians and best fit values are listed in Supplementary Table 2. The vertical dashed line represents the molecular Kβ emission line. Error bars are calculated as the standard error for each energy bin. See Supplementary Note 2 for more details. Source data are provided as a Source data file.
The spectral features in the XPXP signal are fit using Gaussian lineshapes. The best fit is plotted as solid lines in Fig. 3a, b, and the peak amplitudes, positions and linewidths extracted from the fit are listed in Supplementary Table 2. From the fits, we determine that the XPXP transient signals in FeII and FeIII complexes exhibit two 1s → 3p transitions at ~7060 and ~7062 eV. The peaks in the transient difference XPXP spectra from the FeII and FeIII complexes are blue-shifted from the peak of the Kβ X-ray emission signals (Supplementary Fig. 8) by 1.9 and 1.2 eV respectively. The fits reveal that the overall transient signal in the FeII complex is blue shifted by 0.35 eV with respect to the FeIII signal. We note that this shift is within the 0.4 eV resolution of the detection spectrometer. The widths of all the 1s → 3p transmission peaks in the transient XPXP spectra for both samples are ~2 eV. These widths are determined by the non-radiative lifetime of the 3p hole and are narrower compared to the width of the Kβ emission peak, which is determined by the shorter lifetime of the 1s hole. The narrower peaks in the XPXP spectra make it easier to resolve individual core-excited electronic states. We compare the intensities of the spectral features by their integrated peak areas (Supplementary Table 2) and find that for the FeII sample, the peak at ~7060 eV is 15% more intense than the peak at ~7062 eV. In the case of the FeIII sample, the peak at ~7060 eV is 3% more intense than the peak at ~7062 eV. Assuming similar dipole strengths for all 1s → 3p transitions, the integrated area of each peak corresponds to the relative population of that particular 3p state in the sample of interest. We also observe that the transitions in the XPXP FeIII data show overall greater intensity by 10% of the observed transitions relative to the FeII data and we attribute this to an initial additional hole in the 3dt2g orbital. Our observations are in agreement with previous calculations on the generalized case of 3p vacancy dependent M-edge spectroscopy40. In summary, the 1s → 3p XPXP transient absorption spectra for the FeII and FeIII complexes show remarkable similarities in the peak positions, integrated areas, and lineshapes.
To aid the interpretation of the data shown in Fig. 3, we perform TDDFT calculations of 1s → 3p X-ray absorption spectra for a variety of reference electronic configurations on separate geometries for both FeII and FeIII compounds following our previously published computational approach37. Additional computational details are provided in Supplementary Note 5. Figure 4a, b show calculated 1s → 3p transitions in core-excited states with specific electronic configurations. The TDDFT calculation reports that each progressive hole in the 3d valence shell produces a corresponding shift in the 1s → 3p energy gap. From Fig. 4, we note that in the [Ne]3s23p53dt2gy configurations, each additional hole in the t2g orbital corresponds to a + 2 eV shift of the 1s → 3p calculated transition. This shift serves as an indirect reporter on the strength of the 3p–3d interactions in the molecular complex under investigation. The TDDFT calculations reveal that 1s → 3p transitions for states with electronic configurations of the type [Ne]3s23px<53dt2gy are shifted ~6 eV to the blue of the experimental Kβ emission peak. Further, we see that the peak from the 1s → [Ne]3s23p53dt2g6 configuration for the FeII species (starred in Fig. 4a), would be located around the peak of the Kβ emission line.

Calculated shifts in the 1s → 3p transition energy and oscillator strengths for (a) [FeII(CN)6]4− and (b) [FeIII(CN)6]3−, for a selected sample of core-excited electronic states. The starred transition is not observed in the data. Each of the calculated transitions are broadened by 1.5 eV and shifted by 143.8 eV to match the experimental 1s → 3deg pre-edge feature in the Fe K-edge X-ray absorption spectrum of K4FeII(CN)6 (plotted in ref. 38).
Combining the electron cascade (Fig. 2) and the TDDFT (Fig. 4) calculations, we assign the peaks at 7060 and ~7062 eV in the FeII and FeIII XPXP transient absorption spectra (Fig. 3) to 1s → [Ne]3s23p53dt2g5 and 1s → [Ne]3s23p53dt2g4 transitions, respectively. Both the MC-MC electron cascade simulations and the TDDFT calculations confirm that the valence-hole-free state in the FeII complex does not survive past the nominal 1s core lifetime, and the observed transitions in both complexes originate from similar core-excited states. From the integrated area of the peaks in the XPXP data, we observe that the electron cascade in the FeIII complex generates more highly ionized states in the relative populations of t2g5 and t2g4 states compared to the electron cascade in the FeII complex. A comparison of the TDDFT calculations and the XPXP experimental data reveals that excited states with configurations of [Ne]3s23px<53dt2gy are not formed in this experiment, confirmed by the lack of observed transitions above the signal to noise in Fig. 3 for X-ray probe energies greater than 7063 eV. We note that several factors could contribute to the lack of observing 3px<5 holes in this XPXP experiment including (i) data collection at zero nominal delay between the X-ray pump and probe pulses, limiting the timeframe for electron cascade evolution, and (ii) contribution to the electron cascade by the surrounding ligand or solvent electrons resulting in an effective quenching of the solute’s electron cascade signal by decreasing the 3px<5 lifetime. We consider the possibility that the peaks seen in the XPXP experimental signal could arise from the splitting of the 3p5 configurations via spin–orbit interactions. In reference calculations of 3p5 configurations, the 3p spin–orbit coupling was computed to be ~1.3 eV for both FeII and FeIII complexes, in agreement with previously published data on transition metal ions41. Given that the 3p spin–orbit coupling is less than the linewidth and the energy separation of the observed peaks in the XPXP transient spectra shown in Fig. 3, we confirm the assignment of the observed peaks to 1s→[Ne]3p53dt2g5 and 1s→[Ne]3p53dt2g4 transitions in solvated FeII and FeIII complexes. Our combined femtosecond XPXP experimental data and simulation protocol provide a direct experimental measure of the ~2 eV blue-shift of the 1s → 3p dipole transition as a function of the number of 3d holes in solvated Fe complexes.
Discussion
Measuring the time-evolution of core–valence interactions in transition metal complexes is crucial for controlling electronic correlations in molecules and materials being developed for catalytic, magnetic, and information storage applications. The XPXP experiment reported here is sensitive to the 3p–3d Coulomb and valence interactions as a function of the electronic configuration, oxidation, and spin state of the Fe atom in solvated molecules. By directly probing core-to-core, dipole-allowed, 1s → 3p transitions, our data reports on both the 1s relaxation and the 3p–3d interactions (Supplementary Fig. 10).
In the novel core-excited electronic states produced by the X-ray pump, core and valence electrons rearrange and relax due to the constantly changing electrostatic shielding in the Fe atom, which includes shifts from 3p–3d and 3d–3d Coulomb and valence exchange interactions, crystal field interactions and spin–orbit interactions. These interactions are encoded in numerous transitions connecting the typical K- (1s), L- (2p), or M- (3p) edges via X-ray absorption and emission spectra of transition metal complexes measured at synchrotrons, XFELs and with table-top HHG-based sources (see Fig. 5)42. At the K-edge, Kβ fluorescence XES probes the 3p–3d exchange energy through the relative intensities of the spectral features sensitive to spin and 3d occupancy43,44,45. Similarly, L-edge spectroscopy has been used extensively to monitor the covalency, spin, and back-bonding in transition metals46,47,48,49and M-edge spectra directly measure 3p → 3d transitions50. The extraction of core electronic state-specific information from equilibrium and optically-pumped X-ray absorption spectroscopy (XAS) and XES spectral features at the K-, L- and M-edges is limited given the lifetime broadening of 1s and 2p core holes51 and or further complicated by many possible dipole-allowed transitions to a dense continuum of valence states. Furthermore, the temporal resolution of transient XAS is limited by the lifetime of the valence excited state and the pulse duration of the optical pump pulse. In contrast, the time-resolution of XPXP experiments can be shorter given the availability of few fs X-ray pulses at XFELs.

The thick red arrows correspond to dipole allowed transitions in the XPXP experiment and the thin red line represents the quadrupolar transitions probed in a transient XAS experiment. The red ovals indicate holes. XPXP transient absorption spectra measure the progressive blue-shifting of the 1s → 3p transition energy resulting from the unequal stabilization of 1s, 3p and 3d orbitals as a function of the number of holes in the t2g level. Here we probe core-excited 3p states with filled (a) and unfilled (b) and (c) 3dt2g shells and the resultant peaks are spectrally isolated around the Kβ emission energy peak and well-separated from the absorption edge.
In this XPXP study, our ability to extract microscopic information from the 1s → 3p spectral features is theoretically limited by the 3p non-radiative lifetime broadening and experimentally by the spectrometer (0.40 eV)52 resolution and the 10 fs temporal widths of the X-ray pump and X-ray probe pulses. Given that the temporal profiles of the X-ray pulses are much longer than the core-hole-lifetime of the 1s electron ionized by the X-ray pump pulse, the transient XPXP absorption spectra probe a near static population of core-excited states with varying 3d holes. With the generation of tunable attosecond hard X-ray pump and probe pulses, the XPXP experiment described here could measure transient absorption spectra prior to Auger–Meitner decay and would be uniquely sensitive to tracking specific core-excited electronic states, time-dependent 3p−3d electronic correlations, and the time-evolution of pure valence electronic coherences with atomic specificity.
The interpretation of the experimental XPXP transient absorption spectra in this study relies on the MC-MC simulation of the electron cascade in isolated FeII and FeIII atoms to model the effect of the X-ray pump pulse and the TDDFT calculations of the 1s → 3p5 transitions for a select group of core-excited states in [FeII(CN)6]4− and [FeIII(CN)6]3− complexes. Despite the atomic nature of the electron cascade calculations and the single excitation nature of TDDFT calculations, we are able to predict and measure core hole relaxation manifesting in 3p and 3d transition energies as a function of the 3d hole density. We stress that the successful demonstration of a fs XPXP experiment of a solvated molecular system, as shown here, increases the urgency of developing theoretical tools to accurately model multi-pulse X-ray-matter interactions with complex molecules in solution. Such calculations will be crucial for understanding how electronic correlations, spin–orbit, ligand–field, and solute-solvent interactions are manifested in XPXP transient absorption spectra of core-excited molecular complexes in solution.
Along with theoretical developments, experimental developments in generating intense, multi-color, time-delayed, attosecond pulse-pairs in the hard X-ray regime will result in an extension of the fs XPXP spectroscopy presented here to a coherent multidimensional nonlinear X-ray experiment on molecular complexes in solution. An analogy is the development of third-order nonlinear coherent multidimensional optical and IR spectroscopy to measure couplings between excitonic states and anharmonic vibrations, respectively, following the establishment of femtosecond optical pump–probe and IR pump–probe and transient grating experiments. Mukamel and co-workers have proposed several coherent multidimensional techniques in the X-ray regime to elucidate coherent electronic charge and energy transfer pathways53,54,55. With the availability of tunable attosecond hard X-ray pulses, it will be possible to create and collapse coherences within the 1s core-hole-lifetime, generating opportunities for investigating the coherences between specific core-excited electronic states of complex systems in solution.
Methods
Experimental methods
Experiments were conducted in the Coherent X-ray Imaging (CXI) hutch at the Linac Coherent Light Source56. We utilize a pulse generation scheme similar to that used previously36, utilizing the split undulator method57. Two collinear X-ray pulses ( ≈ 60 μJ/pulse, 10 fs) were generated in a series of undulators from a single electron bunch with a chicane delay controlling the experimental time delay (τ) between the two pulses. As shown in Fig. 1 a, the electron bunch is inefficiently lased in the first half of the undulators to produce the 7.20 keV pump pulse. The remaining electron energy is utilized in the second half of the undulators to produce the lower energy 7.06 keV probe pulse. Data shown here are restricted to τ = 0 fs during the overlap of the pump and probe pulses on the sample. The spectrometer calibration was performed by inserting a channel cut monochromator upstream at the XPP endstation58. The monochromator was tuned to multiple energies in the 7.06 keV region which showed up as valleys, missing spectral frequencies, in the resolved probe spectrum on the ePix 10 K detector59.
We measure the transmission of the probe pulse as a function of pump fluence, achieved by measuring two focal conditions of the X-ray beams on the sample. In the first condition, we position the sample jet at the focus of the two beams. The second condition is achieved with the sample positioned upstream (∆z = +2 mm) to reduce the fluence of both pulses by increasing the spot size (~10 Rayleigh lengths from focus)60. As the response to this technique is linear with respect to the fluence of each pulse, small perturbations in the focal area create a quartic drop in signal strength. Calculations estimating the signal strength with this experimental design are detailed in Supplementary Note 1.
The two focal conditions serve as “pump on” and “pump off” conditions as neither pulse can be uniquely blocked or eliminated without drastically impacting the energy, intensity, and temporal profile of the other pulse. The fact that both pulses are generated from the same electron bunch creates an intrinsic link between the character and intensity of the two pulses. Future experiments will take advantage of a non-collinear generation geometry, generating pulses from distinct electron bunches, enabling direct chopping of the pump pulses either by mechanical means or through electron bunch control. Data are collected at 120 Hz, alternating 36,000 shots (5 min) in each focal condition. Total shot counts for each spectrum are provided in Supplementary Table 1.
The complexes, potassium ferrocyanide (K4[FeII(CN)6]) and potassium ferricyanide (K3[FeIII(CN)6]), were purchased from Sigma Aldrich and used without further purification. Aqueous 500 mM solutions were prepared by dissolving these complexes in ultrapure water. An HPLC pump was used to flow the solutions through a 250 µm (inner diameter) capillary. A catcher placed below the capillary refed the pump to enable closed loop recirculation of the sample. The focus spot size was ≈100 nm60,61. Beam throughput was measured to be 3%. Assuming half of the beam is in the focal volume62, the fluence from each pulse at the sample measured 1.1×1018 W/cm2. Data are further filtered based on correlated intensity measures as specified in the Supplementary Note 2.
Computational methods
Theoretical approaches for the on-the-fly Monte Carlo simulation of the electron cascade63,64,65, and TDDFT calculations of the 3p hole state spectral signatures66,67 have been outlined in previous work and are described in the Supplementary Note 5. The present work builds upon previous work via the consideration of the FeIII electron cascade and in contrasting it with that of the FeII cascade.
Data availability
The data shown in Figs. 2 and 3 are provided as Source data files. Source data are provided with this paper.
References
Schoenlein, R. W., Peteanu, L. A., Mathies, R. A. & Shank, C. V. The first step in vision: femtosecond isomerization of rhodopsin. Science 254, 412–415 (1991).
Jimenez, R., Fleming, G. R., Kumar, P. V. & Maroncelli, M. Femtosecond solvation dynamics of water. Nature 369, 471–473 (1994).
Anfinrud, P. A., Han, C. & Hochstrasser, R. M. Direct observations of ligand dynamics in hemoglobin by subpicosecond infrared-spectroscopy. Proc. Natl Acad. Sci. USA 86, 8387–8391 (1989).
Owrutsky, J. C., Raftery, D. & Hochstrasser, R. M. Vibrational-relaxation dynamics in solutions. Annu. Rev. Phys. Chem. 45, 519–555 (1994).
Mukamel, S. Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 51, 691–729 (2000).
Jonas, D. M. Two-dimensional femtosecond spectroscopy. Annu. Rev. Phys. Chem. 54, 425–463 (2003).
Schoenlein, R. et al. Recent advances in ultrafast X-ray sources. Philos. Trans. R. Soc. A 377, 20180384 (2019).
Chen, L. X. et al. Capturing a photoexcited molecular structure through time-domain X-ray absorption fine structure. Science 292, 262–264 (2001).
Saes, M. et al. Observing photochemical transients by ultrafast X-ray absorption spectroscopy. Phys. Rev. Lett. 90, 047403 (2003).
Khalil, M. et al. Picosecond X-ray absorption spectroscopy of a photoinduced iron(II) spin crossover reaction in solution. J. Phys. Chem. A 110, 38–44 (2006).
Lee, T., Jiang, Y., Rose-Petruck, C. G. & Benesch, F. Ultrafast tabletop laser-pump-x-ray probe measurement of solvated Fe(CN)(6)(4-). J. Chem. Phys. 122, 84506 (2005).
Bergmann, U. et al. Using X-ray free-electron lasers for spectroscopy of molecular catalysts and metalloenzymes. Nat. Rev. Phys. 3, 264–282 (2021).
Kraus, P. M., Zürch, M., Cushing, S. K., Neumark, D. M. & Leone, S. R. The ultrafast X-ray spectroscopic revolution in chemical dynamics. Nat. Rev. Chem. 2, 82–94 (2018).
Wernet, P. Chemical interactions and dynamics with femtosecond X-ray spectroscopy and the role of X-ray free-electron lasers. Philos. Trans. R. Soc. A 377, 20170464 (2019).
Chen, L. X. Ultrafast photochemical reaction trajectories revealed by X-ray transient absorption spectroscopy using X-ray free electron laser sources. RSC Energy Environ. Ser. 18, 201–224 (2017).
Chergui, M. & Collet, E. Photoinduced structural dynamics of molecular systems mapped by time-resolved X-ray methods. Chem. Rev. 117, 11025–11065 (2017).
Lemke, H. T. et al. Coherent structural trapping through wave packet dispersion during photoinduced spin state switching. Nat. Commun. 8, 15342 (2017).
Kunnus, K. et al. Vibrational wavepacket dynamics in Fe carbene photosensitizer determined with femtosecond X-ray emission and scattering. Nat. Commun. 11, 634 (2020).
Biasin, E. et al. Direct observation of coherent femtosecond solvent reorganization coupled to intramolecular electron transfer. Nat. Chem. 13, 343–349 (2021).
Liekhus-Schmaltz, C. et al. Femtosecond X-ray spectroscopy directly quantifies transient excited-state mixed valency. J. Phys. Chem. Lett. 13, 378–386 (2022).
Marinelli, A. et al. High-intensity double-pulse X-ray free-electron laser. Nat. Commun. 6, 6369 (2015).
Lutman, A. A. et al. Demonstration of single-crystal self-seeded two-color X-ray free-electron lasers. Phys. Rev. Lett. 113, 254801 (2014).
Hara, T. et al. Two-colour hard X-ray free-electron laser with wide tunability. Nat. Commun. 4, 2919 (2013).
Lu, W. et al. Development of a hard X-ray split-and-delay line and performance simulations for two-color pump-probe experiments at the European XFEL. Rev. Sci. Instrum. 89, 063121 (2018).
Prat, E. et al. Widely tunable two-color X-ray free-electron laser pulses. Phys. Rev. Res. 4, L022025 (2022).
Ilchen, M. et al. Site-specific interrogation of an ionic chiral fragment during photolysis using an X-ray free-electron laser. Commun. Chem. 4, 119 (2021).
Picón, A. et al. Hetero-site-specific X-ray pump-probe spectroscopy for femtosecond intramolecular dynamics. Nat. Commun. 7, 11652 (2016).
Fang, L., Xiong, H., Kukk, E. & Berrah, N. X-ray pump–probe investigation of charge and dissociation dynamics in methyl iodine molecule. Appl. Sci. 7, 529 (2017).
Shinohara, Y. et al. Split-pulse X-ray photon correlation spectroscopy with seeded X-rays from X-ray laser to study atomic-level dynamics. Nat. Commun. 11, 6213 (2020).
Roseker, W. et al. Towards ultrafast dynamics with split-pulse X-ray photon correlation spectroscopy at free electron laser sources. Nat. Commun. 9, 1704 (2018).
Inoue, I. et al. Observation of femtosecond X-ray interactions with matter using an X-ray-X-ray pump-probe scheme. Proc. Natl Acad. Sci. USA 113, 1492–1497 (2016).
Opara, N. L. et al. Demonstration of femtosecond X-ray pump X-ray probe diffraction on protein crystals. Struct. Dyn. 5, 054303 (2018).
Ferguson et al. Transient lattice contraction in the solid-to-plasma transition. Sci. Adv. 2, e1500837 (2016).
Pardini, T. et al. Delayed onset of nonthermal melting in single-crystal silicon pumped with hard X rays. Phys. Rev. Lett. 120, 265701 (2018).
Hartley, N. J. et al. Using diffuse scattering to observe X-ray-driven nonthermal melting. Phys. Rev. Lett. 126, 015703 (2021).
Kroll, T. et al. Observation of seeded Mn Kβ stimulated X-ray emission using two-color X-ray free-electron laser pulses. Phys. Rev. Lett. 125, 037404 (2020).
Liekhus-Schmaltz, C. E. et al. Ultrafast X-ray pump X-ray probe transient absorption spectroscopy: a computational study and proposed experiment probing core-valence electronic correlations in solvated complexes. J. Chem. Phys. 154, 214107 (2021).
Ross, M. et al. Comprehensive experimental and computational spectroscopic study of hexacyanoferrate complexes in water: from infrared to X-ray wavelengths. J. Phys. Chem. B 122, 5075–5086 (2018).
Krause, M. O. Atomic radiative and radiationless yields for K and L shells. J. Phys. Chem. Ref. Data 8, 307–327 (1979).
Davis, L. C. & Feldkamp, L. A. Interpretation of 3p-core-excitation spectra in Cr, Mn, Fe, Co, and Ni. Solid State Commun. 19, 413–416 (1976).
Laan, G. V. D. M2,3 absorption spectroscopy of 3d transition-metal compounds. J. Phys.: Condens. Matter 3, 7443–7454 (1991).
Solomon, E. I. Inorganic Electronic Structure and Spectroscopy (Wiley, 2006).
Tsutsumi, K. The X-ray non-diagram lines Kβ‘ of some compounds of the iron group. J. Phys. Soc. Jpn. 14, 1696–1706 (1959).
Tsutsumi, K. & Nakamori, H. X-ray K emission spectra of chromium in various chromium compounds. J. Phys. Soc. Jpn. 25, 1418–1423 (1968).
Tsutsumi, K., Nakamori, H. & Ichikawa, K. X-ray Mn Kβ emission spectra of manganese oxides and manganates. Phys. Rev. B 13, 929–933 (1976).
Hocking, R. K. et al. Fe L- and K-edge XAS of low-spin ferric corrole: bonding and reactivity relative to low-spin ferric porphyrin. Inorg. Chem. 48, 1678–1688 (2009).
Hocking, R. K. et al. Fe L-edge XAS studies of K4[Fe(CN)6] and K3[Fe(CN)6]: a direct probe of back-bonding. J. Am. Chem. Soc. 128, 10442–10451 (2006).
Hocking, R. K. et al. Fe L-edge X-ray absorption spectroscopy determination of differential orbital covalency of siderophore model compounds: electronic structure contributions to high stability constants. J. Am. Chem. Soc. 132, 4006–4015 (2010).
Wasinger, E. C., de Groot, F. M. F., Hedman, B., Hodgson, K. O. & Solomon, E. I. L-edge X-ray absorption spectroscopy of non-heme iron sites: experimental determination of differential orbital covalency. J. Am. Chem. Soc. 125, 12894–12906 (2003).
de Groot, F. & Kotani, A. Core Level Spectroscopy of Solids (CRC Press, 2008).
Krause, M. O. & Oliver, J. H. Natural widths of atomic K and L levels, Kα X‐ray lines and several KLL Auger lines. J. Phys. Chem. Ref. Data 8, 329–338 (1979).
Yin, L. I. et al. Widths of atomic M-shell vacancy states and quasiatomic aspects of radiationless transitions in solids. Phys. Rev. A 9, 1070–1080 (1974).
Healion, D., Zhang, Y., Biggs, J. D., Govind, N. & Mukamel, S. Entangled valence electron–hole dynamics revealed by stimulated attosecond X-ray Raman scattering. J. Phys. Chem. Lett. 3, 2326–2331 (2012).
Mukamel, S., Healion, D., Zhang, Y. & Biggs, J. D. Multidimensional attosecond resonant X-ray spectroscopy of molecules: lessons from the optical regime. Annu. Rev. Phys. Chem. 64, 101–127 (2013).
Cavaletto, S. M., Nascimento, D. R., Zhang, Y., Govind, N. & Mukamel, S. Resonant stimulated X-ray Raman spectroscopy of mixed-valence manganese complexes. J. Phys. Chem. Lett. 12, 5925–5931 (2021).
Liang, M. et al. The coherent X-ray imaging instrument at the Linac Coherent Light Source. J. Synchrot. Radiat. 22, 514–519 (2015).
Lutman, A. A. et al. Experimental demonstration of femtosecond two-color X-ray free-electron lasers. Phys. Rev. Lett. 110, 134801 (2013).
Chollet, M. et al. The X-ray pump-probe instrument at the Linac Coherent Light Source. J. Synchrot. Radiat. 22, 503–507 (2015).
Blaj, G. et al. X-ray detectors at the Linac Coherent Light Source. J. Synchrot. Radiat. 22, 577–583 (2015).
Seaberg, M. et al. Nanofocus characterization at the coherent X-ray imaging instrument using 2D single grating interferometry. In Proc. SPIE 11038, X-Ray Free-Electron Lasers: Advances in Source Development and Instrumentation V. 110380L (SPIE, 2019).
Nagler, B. et al. Focal spot and wavefront sensing of an X-ray free electron laser using Ronchi shearing interferometry. Sci. Rep. 7, 13698 (2017).
Fransson, T. et al. Effects of x-ray free-electron laser pulse intensity on the Mn Kβ1,3 x-ray emission spectrum in photosystem II—a case study for metalloprotein crystals and solutions. Struct. Dyn. 8, 064302 (2021).
Herman, F. & Skillman, S. Atomic Structure Calculations (Prentice-Hall, 1963).
Ho, P. J., Bostedt, C., Schorb, S. & Young, L. Theoretical tracking of resonance-enhanced multiple ionization pathways in X-ray free-electron laser pulses. Phys. Rev. Lett. 113, 253001 (2014).
Ho, P. J., Kanter, E. P. & Young, L. Resonance-mediated atomic ionization dynamics induced by ultraintense x-ray pulses. Phys. Rev. A 92, 063430 (2015).
Lopata, K., Van Kuiken, B. E., Khalil, M. & Govind, N. Linear-response and real-time time-dependent density functional theory studies of core-level near-edge X-ray absorption. J. Chem. Theory Comput. 8, 3284–3292 (2012).
Aprà, E. et al. NWChem: past, present, and future. J. Chem. Phys. 152, 184102 (2020).
Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division under Awards DE-SC0019277 and DE-SC0023249 (R.B.W., C.E.L.-S., B.I.P., C.M.L., and M.K.), KC-030105172685 and KC-030105180818 (E.B., N.G.), and Contracts DE-AC02-76SF00515 (E.B., R.W.S.) and DE-AC02-06CH11357 (P.J.H). R.B.W. and B.I.P. acknowledge support from the NSF Graduate Research Fellowship Program under Grant No. DGE-1762114. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. This research benefited from computational resources provided by EMSL, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle Memorial Institute for the United States Department of Energy under DOE contract number DE-AC05-76RL1830. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393 and P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Author information
Authors and Affiliations
Contributions
R.B.W., C.E.L.-S., A.A., E.B., B.I.P., D.Z., R.W.S., N.G., and M.K. designed the research and experiments. R.B.W., E.B., R.A.-M., A.A., S.B., F.D.F., T.K., U.B., R.W.S., B.I.P., C.M.L., A.L., and M.K. conducted the experiment at the LCLS (onsite and remotely). R.B.W. analyzed the experimental data. R.B.W., C.E.L.-S., and N.G. performed the TDDFT calculations. P.H. performed the electron cascade calculations. R.B.W., N.G., and M.K. interpreted the results. R.B.W. and M.K. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Majed Chergui and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weakly, R.B., Liekhus-Schmaltz, C.E., Poulter, B.I. et al. Revealing core-valence interactions in solution with femtosecond X-ray pump X-ray probe spectroscopy. Nat Commun 14, 3384 (2023). https://doi.org/10.1038/s41467-023-39165-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39165-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
