
Abstract
The inducible lysine decarboxylase LdcI is an important enterobacterial acid stress response enzyme whereas LdcC is its close paralogue thought to play mainly a metabolic role. A unique macromolecular cage formed by two decamers of the Escherichia coli LdcI and five hexamers of the AAA+ ATPase RavA was shown to counteract acid stress under starvation. Previously, we proposed a pseudoatomic model of the LdcI-RavA cage based on its cryo-electron microscopy map and crystal structures of an inactive LdcI decamer and a RavA monomer. We now present cryo-electron microscopy 3D reconstructions of the E. coli LdcI and LdcC, and an improved map of the LdcI bound to the LARA domain of RavA, at pH optimal for their enzymatic activity. Comparison with each other and with available structures uncovers differences between LdcI and LdcC explaining why only the acid stress response enzyme is capable of binding RavA. We identify interdomain movements associated with the pH-dependent enzyme activation and with the RavA binding. Multiple sequence alignment coupled to a phylogenetic analysis reveals that certain enterobacteria exert evolutionary pressure on the lysine decarboxylase towards the cage-like assembly with RavA, implying that this complex may have an important function under particular stress conditions.
Similar content being viewed by others

Introduction
Enterobacterial inducible decarboxylases of basic amino acids lysine, arginine and ornithine have a common evolutionary origin and belong to the α-family of pyridoxal-5′-phosphate (PLP)-dependent enzymes1,2. They counteract acid stress experienced by the bacterium in the host digestive and urinary tract, and in particular in the extremely acidic stomach3,4. Each decarboxylase is induced by an excess of the target amino acid and a specific range of extracellular pH, and works in conjunction with a cognate inner membrane antiporter. Decarboxylation of the amino acid into a polyamine is catalysed by a PLP cofactor in a multistep reaction1,2 that consumes a cytoplasmic proton and produces a CO2 molecule passively diffusing out of the cell, while the polyamine is excreted by the antiporter in exchange for a new amino acid substrate. Consequently, these enzymes buffer both the bacterial cytoplasm and the local extracellular environment5. These amino acid decarboxylases are therefore called acid stress inducible or biodegradative to distinguish them from their biosynthetic lysine and ornithine decarboxylase paralogs catalysing the same reaction but responsible for the polyamine production at neutral pH.
Inducible enterobacterial amino acid decarboxylases have been intensively studied since the early 19406,7 because the ability of bacteria to withstand acid stress can be linked to their pathogenicity in humans. In particular, the inducible lysine decarboxylase LdcI (or CadA) attracts attention due to its broad pH range of activity and its capacity to promote survival and growth of pathogenic enterobacteria such as Salmonella enterica serovar Typhimurium, Vibrio cholerae and Vibrio vulnificus under acidic conditions5,8,9. Furthermore, both LdcI and the biosynthetic lysine decarboxylase LdcC of uropathogenic Escherichia coli (UPEC) appear to play an important role in increased resistance of this pathogen to nitrosative stress produced by nitric oxide and other damaging reactive nitrogen intermediates accumulating during the course of urinary tract infections (UTI)10,11. This effect is attributed to cadaverine, the diamine produced by decarboxylation of lysine by LdcI and LdcC, that was shown to enhance UPEC colonisation of the bladder11. In addition, the biosynthetic E. coli lysine decarboxylase LdcC, long thought to be constitutively expressed in low amounts, was demonstrated to be strongly upregulated by fluoroquinolones via their induction of RpoS12,13. A direct correlation between the level of cadaverine and the resistance of E. coli to these antibiotics commonly used as a first-line treatment of UTI could be established12. Both acid pH and cadaverine induce closure of outer membrane porins thereby contributing to bacterial protection from acid stress, but also from certain antibiotics, by reduction in membrane permeability14,15,16.
The crystal structure of the E. coli LdcI17 as well as its low resolution characterisation by electron microscopy17,18,19 (EM) showed that it is a decamer made of two pentameric rings. Each monomer is composed of three domains – an N-terminal wing domain (residues 1–129), a PLP-binding core domain (residues 130–563), and a C-terminal domain (CTD, residues 564–715). Monomers tightly associate via their core domains into 2-fold symmetrical dimers with two complete active sites, and further build a toroidal D5-symmetrical structure held by the wing and core domain interactions around the central pore, with the CTDs at the periphery.
Ten years ago19 we showed that the E. coli AAA+ ATPase RavA, involved in multiple stress response pathways19,20,21,22, tightly interacted with LdcI but was not capable of binding to LdcC. We described how two double pentameric rings of the LdcI17 tightly associate with five hexameric rings of RavA21 to form a unique cage-like architecture that enables the bacterium to withstand acid stress even under conditions of nutrient deprivation eliciting stringent response19,21,23. Furthermore, we recently solved the structure of the E. coli LdcI-RavA complex by cryo-electron microscopy (cryoEM) and combined it with the crystal structures of the individual proteins23. This allowed us to make a pseudoatomic model of the whole assembly, underpinned by a cryoEM map of the LdcI-LARA complex (with LARA standing for LdcI associating domain of RavA), and to identify conformational rearrangements and specific elements essential for complex formation23. The main determinants of the LdcI-RavA cage assembly appeared to be the N-terminal loop of the LARA domain of RavA and the C-terminal β-sheet of LdcI23.
In spite of this wealth of structural information, the fact that LdcC does not interact with RavA, although the two lysine decarboxylases are 69% identical and 84% similar19,24, and the physiological significance of the absence of this interaction remained unexplored. To solve this discrepancy, in the present work we provided a three-dimensional (3D) cryoEM reconstruction of LdcC and compared it with the available LdcI and LdcI-RavA structures. Given that the LdcI crystal structures were obtained at high pH where the enzyme is inactive (LdcIi, pH 8.5), whereas the cryoEM reconstructions of LdcI-RavA and LdcI-LARA were done at acidic pH optimal for the enzymatic activity, for a meaningful comparison, we also produced a 3D reconstruction of the LdcI at active pH (LdcIa, pH 6.2). This comparison pinpointed differences between the biodegradative and the biosynthetic lysine decarboxylases and brought to light interdomain movements associated to pH-dependent enzyme activation and RavA binding, notably at the predicted RavA binding site at the level of the C-terminal β-sheet of LdcI. Consequently, we tested the capacity of cage formation by LdcI-LdcC chimeras where we interchanged the C-terminal β-sheets in question. Finally, we performed multiple sequence alignment of 22 lysine decarboxylases from Enterobacteriaceae containing the ravA-viaA operon in their genome. Remarkably, this analysis revealed that several specific residues in the above-mentioned β-sheet, independently of the rest of the protein sequence, are sufficient to define if a particular lysine decarboxylase should be classified as an “LdcC-like” or an “LdcI-like”. Moreover, this classification perfectly agrees with the genetic environment of the lysine decarboxylase genes. This fascinating parallelism between the propensity for RavA binding and the genetic environment of an enterobacterial lysine decarboxylase, as well as the high degree of conservation of this small structural motif, emphasize the functional importance of the interaction between biodegradative enterobacterial lysine decarboxylases and the AAA+ ATPase RavA.
Results and Discussion
CryoEM 3D reconstructions of LdcC, LdcIa and LdcI-LARA
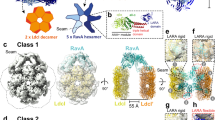
In the frame of this work, we produced two novel subnanometer resolution cryoEM reconstructions of the E. coli lysine decarboxylases at pH optimal for their enzymatic activity – a 5.5 Å resolution cryoEM map of the LdcC (pH 7.5) for which no 3D structural information has been previously available (Figs 1A,B and S1), and a 6.1 Å resolution cryoEM map of the LdcIa, (pH 6.2) (Figs 1C,D and S2). In addition, we improved our earlier cryoEM map of the LdcI-LARA complex from 7.5 Å to 6.2 Å resolution (Figs 1E,F and S3). Based on these reconstructions, reliable pseudoatomic models of the three assemblies were obtained by flexible fitting of either the crystal structure of LdcIi or a derived structural homology model of LdcC (Table S1). Significant differences between these pseudoatomic models can be interpreted as movements between specific biological states of the proteins as described below.

(A,C,E) cryoEM map of the LdcC (A), LdcIa (C) and LdcI-LARA (E) decamers with one protomer in light grey. In the rest of the protomers, the wing, core and C-terminal domains are colored from light to dark in shades of green for LdcC (A), pink for LdcIa (C) and blue for LdcI in LdcI-LARA (E). In (E), the LARA domain density is shown in dark grey. Two monomers making a dimer are delineated. Scale bar 50 Å. (B,D,F) One protomer from the cryoEM map of the LdcC (B), LdcIa (D) and LdcI-LARA (F) in light grey with the pseudoatomic model represented as cartoons and colored as the densities in (A,C,E). Each domain is indicated for clarity. Scale bar 50 Å. See also Figs S1 and S3.
The wing domains as a stable anchor at the center of the double-ring
As a first step of a comparative analysis, we superimposed the three cryoEM reconstructions (LdcIa, LdcI-LARA and LdcC) and the crystal structure of the LdcIi decamer (Fig. 2 and Movie S1). This superposition reveals that the densities lining the central hole of the toroid are roughly at the same location, while the rest of the structure exhibits noticeable changes. Specifically, at the center of the double-ring the wing domains of the subunits provide the conserved basis for the assembly with the lowest root mean square deviation (RMSD) (between 1.4 and 2 Å for the Cα atoms only), whereas the peripheral CTDs containing the RavA binding interface manifest the highest RMSD (up to 4.2 Å) (Table S2). In addition, the wing domains of all structures are very similar, with the RMSD after optimal rigid body alignment (RMSDmin) less than 1.1 Å. Thus, taking the limited resolution of the cryoEM maps into account, we consider that the wing domains of all the four structures are essentially identical and that in the present study the RMSD of less than 2 Å can serve as a baseline below which differences may be assumed as insignificant. This preservation of the central part of the double-ring assembly may help the enzymes to maintain their decameric state upon activation and incorporation into the LdcI-RavA cage.

Superposition of the pseudoatomic models of LdcC, LdcI from LdcI-LARA and LdcIa colored as in Fig. 1, and the crystal structure of LdcIi in shades of yellow. Only one of the two rings of the double toroid is shown for clarity. The dashed circle indicates the central region that remains virtually unchanged between all the structures, while the periphery undergoes visible movements. Scale bar 50 Å.
The core domain and the active site rearrangements upon pH-dependent enzyme activation and LARA binding
Both visual inspection (Fig. 2) and RMSD calculations (Table S2) show that globally the three structures at active pH (LdcIa, LdcI-LARA and LdcC) are more similar to each other than to the structure determined at high pH conditions (LdcIi). The decameric enzyme is built of five dimers associating into a 5-fold symmetrical double-ring17 (two monomers making a dimer are delineated in Fig. 1). As common for the α family of the PLP-dependent decarboxylases17,25, dimerization is required for the enzymatic activity because the active site is buried in the dimer interface (Fig. 3A,B). This interface is formed essentially by the core domains with some contribution of the CTDs. The core domain is built by the PLP-binding subdomain (PLP-SD, residues 184–417) flanked by two smaller subdomains rich in partly disordered loops – the linker region (residues 130–183) and the subdomain 4 (residues 418–563). Zooming in the variations in the PLP-SD shows that most of the structural changes concern displacements in the active site (Fig. 3C–F). The most conspicuous differences between the PLP-SDs can be linked to the pH-dependent activation of the enzymes. The resolution of the cryoEM maps does not allow modeling the position of the PLP moiety and calls for caution in detailed mechanistic interpretations in terms of individual amino acids. Therefore we restrict our analysis to secondary structure elements. In particular, transition from LdcIi to LdcI-LARA involves ~3.5 Å and ~4.5 Å shifts away from the 5-fold axis in the active site α-helices spanning residues 218–232 and 246–254 respectively (Fig. 3C–E). Between these two extremes, the PLP-SDs of LdcIa and LdcC are similar both in the context of the decamer (Fig. 3F) and in terms of RMSDmin = 0.9 Å, which probably reflects the fact that, at the optimal pH, these lysine decarboxylases have a similar enzymatic activity26. In addition, our earlier biochemical observation that the enzymatic activity of LdcIa is unaffected by RavA binding19 is consistent with the relatively small changes undergone by the active site upon transition from LdcIa to LdcI-LARA. Worthy of note, our previous comparison of the crystal structure of LdcIi with that of the inducible arginine decarboxylase AdiA17 revealed high conservation of the PLP-coordinating residues and identified a patch of negatively charged residues lining the active site channel as a potential binding site for the target amino acid substrate17 (Figs S3 and S4 in ref. 17).

(A) LdcIi crystal structure, with one ring represented as a grey surface and the second as a cartoon. A monomer with its PLP cofactor is delineated. The PLP moieties of the cartoon ring are shown in red. (B) The LdcIi dimer extracted from the crystal structure of the decamer. One monomer is colored in shades of yellow as in Figs 1 and 2, while the monomer related by C2 symmetry is grey. The PLP is red. The active site is boxed. (C–F) Close-up views of the active site. The PLP moiety in red is from the LdcIi crystal structure. We did not attempt to model it in the cryoEM maps. The dimer interface is shown as a dashed line and the active site α-helices mentioned in the text are highlighted. (C) Compares LdcIi (yellow) and LdcIa (pink), (D) compares LdcIa (pink) and LdcI-LARA (blue), and (E) compares LdcIi (yellow), LdcIa (pink) and LdcI-LARA (blue) simultaneously in order to show the progressive shift described in the text. (F) Shows the similarity between LdcIa and LdcC at the level of the secondary structure elements composing the active site. Colors are as in the other figures.
Rearrangements of the ppGpp binding pocket upon pH-dependent enzyme activation and LARA binding
An inhibitor of the LdcI and LdcC activity, the stringent response alarmone ppGpp, is known to bind at the interface between neighboring monomers within each ring (Fig. S4). The ppGpp binding pocket is made up by residues from all domains and is located approximately 30 Å away from the PLP moiety17. Whereas the crystal structure of the ppGpp-LdcIi was solved to 2 Å resolution, only a 4.1 Å resolution structure of the ppGpp-free LdcIi could be obtained17. At this resolution, the apo-LdcIi and ppGpp-LdcIi structures (both solved at pH 8.5) appeared indistinguishable except for the presence of ppGpp17 (Fig. S11 in ref. 17). Thus, we speculated that inhibition of LdcI by ppGpp would be accompanied by a transduction of subtle structural changes at the level of individual amino acid side chains between the ppGpp binding pocket and the active site of the enzyme17. All our current cryoEM reconstructions of the lysine decarboxylases were obtained in the absence of ppGpp in order to be closer to the active state of the enzymes under study. While differences in the ppGpp binding site could indeed be visualized (Fig. S4), the level of resolution warns against speculations about their significance. The fact that interaction with RavA reduces the ppGpp affinity for LdcI21 despite the long distance of ~30 Å between the LARA domain binding site and the closest ppGpp binding pocket (Fig. S5) seems to favor an allosteric regulation mechanism. Interestingly, although a number of ppGpp binding residues are strictly conserved between LdcI and AdiA that also forms decamers at low pH optimal for its arginine decarboxylase activity, no ppGpp regulation of AdiA could be demonstrated26.
Swinging and stretching of the CTDs upon pH-dependent LdcI activation and LARA binding
Inspection of the superimposed decameric structures (Figs 2 and S6) suggests a depiction of the wing domains as an anchor around which the peripheral CTDs swing. This swinging movement seems to be mediated by the core domains and is accompanied by a stretching of the whole LdcI subunits attracted by the RavA magnets. Indeed, all CTDs have very similar structures (RMSDmin <1 Å). Yet the superposition of the decamers lays bare a progressive movement of the CTD as a whole upon enzyme activation by pH and the binding of LARA. The LdcIi monomer is the most compact, whereas LdcIa and especially LdcI-LARA gradually extend their CTDs towards the LARA domain of RavA (Figs 2 and 4). These small but noticeable swinging and stretching (up to ~4 Å) may be related to the incorporation of the LdcI decamer into the LdcI-RavA cage.

(A–C) A slice through the pseudoatomic models of the LdcI monomers extracted from the superimposed decamers (Fig. 2) The rectangle indicates the regions enlarged in (D–F). (A) compares LdcIi (yellow) and LdcIa (pink), (B) compares LdcIa (pink) and LdcI-LARA (blue), and (C) compares LdcIi (yellow), LdcIa (pink) and LdcI-LARA (blue) simultaneously in order to show the progressive stretching described in the text. The cryoEM density of the LARA domain is represented as a grey surface to show the position of the binding site and the direction of the movement. (D–F) Inserts zooming at the CTD part in proximity of the LARA binding site. Loop regions are removed for a clearer visual comparison. An arrow indicates a swinging movement.
The C-terminal β-sheet of a lysine decarboxylase as a major determinant of the interaction with RavA
In our previous contribution, based on the fit of the LdcIi and the LARA crystal structures into the LdcI-LARA cryoEM density, we predicted that the LdcI-RavA interaction should involve the C-terminal two-stranded β-sheet of the LdcI23. Our present cryoEM maps and pseudoatomic models provide first structure-based insights into the differences between the inducible and the constitutive lysine decarboxylases. However, at the level of this structural element the two proteins are actually surpisingly similar. Therefore, we wanted to check the influence of the primary sequence of the two proteins in this region on their ability to interact with RavA. To this end, we swapped the relevant β-sheets of the two proteins and produced their chimeras, namely LdcIC (i.e. LdcI with the C-terminal β-sheet of LdcC) and LdcCI (i.e. LdcC with the C-terminal β-sheet of LdcI) (Fig. 5A–C). Both constructs could be purified and could form decamers visually indistinguishable from the wild-type proteins. As expected, binding of LdcI to RavA was completely abolished by this procedure and no LdcIC-RavA complex could be detected. On the contrary, introduction of the C-terminal β-sheet of LdcI into LdcC led to an assembly of the LdcCI-RavA complex. On the negative stain EM grid, the chimeric cages appeared less rigid than the native LdcI-RavA, which probably means that the environment of the β-sheet contributes to the efficiency of the interaction and the stability of the entire architecture (Fig. 5D–F).

(A) A slice through the pseudoatomic models of the LdcIa (purple) and LdcC (green) monomers extracted from the superimposed decamers (Fig. 2). (B) The C-terminal β-sheet in LdcIa and LdcC enlarged from (A,C) Exchanged primary sequences (capital letters) and their immediate vicinity (lower case letters) colored as in (A,B), with the corresponding secondary structure elements and the amino acid numbering shown. (D,E) A gallery of negative stain EM images of (D) the wild type LdcI-RavA cage and (E) the LdcCI-RavA cage-like particles. (F) Some representative class averages of the LdcCI-RavA cage-like particles. Scale bar 20 nm.
The C-terminal β-sheet of a lysine decarboxylase is a highly conserved signature allowing to distinguish between LdcI and LdcC
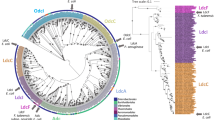
Alignment of the primary sequences of the E. coli LdcI and LdcC shows that some amino acid residues of the C-terminal β-sheet are the same in the two proteins, whereas others are notably different in chemical nature. Importantly, most of the amino acid differences between the two enzymes are located in this very region. Thus, to advance beyond our experimental confirmation of the C-terminal β-sheet as a major determinant of the capacity of a particular lysine decarboxylase to form a cage with RavA, we set out to investigate whether certain residues in this β-sheet are conserved in lysine decarboxylases of different enterobacteria that have the ravA-viaA operon in their genome. We inspected the genetic environment of lysine decarboxylases from 22 enterobacterial species referenced in the NCBI database, corrected the gene annotation where necessary (Tables S3 and S4), and performed multiple sequence alignment coupled to a phylogenetic analysis (see Methods). This procedure yielded several unexpected and exciting results. First of all, consensus sequence for the entire lysine decarboxylase family was derived. Second, the phylogenetic analysis clearly split the lysine decarboxylases into two groups (Fig. 6A). All lysine decarboxylases predicted to be “LdcI-like” or biodegradable based on their genetic environment, as for example their organization in an operon with a gene encoding the CadB antiporter (see Methods), were found in one group, whereas all enzymes predicted as “LdcC-like” or biosynthetic partitioned into another group. Thus, consensus sequences could also be determined for each of the two groups (Figs 6B,C and S7). Inspection of these consensus sequences revealed important differences between the groups regarding charge, size and hydrophobicity of several residues precisely at the level of the C-terminal β-sheet that is responsible for the interaction with RavA (Fig. 6B–D). For example, in our previous study23, site-directed mutations identified Y697 as critically required for the RavA binding. Our current analysis shows that Y697 is strictly conserved in the “LdcI-like” group whereas the “LdcC-like” enzymes always have a lysine in this position; it also uncovers several other residues potentially essential for the interaction with RavA which can now be addressed by site-directed mutagenesis. The third and most remarkable finding was that exactly the same separation into “LdcI-like” and “LdcC”-like groups can be obtained based on a comparison of the C-terminal β-sheets only, without taking the rest of the primary sequence into account. Therefore the C-terminal β-sheet emerges as being a highly conserved signature sequence, sufficient to unambiguously discriminate between the “LdcI-like” and “LdcC-like” enterobacterial lysine decarboxylases independently of any other information (Figs 6 and S7). Our structures show that this motif is not involved in the enzymatic activity or the oligomeric state of the proteins. Thus, enterobacteria identified here (Fig. 6, Table S4) appear to exert evolutionary pressure on the biodegradative lysine decarboxylase towards the RavA binding. One of the elucidated roles of the LdcI-RavA cage is to maintain LdcI activity under conditions of enterobacterial starvation by preventing LdcI inhibition by the stringent response alarmone ppGpp21. Furthermore, the recently documented interaction of both LdcI27 and RavA22 with specific subunits of the respiratory complex I, together with the unanticipated link between RavA and maturation of numerous iron-sulfur proteins, tend to suggest an additional intriguing function for this 3.5 MDa assembly. The conformational rearrangements of LdcI upon enzyme activation and RavA binding revealed in this work, and our amazing finding that the molecular determinant of the LdcI-RavA interaction is the one that straightforwardly determines if a particular enterobacterial lysine decarboxylase belongs to “LdcI-like” or “LdcC-like” proteins, should give a new impetus to functional studies of the unique LdcI-RavA cage. Besides, the structures and the pseudoatomic models of the active ppGpp-free states of both the biodegradative and the biosynthetic E. coli lysine decarboxylases offer an additional tool for analysis of their role in UPEC infectivity. Together with the apo-LdcI and ppGpp-LdcIi crystal structures, our cryoEM reconstructions provide a structural framework for future studies of structure-function relationships of lysine decarboxylases from other enterobacteria and even of their homologues outside Enterobacteriaceae. For example, the lysine decarboxylase of Eikenella corrodens is thought to play a major role in the periodontal disease and its inhibitors were shown to retard gingivitis development28,29,30. Finally, cadaverine being an important platform chemical for the production of industrial polymers such as nylon, structural information is valuable for optimisation of bacterial lysine decarboxylases used for its production in biotechnology31,32,33.

(A) Maximum likelihood tree with the “LdcC-like” and the “LdcI-like” groups highlighted in green and pink, respectively. Only nodes with higher values than 95% are shown (500 replicates of the original dataset, see Methods for details). Scale bar indicates the average number of substitutions per site. (B) Analysis of consensus “LdcI-like” and “LdcC-like” sequences around the first and second C-terminal β-strands. The height of the bars and the letters representing the amino acids reflects the degree of conservation of each particular position is in the alignment. Amino acids are colored according to a polarity color scheme with hydrophobic residues in black, hydrophilic in green, acidic in red and basic in blue. Numbering as in E. coli. (C) Signature sequences of LdcI and LdcC in the C-terminal β-sheet. Polarity differences are highlighted. (D) Position and nature of these differences at the surface of the respective cryoEM maps with the color code as in B. See also Fig. S7 and Tables S3 and S4.
Methods
Protein expression and purification
LdcI and LdcC were expressed and purified as described19,23,26 from an E. coli strain that cannot produce ppGpp (MG1655 ΔrelA ΔspoT strain). LdcI was stored in 20 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, 0.1 mM PLP, pH 6.8 (buffer A) and LdcC in 20 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, 0.1 mM PLP, pH 7.5 (buffer B).
Chimeric LdcIC and LdcCI were constructed, expressed and purified as follows. The chimeras were designed by exchange, between LdcI and LdcC, of residues from 631 to 640 and from 697 to the C-terminus, corresponding to the regions around the two strands of the C-terminal β-sheet (Fig. 5B,C), while leaving the rest of the sequence unaltered. The synthetic ldcIC and ldcCI genes (2148 bp and 2154 bp respectively), provided within a pUC57 vector (GenScript) were subcloned into pET-TEV vector based on pET-28a (Invitrogen) containing an N-terminal TEV-cleavable 6x-His-Tag. Proteins were expressed in Rosetta 2 (DE3) cells (Novagen) in LB medium supplemented with kanamycin and chloramphenicol at 37 °C, upon induction with 0.5 mM IPTG at 18 °C. Cells were harvested by centrifugation, the pellet resuspended in a 50 mM Tris-HCl, 150 mM NaCl, pH 8 buffer supplemented with Complete EDTA free (Roche) and 0.1 mM PMSF (Sigma), and disrupted by sonication at 4 °C. After centrifugation at 75000 g at 4 °C for 20 min, the supernatant was loaded on a Ni-NTA column. The eluted protein-containing fractions were pooled and the His-Tag removed by incubation with the TEV protease at 1/100 ratio and an extensive dialysis in a 50 mM Tris-HCl, 150 mM NaCl, 1 mM DTT, 5 mM EDTA, pH 8 buffer. After a second dialysis in a 50 mM Tris-HCl, 150 mM NaCl, pH 8 buffer supplemented with 10 mM imidazole, the sample was loaded on a Ni-NTA column in the same buffer, which allowed to separate the TEV protease and LdcCI/LdcIC. Finally, the pure proteins were obtained by size exclusion chromatography on a Superdex-S200 column in buffer A.
LdcIa -cryoEM data collection and 3D reconstruction
LdcI was prepared at 2 mg/ml in a buffer containing 25 mM MES, 100 mM NaCl, 0.2 mM PLP, 1 mM DTT, pH 6.2. 3 μl of sample were applied to glow-discharged quantifoil grids 300 mesh 2/1 (Quantifoil Micro Tools GmbH, Germany), excess solution was blotted during 2.5 s with a Vitrobot (FEI) and the grid frozen in liquid ethane34. Data collection was performed on a FEI Polara microscope operated at 300 kV under low dose conditions. Micrographs were recorded on Kodak SO-163 film at 59,000 magnification, with defocus ranging from 0.6 to 4.9 μm. Films were digitized on a Zeiss scanner (Photoscan) at a step size of 7 μm giving a pixel size of 1.186 Å. The contrast transfer function (CTF) for each micrograph was determined with CTFFIND335.
Initially ~2500 particles of 256 × 256 pixels were extracted manually, binned 4 times and subjected to one round of multivariate statistical analysis and classification using IMAGIC36. Representative class averages corresponding to projections in different orientations were used as input for an ab-initio 3D reconstruction by RICOserver (rico.ibs.fr/)37. The resulting 3D reconstruction was refined using RELION38, which yielded an 18 Å resolution map. Using projections of this model as a template, particles of size 256 × 256 pixels were semi-automatically selected from all the micrographs using the Fast Projection Matching (FPM) algorithm39. The resulting dataset of ~46000 particles was processed in RELION with the previous map as an initial model and with a full CTF correction after the first peak. The final map comprised 44207 particles with a resolution of 7.4 Å as per the gold-standard FSC = 0.143 criterion40. It was sharpened with EMBfactor41 using calculated B-factor of −350 Å2 and masked with a soft mask to obtain a final map with a resolution of 6.1 Å (Fig. S3, Table S1).
LdcC - cryoEM data collection and 3D reconstruction
LdcC was prepared at 2 mg/ml in a buffer containing 25 mM HEPES, 100 mM NaCl, 0.2 mM PLP, 1 mM DTT, pH 7.2. Grids were prepared and sample imaged as LdcIa. Data were processed essentially as LdcIa described above. Briefly, an initial ~20 Å resolution model was generated by angular reconstitution after manual picking of circa 3000 particles from the first micrographs, filtered to 60 Å resolution, refined with RELION and used for a semi-automatic selection with FPM. The dataset was processed in RELION with a full CTF correction to yield a final reconstruction comprising 61000 particles. The map was sharpened with Relion postprocessing, using a soft mask and automated B-factor estimation (−690 Å2), yielding a map at 5.5 Å resolution (Fig. S1, Table S1).
LdcI-LARA - 3D reconstruction
In our first study23, the dataset was processed in SPIDER and the CTF correction involved a simple phase-flipping. Therefore, for consistency with the present work, we revisited the dataset and processed it in RELION with a full CTF correction after the first peak. It was sharpened with EMBfactor41 using calculated B-factor of −350 Å2 and masked with a soft mask to obtain a final map with a resolution of 6.2 Å (Fig. S2). This reconstruction is of a slightly better quality in terms of the continuity of the internal density. Therefore we used this improved map for fitting of the atomic model and further analysis (Fig. S2, Table S1).
Additional image processing
As a crosscheck, each data set was also refined either from other initial models: a “featureless donut” with approximate dimensions of the decamer, and low pass-filtered reconstructions from the two other data sets (i.e. the LdcC reconstruction was used as a model for the LdcIa and LdcI-LARA data sets, etc). All refinements converged to the same solutions independently of the starting model. Local resolution of all maps was determined with ResMap42.
LdcCI and LdcIC chimeras —negative stain EM and 2D image analysis
0.4 mg/ml of RavA (in a 20 mM Tris-HCl, 500 mM NaCl, 10 mM MgCl2, 1 mM DTT, 5% glycerol, pH 6.8 buffer) was mixed with 0.3 mg/ml of either LdcI, LdcC, LdcCI or LdcIC in the presence of 2 mM ADP and 10 mM MgCl2 in a buffer containing 20 mM Hepes and 150 mM NaCl at pH 7.4. After 10 minutes incubation at room temperature, 3 μl of mixture were applied to the clear side of the carbon on a carbon-mica interface and negatively stained with 2% uranyl acetate. Images were recorded with a JEOL 1200 EX II microscope at 100 kV at a nominal magnification of 15000 on a CCD camera yielding a pixel size of 4.667 Å. No complexes between RavA and LdcC or LdcIC could be observed, whereas the LdcCI-RavA preparation manifested cage-like particles similar to the previously published LdcI-RavA19, but also unbound RavA and LdcCI, which implies that the affinity of RavA to the LdcCI chimera is lower than its affinity to the native LdcI. 1260 particles of 96 × 96 pixels were extracted interactively from several micrographs. 2D centering, multivariate statistical analysis and classification were performed using IMAGIC36. Class-averages similar to the cage-like LdcI-RavA complex were used as references for multi-reference alignment followed by multivariate statistical analysis and classification.
Fitting of atomic models into cryoEM maps
A homology model of LdcC was obtained using the atomic coordinates of the LdcI monomer (3N75) as the template in SWISS-MODEL server43. The RMSD between the template and the resulting model was 0.26 Å. The LdcC model was then fitted as a rigid body into the LdcC cryoEM map using the fit-in-map module of UCSF Chimera44. This rigid fit indicated movements of several parts of the protein. Therefore, the density corresponding to one LdcC monomer was extracted and flexible fitting was performed using IMODFIT45 at 8 Å resolution. This monomeric model was then docked into the decameric cryoEM map with URO46 and its graphical version VEDA47 that use symmetry information for fitting in Fourier space. The Cα RMSDmin between the initial model of the LdcC monomer and the final IMODFIT LdcC model is 1.2 Å. In the case of LdcIa, the density corresponding to an individual monomer was extracted and the fit performed similarly to the one described above, with the final model of the decameric particle obtained with URO and VEDA. The Cα RMSDmin between the LdcIi monomer and the final IMODFIT model is 1.4 Å. For LdcI-LARA, the density accounting for one LdcI monomer bound to a LARA domain was extracted and further separated into individual densities corresponding to LdcI and to LARA. The fit of LdcI was performed as for LdcC and LdcIa, while the crystal structure of LARA was docked into the monomeric LdcI-LARA map as a rigid body using SITUS. The resulting pseudoatomic models were used to create the final model of the LdcI-LARA decamer with URO and VEDA. The Cα RMSDmin between the LdcIi monomer and the final IMODFIT model is 1.4 Å. A brief summary of relevant parameters is provided in Table S1.
Sequence analysis
Out of a non-exhaustive list of 50 species of Enterobacteriaceae (Table S3), 22 were found to contain genes annotated as ldcI or ldcC and containing the ravA-viaA operon (Table S4). An analysis using MUSCLE48 with default parameters showed that these genes share more than 65% identity. To verify annotation of these genes, we compared their genetic environment with that of E. coli ldcI and ldcC. Indeed, in E. coli the ldcI gene is in operon with the cadB gene encoding a lysine-cadaverine antiporter, whereas the ldcC gene is present between the accA gene (encoding an acetyl-CoA carboxylase alpha subunit carboxyltransferase) and the yaeR gene (coding for an unknown protein belonging to the family of Glyoxalase/Dioxygenase/Bleomycin resistance proteins). Compared with this genetic environment, the annotation of several ldcI and ldcC genes in enterobacteria was found to be inconsistent (Table S4). For example, several strains contain genes annotated as ldcC in the genetic background of ldcI and vice versa, as in the case of Salmonella enterica and Trabulsiella guamensi. Furthermore, the gene with an “ldcC-like” environment was found to be annotated as cadA in particular strains of Citrobacter freundii, Cronobacter sakazakii, Enterobacter cloacae subsp. Cloaca, Erwinia amylovora, Pantoea agglomerans, Rahnella aquatilis, Shigella dysenteriae, and Yersinia enterocolitica subsp. enterocolitica, whereas in Hafnia alvei, Kluyvera ascorbata, and Serratia marcescens subsp. marcescens, the gene with an “ldcI-like” environment was found to be annotated as ldcC. In addition, as far as the genetic environment is concerned, Plesiomonas appears to have two ldc genes with the organization of the E. coli ldcI (operon cadA-cadB). Consequently, we corrected for gene annotation consistent with the genetic environment and made multiple sequence alignments using version 8.0.1 of the CLC Genomics Workbench software. A phylogenetic tree was generated based on Maximum Likelihood and following the Neighbor-Joining method with the WAG protein substitution model49. The reliability of the generated phylogenetic tree was assessed by bootstrap analysis. The presented unrooted phylogenetic tree shows the nodes that are reliable over 95% (Fig. 6A). Remarkably, the multiple sequence alignment and the resulting phylogenetic tree clearly grouped together all sequences annotated as ldcI on the one side, and all sequences annotated as ldcC on the other side. Thus, we conclude that all modifications in gene annotations that we introduced for the sake of consistency with the genetic environment are perfectly corroborated by the multiple sequence alignment and the phylogenetic analysis. Since the regulation of the lysine decarboxylase gene (i.e. inducible or constitutive) cannot be assessed by this analysis, we call the resulting groups “ldcI-like” and “ldcC-like” as referred to the E. coli enzymes, and make a parallel between the membership in a given group and the ability of the protein to form a cage complex with RavA.
Additional Information
Accession codes: CryoEM maps and corresponding fitted atomic structures (main chain atoms) have been deposited in the Electron Microscopy Data Bank and Protein Data Bank, respectively, with accession codes EMD-3205 and 5FKZ for LdcC, EMD-3204 and 5FKX for LdcIa and EMD-3206 and 5FL2 for LdcI-LARA.
How to cite this article: Kandiah, E. et al. Structural insights into the Escherichia coli lysine decarboxylases and molecular determinants of interaction with the AAA+ ATPase RavA. Sci. Rep. 6, 24601; doi: 10.1038/srep24601 (2016).
References
Christen, P. & Mehta, P. K. From cofactor to enzymes. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Chem. Rec. N. Y. N 1, 436–447 (2001).
Eliot, A. C. & Kirsch, J. F. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415 (2004).
Zhao, B. & Houry, W. A. Acid stress response in enteropathogenic gammaproteobacteria: an aptitude for survival. Biochem. Cell Biol. Biochim. Biol. Cell. 88, 301–314 (2010).
Kanjee, U. & Houry, W. A. Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 67, 65–81 (2013).
Viala, J. P. M. et al. Sensing and adaptation to low pH mediated by inducible amino acid decarboxylases in Salmonella. PloS One 6, e22397 (2011).
Gale, E. F. & Epps, H. M. The effect of the pH of the medium during growth on the enzymic activities of bacteria (Escherichia coli and Micrococcus lysodeikticus) and the biological significance of the changes produced. Biochem. J. 36, 600–618 (1942).
Gale, E. F. & Epps, H. M. Studies on bacterial amino-acid decarboxylases: 1. l(+)-lysine decarboxylase. Biochem. J. 38, 232–242 (1944).
Merrell, D. S. & Camilli, A. The cadA gene of Vibrio cholerae is induced during infection and plays a role in acid tolerance. Mol. Microbiol. 34, 836–849 (1999).
Kim, J.-S., Choi, S. H. & Lee, J. K. Lysine decarboxylase expression by Vibrio vulnificus is induced by SoxR in response to superoxide stress. J. Bacteriol. 188, 8586–8592 (2006).
Bower, J. M. & Mulvey, M. A. Polyamine-mediated resistance of uropathogenic Escherichia coli to nitrosative stress. J. Bacteriol. 188, 928–933 (2006).
Bower, J. M., Gordon-Raagas, H. B. & Mulvey, M. A. Conditioning of uropathogenic Escherichia coli for enhanced colonization of host. Infect. Immun. 77, 2104–2112 (2009).
Akhova, A. V. & Tkachenko, A. G. Lysine Decarboxylase Activity as a Factor of Fluoroquinolone Resistance in Escherichia coli. Microbiology 78, 575–579 (2009).
Kikuchi, Y., Kurahashi, O., Nagano, T. & Kamio, Y. RpoS-dependent expression of the second lysine decarboxylase gene in Escherichia coli. Biosci. Biotechnol. Biochem. 62, 1267–1270 (1998).
Samartzidou, H., Mehrazin, M., Xu, Z., Benedik, M. J. & Delcour, A. H. Cadaverine inhibition of porin plays a role in cell survival at acidic pH. J. Bacteriol. 185, 13–19 (2003).
Bekhit, A., Fukamachi, T., Saito, H. & Kobayashi, H. The role of OmpC and OmpF in acidic resistance in Escherichia coli. Biol. Pharm. Bull. 34, 330–334 (2011).
Tkachenko, A. G., Pozhidaeva, O. N. & Shumkov, M. S. Role of polyamines in formation of multiple antibiotic resistance of Escherichia coli under stress conditions. Biochem. Biokhimiia 71, 1042–1049 (2006).
Kanjee, U. et al. Linkage between the bacterial acid stress and stringent responses: the structure of the inducible lysine decarboxylase. EMBO J. 30, 931–944 (2011).
Sabo, D. L., Boeker, E. A., Byers, B., Waron, H. & Fischer, E. H. Purification and physical properties of inducible Escherichia coli lysine decarboxylase. Biochemistry (Mosc.) 13, 662–670 (1974).
Snider, J. et al. Formation of a distinctive complex between the inducible bacterial lysine decarboxylase and a novel AAA+ ATPase. J. Biol. Chem. 281, 1532–1546 (2006).
Wong, K. S. & Houry, W. A. Novel structural and functional insights into the MoxR family of AAA+ ATPases. J. Struct. Biol. 179, 211–221 (2012).
El Bakkouri, M. et al. Structure of RavA MoxR AAA+ protein reveals the design principles of a molecular cage modulating the inducible lysine decarboxylase activity. Proc. Natl. Acad. Sci. USA 107, 22499–22504 (2010).
Wong, K. S. et al. The MoxR ATPase RavA and its cofactor ViaA interact with the NADH:ubiquinone oxidoreductase I in Escherichia coli. PloS One 9, e85529 (2014).
Malet, H. et al. Assembly principles of a unique cage formed by hexameric and decameric E. coli proteins. eLife 3, e03653 (2014).
Yamamoto, Y., Miwa, Y., Miyoshi, K., Furuyama, J. & Ohmori, H. The Escherichia coli ldcC gene encodes another lysine decarboxylase, probably a constitutive enzyme. Genes Genet. Syst. 72, 167–172 (1997).
Käck, H., Sandmark, J., Gibson, K., Schneider, G. & Lindqvist, Y. Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J. Mol. Biol. 291, 857–876 (1999).
Kanjee, U., Gutsche, I., Ramachandran, S. & Houry, W. A. The enzymatic activities of the Escherichia coli basic aliphatic amino acid decarboxylases exhibit a pH zone of inhibition. Biochemistry (Mosc.) 50, 9388–9398 (2011).
Erhardt, H. et al. Disruption of individual nuo-genes leads to the formation of partially assembled NADH:ubiquinone oxidoreductase (complex I) in Escherichia coli. Biochim. Biophys. Acta 1817, 863–871 (2012).
Lohinai, Z. et al. Biofilm Lysine Decarboxylase, a New Therapeutic Target for Periodontal Inflammation. J. Periodontol. 86, 1176–1184 (2015).
Peters, J. L. et al. Effects of immunization with natural and recombinant lysine decarboxylase on canine gingivitis development. Vaccine 30, 6706–6712 (2012).
Lohinai, Z. et al. Bacterial lysine decarboxylase influences human dental biofilm lysine content, biofilm accumulation, and subclinical gingival inflammation. J. Periodontol. 83, 1048–1056 (2012).
Kim, H. J. et al. Optimization of Direct Lysine Decarboxylase Biotransformation for Cadaverine Production with Whole-Cell Biocatalysts at High Lysine Concentration. J. Microbiol. Biotechnol. 25, 1108–1113 (2015).
Ma, W. et al. Enhanced cadaverine production from L-lysine using recombinant Escherichia coli co-overexpressing CadA and CadB. Biotechnol. Lett. 37, 799–806 (2015).
Li, N., Chou, H., Yu, L. & Xu, Y. Cadaverine production by heterologous expression of Klebsiella oxytoca lysine decarboxylase. Biotechnol. Bioprocess Eng. 19, 965–972 (2014).
Dubochet, J. et al. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 21, 129–228 (1988).
Mindell, J. A. & Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003).
van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R. & Schatz, M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 116, 17–24 (1996).
Navaza, J. On the three-dimensional reconstruction of icosahedral particles. J. Struct. Biol. 144, 13–23 (2003).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Estrozi, L. F. & Navaza, J. Ab initio high-resolution single-particle 3D reconstructions: the symmetry adapted functions way. J. Struct. Biol. 172, 253–260 (2010).
Scheres, S. H. W. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Fernández, J. J., Luque, D., Castón, J. R. & Carrascosa, J. L. Sharpening high resolution information in single particle electron cryomicroscopy. J. Struct. Biol. 164, 170–175 (2008).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Schwede, T., Kopp, J., Guex, N. & Peitsch, M. C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 (2003).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Lopéz-Blanco, J. R. & Chacón, P. iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 184, 261–270 (2013).
Navaza, J., Lepault, J., Rey, F. A., Alvarez-Rúa, C. & Borge, J. On the fitting of model electron densities into EM reconstructions: a reciprocal-space formulation. Acta Crystallogr. D Biol. Crystallogr. 58, 1820–1825 (2002).
Siebert, X. & Navaza, J. UROX 2.0: an interactive tool for fitting atomic models into electron-microscopy reconstructions. Acta Crystallogr. D Biol. Crystallogr. 65, 651–658 (2009).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001).
Acknowledgements
We thank Guy Schoehn for establishing and managing the cryo-electron microscopy platform and for providing training and support. We also thank Pawel Penczek and Leandro Estrozi for helpful discussions. For electron microscopy, this work used the platforms of the Grenoble Instruct center (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The electron microscope facility (Polara electron microscope) is supported by the Rhône-Alpes Region (CIBLE and FEDER), the FRM, the CNRS, the University of Grenoble Alpes and the GIS-IBISA. DC was the recipient of the Grenoble Alliance for Integrated Structural and Cell Biology (GRAL) PhD fellowship. KL was the recipient of the Ontario Graduate Scholarship and the Life Sciences Award from the University of Toronto. SWSC was the recipient of the National Sciences and Engineering Research Council of Canada (NSERC) Undergraduate Student Research Award. This work was supported by the ANR-12-JSV8-0002 and the ERC 647784 grants to IG, the CIHR MOP-130374 to WAH, the ANR-10-LABX-49-01 and the ANR-11-LABX-0003-01.
Author information
Authors and Affiliations
Contributions
E.K., H.M. and I.G. carried out EM data collection with assistance of M.B. and analyzed the data. D.C. performed cloning, multiple sequence alignment and phylogenetic analysis under the direction of S.E. and I.G., J.P. cloned and purified chimeric proteins under the direction of S.O.C., K.L. and S.W.S.C. purified LdcI, LdcC and LARA under the direction of W.A.H., I.G. conceived and directed the studies and wrote the manuscript with input from E.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kandiah, E., Carriel, D., Perard, J. et al. Structural insights into the Escherichia coli lysine decarboxylases and molecular determinants of interaction with the AAA+ ATPase RavA. Sci Rep 6, 24601 (2016). https://doi.org/10.1038/srep24601
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24601
This article is cited by
-
Structural and functional analysis of the Francisella lysine decarboxylase as a key actor in oxidative stress resistance
Scientific Reports (2021)
-
Adaptational changes in physiological and transcriptional responses of Bifidobacterium longum involved in acid stress resistance after successive batch cultures
Microbial Cell Factories (2019)
-
Tailoring the properties of (catalytically)-active inclusion bodies
Microbial Cell Factories (2019)
-
Catalytically active inclusion bodies of L-lysine decarboxylase from E. coli for 1,5-diaminopentane production
Scientific Reports (2018)
-
Enhancement of the thermal and alkaline pH stability of Escherichia coli lysine decarboxylase for efficient cadaverine production
Biotechnology Letters (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
