
Abstract
Little is known about the major histocompatibility complex (MHC) in the genome of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis) (YFP) or other cetaceans. In this study, a high-quality YFP bacterial artificial chromosome (BAC) library was constructed. We then determined the organization and characterization of YFP MHC class II region by screening the BAC library, followed by sequencing and assembly of positive BAC clones. The YFP MHC class II region consists of two segregated contigs (218,725 bp and 328,435 bp respectively) that include only eight expressed MHC class II genes, three pseudo MHC genes and twelve non-MHC genes. The YFP has fewer MHC class II genes than ruminants, showing locus reduction in DRB, DQA, DQB and loss of DY. In addition, phylogenic and evolutionary analyses indicated that the DRB, DQA and DQB genes might have undergone birth-and-death evolution, whereas the DQB gene might have evolved under positive selection in cetaceans. These findings provide an essential foundation for future work, such as estimating MHC genetic variation in the YFP or other cetaceans. This work is the first report on the MHC class II region in cetaceans and offers valuable information for understanding the evolution of MHC genome in cetaceans.
Similar content being viewed by others


Introduction
The major histocompatibility complex (MHC) plays a vital role in the vertebrate immune system, recognizing and presenting pathogen-derived peptides to receptors on CD8+ and CD4+ T lymphocytes1. MHC genes are mainly classified as class I and class II genes. The class I genes are primarily involved in immune defense against intracellular pathogens, whereas the class II genes are predominantly responsible for monitoring the extracellular environment for foreign peptides2. The MHC genomic structure of some mammals (e.g., human, rat, dog, cat, horse, pig, sheep and cattle), particularly the MHC class II regions, have been studied extensively3,4,5,6,7,8,9. This region displays highly conserved organization in a wide range of mammals10 and MHC class II genes usually exhibit high intraspecific diversity, possibly reflecting the need to respond to foreign peptides derived from parasites11,12.
However, little is known about the content and organization of the MHC class II region in cetaceans. Sequence variation and polymorphism analyses of exon 2 of DRB and DQB have been performed for some cetacean species13,14,15,16,17,18,19. Though, studies about the expression of DRB and DQB genes have been reported in a few cetaceans20,21,22, the expression and functionality of most MHC class II genes has not been considered in most studies14,16,17,23,24 and gene duplication must be confirmed. For example, only a single DQB locus had been reported in finless porpoises (Neophocaena phocaenoides) inhabiting Japanese waters18 and in some other toothed whales15,19,20. By contrast, multiple DQB loci have been described in the finless porpoise populations living in Chinese waters23,25, which were reclassified in 2009 as two different species: the Indo-Pacific finless porpoise (N. phocaenoides) and the narrow-ridged finless porpoise (N. asiaeorientalis)26,27,28.
The Yangtze finless porpoise (N. a. asiaeorientalis) (YFP) is the sole freshwater subspecies of N. asiaeorientalis and is found in the middle and lower reaches of the Yangtze River and the adjoining Poyang and Dongting lakes29,30. Due to its small population size, sharply declining population and high probability of extinction, the YFP was recently reclassified as a critically endangered population on the IUCN (International Union for Conservation of Nature) Red List29,30. In most mammals, MHC loci are highly variable, which may be an adaptive strategy against the large number of pathogens encountered by natural populations13,31. Thus, species or populations with a low level of MHC diversity might be particularly vulnerable to infectious diseases, leading to a decline of population viability32. A previous study revealed abundant genetic variations in DQB exon 2 and suggested that the YFP population had a much higher level of DQB diversity than marine populations23. However, such estimation of MHC diversity for this population may be inaccurate, because multiple DQB loci might have been amplified simultaneously and been analyzed as a single DQB locus, besides only a single MHC class II gene was analyzed in this study, which could not well reflect genetic variations of MHC genes in YFP23. Thus, determining the organization and characterization of the YFP MHC class II region will provide an essential foundation for estimating MHC genetic diversity and resistance/susceptibility to pathogens, exploring the mechanism of mate choice and other conservation genetics studies based on MHC markers in the YFP. In addition, during the evolutionary transition from land to a marine environment approximately 50 million years ago, cetaceans have undergone numerous critical challenges33,34 and their immune system genes (such as MHC genes) may reflect this shift in habitat. Thus, characterizing the YFP MHC class II region is also of significant importance for exploring adaptive evolution in the cetacean MHC.
In this study, a high-quality bacterial artificial chromosome (BAC) library was constructed for the YFP for in vitro genetic resource conservation and to characterize the YFP MHC genomic structure. We investigated the genomic organization of the YFP MHC class II region in this work and conducted comparative and phylogenic analyses of the MHC class II genes in this region. Determining the organization and characteristics of the YFP MHC class II genes will provide a solid foundation for evaluating MHC genetic diversity in future work and support valid evolutionary inferences about non-model species such as cetaceans. In addition, this work provides the first genomic sequence map of MHC class II region in Cetacea and may be a valuable resource for further cetacean MHC genomic research.
Results
YFP BAC library construction
A BAC library including approximately 440,000 clones was successfully constructed for the YFP with an average insert size of approximately 113 kb and an empty vector rate of approximate 5.4% (Fig. S1). The library contained 14.8-fold genome equivalents based on the YFP genome size of 3.17 × 109 bp35. This library thus has applications in in vitro conservation of genetic resources and as a tool for MHC genomic research.
Sequencing and assembly of the YFP MHC class II contigs
Two BAC clone-based contigs bearing MHC class II genes were identified, consisting of two (588H3 and 1974B12) and four (271G8, 612G6, 951A10 and 685H5) overlapping BAC clones, respectively (Fig. 1). These six clones were sequenced using the Illumina Hiseq 2000 platform. One billion bases were generated from each BAC clone, which were then assembled and aligned into two consensus sequences of 218,725 bp and 328,435 bp without any gap (Fig. 1, Table 1). These six BAC clone sequences and the two consensus sequences have been deposited into GenBank with accession numbers KP114539 – KP114544 and KT804703 – KT804704. In this study, the YFP MHC was designated as MhcNeas (in which Neas is composed of the first two letters of the genus name Neophocaena and the first two letters of the species name asiaeorientalis) according to the proposal for MHC nomenclature36.

Genomic structure and content of the Yangtze finless porpoise MHC class II region, which was divided into two segregated subregions (MHC IIa and MHC IIb).
Black boxes suggest pseudogenes. RI indicates RING1; HS indicates HSD17B18; SL indicates SLC39A7; RX indicates RXRB; ψ indicates pseudogene. The black bold genes indicate MHC class II genes in the Yangtze finless porpoise MHC class II region. The light green genes represent non-MHC genes in the Yangtze finless porpoise MHC class II region. The red vertical bars in BAC clones represent the binding sites for DRA and DMA primers of screening library, the black vertical bars representing the binding sites for end-primers of screening library.
Identification of the YFP MHC class II genes
After repeat masking, gene prediction was conducted using the Genscan and Fgenesh programs and BLAST alignment. A total of 23 genes were identified in the MHC class II region of the YFP. The contig sequence of 218,725 bp included six genes (BTNL2, DRA, DRB1, ψDRB2, DQA and DQB) and 17 genes were detected in the other contig sequence of 328,435 bp, including RING1, HSD17B8, SLC39A7, RXRB, COL11A2, ψDPB, DOA, BRD2, DMA, DMB, PSMB9, TAP1, PSMB8, TAP2, DOB, ψDRB3 and GCLC (the bold text indicates YFP MHC class II genes, the others are non-MHC genes) (Fig. 1). Eight MHC class II genes (DRA, DRB1, DQA, DQB, DMA, DMB, DOA and DOB) were further verified as expressed genes by cDNA PCR. The mRNA sequences of these eight genes have been submitted to GenBank with accession numbers KP114553 – KP114560.
Comparative genomics analysis of the YFP MHC class II region
The dot-plot analysis of the YFP MHC class II region itself showed three intra-MHC repeats corresponding to DRB1, ψDRB2 and ψDRB3 regions (Fig. 2). Additionally, another three relatively small repeat regions were yielded by the self-dot plot of Neas, which were probably specific repeat sequences in Yangtze finless porpoise MHC region (Fig. 2). In addition, the genomic organizations of the MHC class II regions of YFP (Neas), human (HLA), cattle (BoLA), sheep (OLA), horse (ELA), dog (DLA) and cat (FLA) were compared using VISTA with the LAGAN alignment program. The VISTA plot revealed two highly variable segments in the mammalian MHC II regions, namely the DRB1-DQB and GCLC-ψDRB3 subregions (Fig. 3).

Dot matrix analysis of the Yangtze finless porpoise MHC class II nucleotide sequences itself.
The dot plot was performed using Pipmaker.

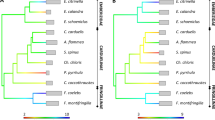
Phylogenetic relationship and VISTA plot of the MHC class II nucleotide sequences from eight mammal species: human (Homo sapiens), cattle (Bos taurus), sheep (Ovis aries), pig (Sus scrofa), horse (Equus caballus), dog (Canis familiaris), cat (Felis catus) and Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis).
The corresponding MHC class II regions are labeled HLA, BoLA, OLA, SLA, ELA, DLA, FLA and Neas, respectively. For the sake of comparative sequence analysis, the orientations of the MHC class IIb regions from Yangtze finless porpoise, cattle and sheep were inverted. The height of the bars represents the repeats distributing in Neas. Different color bar represents different kind of repeat as that showed on Fig. 1.
To explore the changes in the two highly variable segments in YFP after the separation of Cetacea and Artiodactyla, MHC class II gene numbers and their orders in Neas, BoLA, OLA and SLA were compared (Fig. 4). And based on the locus distribution of MHC class II genes, the MHC class II regions among the four species could be divided into five parts, including two conserved segments (A and D) that contain stable number of genes and three highly variable segments (B, C and E) that contain different number of MHC class II genes (Fig. 4). A and D corresponded to BTNL2-DRA and DOB-RING1, respectively, while B, C and E corresponded to the DRB, DQ and DY subregions, respectively (Fig. 4). In the highly variable subregions (B and C), the major differences among the four species were the numbers of DRB and DQ loci. Because DSB evolved from a DRB-like sequence5 and thus, in the highly variable E subregion, DRB is expressed in BoLA but it is a pseudogene in both Neas and SLA. The DY genes (DYA and DYB) have been lost in Neas. By comparison, the loci of MHC class II genes have decreased in the YFP (Fig. 4).

Schematic comparison of the MHC class II region organization of cattle (BoLA), sheep (OLA), pig (SLA) and Yangtze finless porpoise (Neas).
Different colors indicate different categories: white, (A); yellow, (B); purple, (C); green, (D); and blue, (E). Dotted lines indicate breakpoints. Unfilled boxes and ψ indicate pseudogenes. The structure of the BoLA class II region is depicted according to the genome sequence of Bos taurus chromosome 23 (NCBI Reference Sequence: AC_000180.1), the structure of the OLA referring to Gao et al.9 and the structure of the SLA class II region according to the genomic sequence of SLA7.
Evolutionary process and selection pressure analyses
Phylogenetic trees were constructed to explore the evolutionary process of the YFP or cetacean MHC class II genes. First, we constructed a phylogenetic tree based on the entire sequence of the MHC class II region in Neas, BoLA, OLA, SLA, ELA, FLA, DLA and HLA (Fig. 3). This tree revealed that the order of species divergence is consistent with the phylogenetic analyses based on other nucleotide sequences, supporting Cetacea is closer to Ruminantia (cattle and sheep) than Suidae (pig)37,38. The birth-and-death process is a very important model for the evolution of mammalian MHC class II genes10,39. Thus, to further explore the evolutionary history of the YFP MHC class II genes, phylogenetic trees were also constructed using the coding region sequences (CDSs) of DRA, DRB, DQA, DQB, DMA, DMB, DOA and DOB from YFP, cattle, sheep and pig, with human as the outgroup (Fig. 5). The phylogenetic relationships of the eight genes were not consistent. The phylogeny of CDSs for DRA, DMA, DMB, DOA and DOB shared the same tree topology as the tree constructed using the entire MHC class II region (Fig. 3), whereas the phylogenetic trees for the CDSs of DRB, DQA and DQB had quite different tree topologies (Fig. 5).

Phylogenetic trees based on the coding sequences of eight MHC class II genes from cattle (BoLA), sheep (OLA), pig (SLA) and Yangtze finless porpoise (Neas).
MrBayes and PhyML software yielded phylogenetic trees with similar topologies and thus only Bayesian trees are provided. Numbers near nodes indicate Bayesian posterior probabilities.
When evolutionary selection analyses were conducted on the CDSs of the eight MHC class II loci (DRA, DRB, DQA, DQB, DMA, DMB, DOA and DOB) in mammals (human, cattle, sheep, pig and seven cetacean species) (Table S5), all dN/dS ratios were less than 1 (Table 2). However, when evolutionary selection analysis was conducted on the eight MHC class II loci in the seven cetacean species, the ratio of dN/dS was significantly larger than 1 for the DQB locus (dN/dS = 2.125, P = 6.36E-4) (Table 2), indicating positive selection at the DQB locus. In addition, MHC class II genes are classified into classical MHC class II genes (DR, DQ and DP) and non-classical MHC class II genes (DM and DO). The non-classical MHC class II proteins’ roles are known in antigen-presenting cells, which do not directly bind antigens40. However, the classical MHC class II proteins bind antigens with peptide-binding region, which is mainly coded by the second exon41. And thus, the dN/dS ratios of exon 2 and the rest CDS (except exon 2) of classical MHC class II genes (DR and DQ) were respectively estimated in mammals and in cetaceans to explore which part is mainly subjected to positive selection. The result also showed that only the dN/dS of exon 2 of DQB in cetaceans is significantly larger than 1 (Table S6). YFP inhabits in the freshwater environment, which is quite different from the marine environment. And thus, to eliminate the possible effect from YFP, the dN/dS ratios of exon 2 of DQB was further estimated only in marine cetaceans (excluding YFP), which is also significantly larger than 1 (dN/dS = 2.58, P = 3.75E-4).
Discussion
The population genetic structure of the YFP, as revealed by mitochondrial and microsatellite DNA analyses, indicates that the YFP is becoming a genetically fragmented population and emphasizes the need for genetic conservation of this critically endangered population42. Thus, the genetic diversity of the YFP must be further investigated, particularly regarding the variability of MHC genes, to evaluate its long-term survival probability. However, little is known about the MHC genomic structure of the YFP or any other cetacean species, limiting the study of MHC genetic diversity. Therefore, in this work, we first constructed a high-quality BAC library for the YFP and then determined the organization and characteristics of the MHC class II genes.
Based on the high-quality YFP BAC library we constructed, two contigs (MHC IIa and IIb) bearing the YFP MHC class II genes were mapped, sequenced, assembled and annotated (Fig. 1). According to the arrangement of MHC class II genes in YFP (Fig. 4), it is very possible that the MHC class II region has been also interrupted and divided into two segregated subregions (MHC IIa and MHC IIb) in Cetacea like ruminants9,43; in cattle and sheep there are approximate 18.5 Mb between these two segregated suregions9. There are three evidences for this inference. First, a sequence of approximately 80 kb after the DQB locus did not contain any MHC class II gene and there was an overlapping fragment (approximately 40 kb) between BAC clones 588H3 and 1974B12. This indicates that the YFP MHC class IIa region obtained in this study was complete and authentic. Second, the location of the GCLC (glutamate-cysteine ligase, catalytic subunit) gene is far from the MHC class II region in those mammals (e.g., human, dog, cat, horse and pig), of which the genomes were well sequenced and studied. For example, according to available genome data in NCBI, the distance is approximately 20 Mb in human, dog, cat and horse and approximately 2 Mb in pig. However, the GCLC gene is very close to the MHC class II region in the YFP, cattle5 and sheep9 (Fig. 4). The results also suggested that the structure of the YFP MHC class II region is similar to those in cattle and sheep. Third, genome data in NCBI indicate that the MHC class II region in killer whale (Orcinus orca) (NW_004438672, NW_004438437) is also interrupted by other sequences. Taken together, these support the inference that the YFP MHC class II region might be interrupted into two segregated subregions, possibly as the hypothesis in BoLA and OLA that a large inversion of the ancestral chromosome has disrupted the ancestral MHC class II region into IIa and IIb sub-regions9. However, the reported swine MHC class II region is not interrupted by other DNA fragments7. From the phylogenetic trees constructed using the whole MHC class II sequences (Fig. 3), we can infer that the breakpoint in the MHC class II region might have occurred after the divergence of swine and Cetartiodactyla but before the divergence of Cetacea and ruminants.
When gene prediction was conducted on the MHC class II region of the YFP, only 11 MHC class II genes were identified (DRA, DRB1, ψDRB2, DQA and DQB in MHC IIa; ψDPB, DOA, DMA, DMB, DOB and ψDRB3 in MHC IIb) (Fig. 1). Of these, only eight MHC class II genes (DRA, DRB1, DQA, DQB, DMA, DMB, DOA and DOB) were further verified as expressed genes by cDNA PCR. Comparative analyses of this MHC class II region clearly indicated that the YFP has fewer MHC class II loci than cattle and sheep, particularly in the three highly variable segments (B, C, E) (Fig. 4).
To explore the deleting process of MHC class II loci in the YFP, the evolutionary history of the eight expressed MHC class II genes (DRA, DRB1, DQA, DQB, DMA, DMB, DOA, DOB) after the divergence of Cetacea and ruminants was investigated through comparative analyses of their phylogenetic trees constructed for Neas, BoLA, OLA and SLA, using HLA as the outgroup. The phylogenetic trees for the DRA, DOB, DOA, DMB and DMA genes showed similar topology (Fig. 5). By contrast, phylogenetic analysis of the DRB, DQA and DQB genes indicated that the sequences might be clustered according to gene categories (e.g. orthologous or paralogous genes) than species (Fig. 5). But the tree topologies among them were not similar, which may reflect divergent birth-and-death evolution of the DRB, DQA and DQB genes.
In the phylogenetic tree of the DRB genes, DRB genes were clustered into three clades according to gene categories among the YFP, pig, cattle and sheep (Fig. 5). Neas-DRB and OLA-DRB2 were clustered into a clade that was older than the other two clades. SLA-DRB1 was clustered with BoLA-DRB3, BoLA-DRB1 and OLA-DRB1 into a younger clade. However, from the phylogenetic relationship showed in Fig. 3, we can see that SLA was an older clade than cetaceans and ruminants. And thus, based on the three clades showed in Fig. 5, we could infer that at least three DRB loci were present in their ancestral MHC class II region before the divergence of Cetacea and ruminants, but as shown in the Neas B subregion of Fig. 4, at least one DRB locus was entirely deleted in the evolutionary process.
The phylogenetic tree of the DQA genes revealed that the DQA genes were also grouped according to gene categories among the YFP, cattle and sheep, producing two clades (Fig. 5). Thus, there were at least two DQA loci before the divergence of Cetacea and ruminants and the YFP lost one DQA locus thereafter (Fig. 4). The phylogenetic tree of the DQB genes revealed that DQB genes were clustered into two groups after the divergence of Cetacea and ruminants. The DQB phylogenetic tree includes only one DQB locus in the YFP, but the cetacean and ruminants DQB loci may be paralogous genes, as Yang et al. presumed that the sequences of cetaceans and artiodactyls (including ruminants) were paralogous in the DQB genes44. Yang et al. hypothesized that gene-duplication events occurred at least once in the DQB genes prior to the cetacean/artiodactyl divergence44. Gene duplication could give rise to two sets of paralogous genes (the A set and B set). The A sets of all artiodactyls have survived to the present day, whereas the B sets were lost or became pseudogenes. By contrast, the B sets in cetaceans were preserved, whereas the A sets in cetaceans were lost44. As shown in the C subregion in Fig. 4, at least one DQB locus and one DQA locus have been lost since the cetacean/ruminants (BoLA and OLA) divergence.
In comparison with the BoLA II and OLA II genes, the YFP has fewer DRB, DQA and DQB loci in the MHC class II region. Thus, this region underwent the birth-and-death evolutionary process and at least one locus might have been lost among the three genes in the evolutionary process of cetaceans. The DYA and DYB genes were also lost in Neas (Fig. 4). Based on the phylogenetic tree of the eight species (Fig. 3), we inferred that DY genes were lost in cetaceans after the divergence of Cetacea and ruminants. These changes in cetacean MHC regions might be related to their ancestral shift in habitat from land to a marine environment. Due to the lower prevalence of infectious diseases in the marine environment than the land environment, these genes, which play an essential role in defense against foreign peptides derived from parasites, might have been under less evolutionary selection pressure45.
The ratio of dN/dS indicates the selection pressure under which the coding sequence evolved: dN/dS > 1 is interpreted as signifying positive selection, whereas dN/dS < 1 indicates purifying selection46. Based on Table 2 and Table S6, we determined that the eight expressed MHC II genes in mammals have evolved under purifying selection, with the exception of the DQB locus in cetaceans. In cetaceans, the DQB subjected to positive selection mainly focuses on the exon 2, which showed the importance of the exon 2 as peptide-binding region. The evolutionary selection analyses suggested that the eight expressed MHC class II genes in cetaceans might have evolved under purifying selection when the ancestor of cetaceans shifted from land to a marine environment; however, after the cetaceans fully adapted to aquatic life, the DQB locus have evolved under positive selection to defend against different marine pathogens encountered during the process of speciation or population expansion. This result is consistent with a previous study suggested positive selection pressure on the exon 2 of DQB in bottlenose dolphin19, further indicating that the DQB locus may play a key role in the cetacean evolutionary process. Thus, sequence variation at the exon 2 of DQB locus should be examined more closely in future evaluations of genetic variability in the YFP or any other cetacean species.
In this work, only one DQB locus was identified in the YFP, whereas Xu et al. reported that at least three copies of the DQB gene are present in the finless porpoise25. However, a single DQB locus in the YFP is consistent the presence of a single DQB locus in many toothed whales15,19,20. Although there are three DRB genes in the YFP MHC class II region (Fig. 1), only the DRB1 gene is functional, whereas ΨDRB2 has lost exon 1, a part of exon 2 and exon 6 (Fig. S2) and ψDRB3 has retained only exon 3. Exon 2 of the DRB locus has typically been investigated in previous studies of DRB genetic variability14,17,24. Because exon 2 of ψDRB2 in YFP has only lost the first fourteen bases (Fig. S2), inappropriate primer design could lead to the amplification of this exon 2 and consequently, result in overestimation of DRB genetic variability in the YFP. Given the critically endangered status of the YFP, reevaluation of the population genetic diversity of MHC class II genes is urgently needed. Because the DQB locus is under positive selection in cetaceans, assessing the genetic variability of the DQB locus in the YFP population is particularly important. In addition, most studies investigating MHC variability have focused on a single or very few MHC loci due to the lack of valid MHC markers, hindering the elucidation of the intrinsic molecular mechanism12. Thus, this exploration of organization and characteristics of the YFP MHC class II region provides an essential foundation for future work, such as estimating MHC genetic variation in the YFP. This work is also the first report on the MHC class II region in cetaceans and offers valuable information for understanding the evolution of the MHC genome in cetaceans.
Materials and Methods
Tissue collection
To provide high quality genomic DNA for BAC library construction in this study, liver tissue sample was collected from a female YFP that was accidentally killed by a fishing boat in the main stream of the Yangtze River. In addition, to obtain mRNA to verify the functionality of the MHC class II gene identified in this study, blood sample was drawn from the vein in the fluke of a captive YFP at Wuhan Baiji Dolphinarium by using one-off syringe. Necropsy and sampling were conducted systematically in accordance with all ethical guidelines and legal requirements in China. The protocol of this study was approved by the Institutional Review Board of the Institute of Hydrobiology, Chinese Academy of Sciences.
BAC library construction and positive clone screening
Liver tissue was ground with liquid nitrogen and resuspended in ice-cold phosphate-buffered saline. High-molecular-weight DNA (120–170 kb) was extracted following the protocol described by Osoegawa47 and Zeng48. A BAC library was constructed for this YFP using the Copy Control BAC Cloning Kit and TransforMax EPI300 Electrocompetent E. coli according to the manufacturer’s protocol (Epicentre, Madison, USA) and a previously reported protocol for BAC library construction47. To conserve space, each two BAC clones were preserved in one well of a 96-well plate. The BAC library consisted of approximately 440,000 clones and was preserved in 2,290 96-well plates.
Previous studies suggested that the MHC class II regions have a conservation of synteny among certain mammals7,8,10, so we employed a walking method to construct the YFP MHC class II contig(s) in this study. Library screening was performed using a 4D-PCR method49. Firstly, two pairs of PCR primers (DRA and DMA) (Table S1) were used to screen the BAC library. To confirm positive clones containing amplicons of DRA and DMA, the amplified products were cloned and sequenced and then identified by BLAST sequences in NCBI database. And then, end-sequence all real positive BAC clones, primer design of their end sequences and amplifying these positive BAC clones were performed according to reported method48. The positive BAC clone, which had the longest insert size and also had effective end-primer pair at both ends, would be chosen as the best positive BAC clone for the two primer pairs (DRA and DMA) and also as the starting point of next step. Secondly, library screening, positive clone end-sequencing and primer design were repeated to construct the contig until boundaries of MHC class II region were included in certain BAC clones. All the primer pairs that had been used to screen positive clones and to construct the contigs of YFP MHC class II region were listed in Table S1.
Positive BAC clones sequencing and assembly
DNA was extracted from each positive BAC clone using the AxyPrepTM Plasmid Kit (Axygen Biosciences, China) to avoid E. coli genomic contamination. The extracted DNA was sent to Shanghai Majorbio Bio-pharm Technology Company for high-throughput sequencing on the Illumina Hiseq 2000 platform. Clean reads were obtained by removing reads containing adapter sequences, poly-N reads and low-quality reads (containing >50% bases with sequence quality <15) from the raw reads. The clean reads were assembled using SOAPdenovo software50. A primer-walking method was applied to fill any gaps in the assembled sequences.
Gene identification and verification
The complete sequences of BAC clones were aligned into two consensus sequences using Blast2Seq51. Two full-length sequences of the MHC class II region were analyzed by RepeatMasker in a Linux operating system, with Cetacea as the organism-specific parameter. Genes were identified using two different gene prediction programs: Genscan and Fgenesh52,53. In the Genscan and Fgenesh algorithms, the organism-specific parameters were set as vertebrate and cow, respectively. The predicted gene and protein sequences were aligned with NCBI reference RNA sequences using BLASTN54 and non-redundant protein sequences with BLASTP55, respectively, to identify the boundaries of exons and expressed genes or pseudogenes.
To further verify the functionality of the predicted MHC class II genes, cDNA PCR was performed. Firstly, mRNA was extracted from the blood sample of a captive YFP by using RNAprep Pure Kit (TIANGEN, Beijing, China). And then, after quality check, mRNA was reversely transcribed into cDNA by using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, made in EU). Finally, the obtained cDNA was used as template to conduct PCR amplification. At the same time, based on the YFP MHC class II region sequence obtained in this study and those homologous sequences of closely related species downloaded from GenBank, eight primer pairs were designed for the eight MHC class II genes by using Primer Premier 5.0 (Table S2). The PCR products were cloned and two clones from each primer were chosen to sequence according to previously reported methods56.
Comparative genomics analysis
The self-dot matrix of the YFP MHC class II region sequence was created using PipMaker57. For comparative analysis, we downloaded the homologous MHC class II region sequences of human (Homo sapiens), cattle (Bos taurus), sheep (Ovis aries), pig (Sus scrofa), horse (Equus caballus), dog (Canis familiaris) and cat (Felis catus) from GenBank (Table S3). The annotated MHC class II region sequence of the YFP and the trimmed corresponding sequences of mammals from previous reports were aligned using VISTA58 with the LAGAN alignment program. LAGAN is a system for global pair-wise and multiple alignment of genomic sequences and may be more suitable than other pairwise alignments for aligning conserved exons between distant species59.
Evolutionary process and selection pressure analyses
To construct the phylogenetic tree, the MHC class II region sequences of the YFP and the aforementioned seven mammals were aligned using the programs Mauve 2.3.160 and MAFFT (version 7)61 and checked manually. The molecular evolution model of the sequences was generated by MrModeltest 2.362 and the phylogenetic tree was constructed by MrBayes 3.2.263 with 1,000,000 generations.
To explore the evolutionary history of the eight YFP MHC II genes, the coding sequences of the MHC II genes except DMB were completely amplified by using the eight primers listed in Table S2 and the cDNA from a captive YFP and sequenced. The corresponding sequences of human, cattle, sheep and pig were downloaded from GenBank (Table S4). In this process, we collected as many expressed or predicted genes as possible to construct phylogenetic trees. The coding sequences were also aligned using MAFFT software. Phylogenetic trees were constructed using the protocol described above for the entire MHC II gene sequences. In addition, another phylogenetic tree was constructed using the software PhyML (version 3.1) with 1000 bootstrapping replicates64. The molecular evolution model of the nucleotide sequence was estimated by Modeltest 3.765.
Because the ratio of non-synonymous and synonymous substitutions (dN/dS) indicates the selection pressure under which the coding sequence evolved, dN/dS was calculated in MEGA5.0 and the Z-test statistical method was performed66. To strengthen the reliability, before calculating dN/dS, the substitution saturation was measured using DAMBE (version 5.3.108)67. The overall dN/dS ratios of the eight MHC class II genes CDS in mammals and in cetaceans were estimated (Table S5). In addition, due to the exon 2 of classical MHC class II genes (DRA, DRB, DQA and DQB) mainly involving in the peptide-binding region, the dN/dS ratios for the exon 2 and the rest CDS (excluding exon2) of the four classical MHC class II genes were also estimated in mammals and in cetaceans to verify the results obtained based on CDS.
Additional Information
How to cite this article: Ruan, R. et al. Organization and characteristics of the major histocompatibility complex class II region in the Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Sci. Rep. 6, 22471; doi: 10.1038/srep22471 (2016).
References
Wiley, D. C. A hypothetical model of the foreign antigen binding site of class II histocompatibility molecules. Nature 332, 845–850 (1989).
Hughes, A. L. & Yeager, M. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 32, 415–435 (1998).
Beck, S. & Trowsdale, J. The human major histocompatibility complex: lessons from the DNA sequence. Annu. Rev. Genomics Hum. Genet. 1, 117–137 (2000).
Hurt, P. et al. The genomic sequence and comparative analysis of the rat major histocompatibility complex. Genome Res. 14, 631–639 (2004).
Childers, C. P. et al. Comparative analysis of the bovine MHC class IIb sequence identifies inversion breakpoints and three unexpected genes. Anim. Genet. 37, 121–129 (2005).
Tallmadge, R. L., Lear, T. L. & Antczak, D. F. Genomic characterization of MHC class I genes of the horse. Immunogenetics 57, 763–774 (2005).
Renard, C. et al. The genomic sequence and analysis of the swine major histocompatibility complex. Genomics 88, 96–110 (2006).
Yuhki, N., Beck, T., Stephens, R., Neelam, B. & O’Brien, S. J. Comparative genomic structure of human, dog and cat MHC: HLA, DLA and FLA. J. Hered. 98, 390–399 (2007).
Gao, J. F. et al. A complete DNA sequence map of the ovine major histocompatibility complex. BMC Genomics 11, 466 (2010).
Wan, Q. H., Zeng, C. J., Ni, X. W., Pan, H. J. & Fang, S. G. Giant panda genomic data provide insight into the birth-and-death process of mammalian major histocompatibility complex class II genes. Plos One 4, e4147 (2009).
Dengjel, J. et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 102, 7922–7927 (2005).
Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2, 16 (2005).
Murray, B. W., Malik, S. & White, B. N. Sequence variation at the major histocompatibility complex locus DQB in beluga whales (Delphinaterus leucas). Mol. Biol. Evol. 12, 582–593 (1995).
Murray, B. W. & White, B. N. Sequence variation at the major histocompatibility complex DRB loci in beluga (Delphinapterus leucas) and narwhal (Monodon monoceros). Immunogenetics 48, 242–252 (1998).
Hayashi, K. et al. Sequence variation of the DQB allele in the cetacean MHC. Mamm. Study 28, 89–96 (2003).
Yang, G., Yan, J., Zhou, K. Y. & Wei, F. Sequence variation and gene duplication at MHC DQB loci of baiji (Lipotes vexillifer), a Chinese river dolphin. J. Hered. 96, 310–317 (2005).
Baker, C. S. et al. Diversity and duplication of DQB and DRB-like genes of the MHC in baleen whales (suborder: Mysticeti). Immunogenetics 58, 283–296 (2006).
Hayashi, K. et al. Genetic variation of the MHC DQB locus in the finless porpoise (Neophocaena phocaenoides). Zool. Sci. 23, 147–153 (2006).
Yang, W. C., Hu, J. M. & Chou, L. S. Sequence variation of MHC class II DQB gene in bottlenose dolphin (Tursiops truncatus) from Taiwanese waters. Taiwania 53, 42–50 (2008).
Yang, W. C., Chou, L. S. & Hu, J. M. Molecular characterization of major histocompatibility complex class II DQB and DRB genes in bottlenose dolphins (Tursiops truncatus and T. aduncus) from the western Pacific. Zool. Stud. 46, 664 (2007).
Heimeier, D. et al. Confirmed expression of MHC class I and class II genes in the New Zealand endemic Hector’s dolphin (Cephalorhynchus hectori). Mar. Mamm. Sci. 25, 68–90 (2009).
Heinzelmann, L., Tavares, M., Ott, P. H., Moreno, I. & Chies, J. A. B. MHC class II expression in skin biopsies from the franciscana dolphin Pontoporia blainvillei and the southern right whale Eubalaena australis. J. Mar. Biol. Assoc. UK 89, 1009–1013 (2009).
Du, H. J., Zheng, J. S., Wu, M., Zhao, Q. Z. & Wang, D. High MHC DQB variation and asymmetric allelic distribution in the endangered Yangtze finless porpoise, Neophocaena phocaenoides asiaeorientalis. Biochem. Genet. 48, 433–449 (2010).
Xu, S. X., Ren, W. H., Zhou, X. M., Zhou, K. Y. & Yang, G. Sequence polymorphism and geographical variation at a positively selected MHC-DRB gene in the finless porpoise (Neophocaena phocaenoides): implication for recent differentiation of the Yangtze Finless porpoise? J. Mol. Evol. 71, 6–22 (2010).
Xu, S. X., Sun, P., Zhou, K. Y. & Yang, G. Sequence variability at three MHC loci of finless porpoises (Neophocaena phocaenoides). Immunogenetics 59, 581–592 (2007).
Committee on Taxonomy. List of marine mammal species and subspecies. Society for Marine Mammalogy, www.marinemammalscience.org, consulted on Nov 25 (2014).
Wang, J. Y., Frasier, T. R., Yang, S. C. & White, B. N. Detecting recent speciation events: the case of the finless porpoise (genus Neophocaena). Heredity 101, 145–155 (2008).
Wang, J. Y., Yang, S. C., Wang, B. J. & Wang, L. S. Distinguishing between two species of finless porpoises (Neophocaena phocaenoides and N. asiaeorientalis) in areas of sympatry. Mammalia 74, 305–310 (2010).
Mei, Z. G. et al. Accelerating population decline of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Biol. Conserv. 153, 192–200 (2012).
Wang, D., Turvey, S. T., Zhao, X. & Mei, Z. Neophocaena asiaeorientalis ssp. asiaeorientalis. In IUCN 2013. IUCN Red List of Threatened Species. Version 2013.1. www.iucnredlist.org. Downloaded on 02 September 2013.
Klein, J. & Takahata, N. The major histocompatibility complex and the quest for origins. Immunol. Rev. 113, 5–25 (1990).
O’Brien, S. J. & Yuhki, N. Comparative genome organization of the major histocompatibility complex: lessons from the Felidae. Immunol. Rev. 167, 133–144 (1999).
Gingerich, P. D., Ul Haq, M., Zalmout, I. S., Khan, I. H. & Malkani, M. S. Origin of whales from early artiodactyls: hands and feet of Eocene Protocetidae from Pakistan. Science 293, 2239–2242 (2001).
Thewissen, J. G., Cooper, L. N., Clementz, M. T., Bajpai, S. & Tiwari, B. Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 450, 1190–1194 (2007).
Du, B., Wang, D., Zhang, X. F., Guo, Z. & Zhang, J. Genome size determination of Yangtze finless porpoise, Neophocaena phocaenoides asiaeorientalis. Curr. Zool. 52, 731–737 (2006).
Klein, J. et al. Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31, 217–219 (1990).
Springer, M. S., Murphy, W. J., Eizirik, E. & O’Brien, S. J. Placental mammal diversification and the Cretaceous–Tertiary boundary. Proc. Natl. Acad. Sci. USA 100, 1056–1061 (2003).
Murphy, W. J., Pringle, T. H., Crider, T. A., Springer, M. S. & Miller, W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 17, 413–421 (2007).
Takahashi, K., Rooney, A. & Nei, M. Origins and divergence times of mammalian class II MHC gene clusters. J. Hered. 91, 198–204 (2000).
Mellins, E. D. & Stern, L. J. HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr. Opin. Immunol. 26, 115–122 (2014).
Brown, J. H. et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364, 33–33 (1993).
Chen, M. M. et al. Genetic diversity and population structure of the critically endangered yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis) as revealed by mitochondrial and microsatellite DNA. Int. J. Mol. Sci. 15, 11307–11323 (2014).
Andersson, L., Lundén, A., Sigurdardottir, S., Davies, C. J. & Rask, L. Linkage relationships in the bovine MHC region. High recombination frequency between class II subregions. Immunogenetics 27, 273–280 (1988).
Yang, W. C., Hu, J. M. & Chou, L. S. Phylogenetic analyses of MHC class II genes in bottlenose dolphins and their terrestrial relatives reveal pathogen-driven directional selection. Zool. Stud. 49, 132–151 (2010).
Slade, R. W. Limited MHC polymorphism in the southern elephant seal: implications for MHC evolution and marine mammal population biology. Proc. R. Soc. B-Biol. Sci. 249, 163–171 (1992).
Miyata, T. & Yasunaga, T. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J. Mol. Evol. 16, 23–36 (1980).
Osoegawa, K., Jong, P. J., Frengen, E. & Ioannou, P. A. In Current Protocols in Molecular Biology (eds Ausubel, F. M. et al.) Ch. 5, 5.9. 1-5.9. 33 (Wiley, 2001).
Zeng, C. J. et al. Giant panda BAC library construction and assembly of a 650-kb contig spanning major histocompatibility complex class II region. BMC Genomics 8, 315 (2007).
Asakawa, S. et al. Human BAC library: construction and rapid screening. Gene 191, 69–79 (1997).
Luo, R. B. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18 (2012).
Tatusova, T. A. & Madden, T. L. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174, 247–250 (1999).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Evol. 268, 78–94 (1997).
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522 (2000).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Evol. 215, 403–410 (1990).
Gish, W. & States, D. J. Identification of protein coding regions by database similarity search. Nature Genet. 3, 266–272 (1993).
Herrmann, L. M., Brown, W. C., Lewis, G. S. & Knowles, D. P. Identification and phylogenetic analysis of 15 MHC class II DRB1 β1 expressed alleles in a ewe–lamb flock. Immunogenetics 57, 855–863 (2005).
Schwartz, S. et al. PipMaker—a web server for aligning two genomic DNA sequences. Genome Res. 10, 577–586 (2000).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Brudno, M. et al. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731 (2003).
Darling, A. C., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nylander, J. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University (2004).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Guindon, S. & Gascuel, O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Posada, D. & Crandall, K. A. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics. (Oxford University Press, 2000).
Xia, X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30, 1720–1728 (2013).
Acknowledgements
We sincerely thank Professor Sheng-Guo Fang and Qiu-Hong Wan at Zhejiang University for offering invaluable assistance with BAC library construction for the Yangtze finless porpoise. We also thank our colleague Mr. Qing-Zhong Zhao for his assistance in sample collection. This work was supported by grants from the National Natural Science Foundation of China (No. 31430080; No. 31000168), the National Basic Research Program of China (No. 2007CB411600), the Knowledge Innovation Program of the Chinese Academy of Sciences (No. Y05E011101) and the Experimental Platform Program of the Chinese Academy of Sciences (No. CZBZX-1).
Author information
Authors and Affiliations
Contributions
D.W. and J.S.Z. designed this research. J.S.Z. and M.M.C. collected the samples. R.R. constructed the Yangtze finless porpoise BAC library and screened the BAC clones bearing MHC class II genes. J.R. assembled the sequences of the positive BAC clones sequenced by Illumina Hiseq 2000. R.R., X.L.W., Y.Z. and M.M.C. designed the primer pairs, filled the gaps in the assembled sequences and performed cDNA PCR to verify the functionality of the MHC class II genes. R.R. performed sequence analyses. R.R., D.W. and J.S.Z. wrote the manuscript. All authors contributed to discussions and revisions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ruan, R., Ruan, J., Wan, XL. et al. Organization and characteristics of the major histocompatibility complex class II region in the Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Sci Rep 6, 22471 (2016). https://doi.org/10.1038/srep22471
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22471
This article is cited by
-
A high-density BAC physical map covering the entire MHC region of addax antelope genome
BMC Genomics (2019)
-
Distinct evolution of toll-like receptor signaling pathway genes in cetaceans
Genes & Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
