
Abstract
In the context of the global action plan to reduce the dissemination of antibiotic resistances it is of utmost importance to understand the population structure of resistant endemic bacterial lineages and to elucidate how bacteria acquire certain resistance determinants. Vancomycin resistant enterococci represent one such example of a prominent nosocomial pathogen on which nation-wide population analyses on prevalent lineages are scarce and data on how the bacteria acquire resistance, especially of the vanB genotype, are still under debate. With respect to Germany, an increased prevalence of VRE was noted in recent years. Here, invasive infections caused by sequence type ST192 VRE are often associated with the vanB-type resistance determinant. Hence, we analyzed 49 vanB-positive and vanB-negative E. faecium isolates by means of whole genome sequencing. Our studies revealed a distinct population structure and that spread of the Tn1549-vanB-type resistance involves exchange of large chromosomal fragments between vanB-positive and vanB-negative enterococci rather than independent acquisition events. In vitro filter-mating experiments support the hypothesis and suggest the presence of certain target sequences as a limiting factor for dissemination of the vanB element. Thus, the present study provides a better understanding of how enterococci emerge into successful multidrug-resistant nosocomial pathogens.
Similar content being viewed by others

Introduction
Regarded as a gut commensal organism of animals and humans alike, with the potential to incidentally invade underlying tissue and being capable to withstand harsh environmental condition, enterococci have long been recognized as an important nosocomial pathogen. Life-threatening diseases such as endocarditis or sepsis can be elicited mostly in immunocompromised patients, elderly or neonates. Due to their often multi-drug resistant phenotype only a few therapeutic options are left to treat infections caused by vancomycin-resistant enterococci (VRE). Although VRE are of generally low prevalence in European clinics compared to U.S. hospitals1,2, surveillance data from the European Antimicrobial Resistance Surveillance Network (EARS-Net) clearly demonstrate that VRE are on the rise in European countries (http://ecdc.europa.eu). please change to: It was observed a steady increase of VRE in Germany in recent years. As for instance 14.5% of all invasive E. faecium isolates were tested vancomycin-resistant in 2013.
Vancomycin resistance is mediated through expression of a gene cluster of which nine different genotypes, VanA to VanN, can be distinguished on the basis of the central ligase3. VanA and VanB account for the majority of clinically important VR-E. faecalis and VR-E. faecium cases and are usually associated with mobile genetic elements (MGE) such as transposons4,5. While vanA is dominant in the USA and in most of Europe6, the National Reference Centre for Staphylococci and Enterococci in Germany has recognized a steady increase of vanB-type VRE since 2010 (EARS-Net annual report 2013: http://ecdc.europa.eu/en/publications/_layouts/forms/Publication_DispForm.aspx?List=4f55ad51-4aed-4d32-b960-af70113dbb90&ID=1205#sthash.M5euPg56.dpuf). We recently reported about the ongoing high prevalence of vanB-type E. faecium of sequence type (ST) 192 on a neonatal care unit7. As recognized by the German NRC between 2011 and 2014, ST192 (33%) represents the most prevalent ST besides ST117 (25%) and ST17 (16%) isolated from bloodstream infections and is primarily associated with the vanB genotype (NRC annual reports).
The vanB locus is mainly encoded by a conjugative transposon of the Tn1549-/Tn5382-subtype and of chromosomal origin. However, localization on pRUM-like plasmids has also been reported from Sweden, Spain or Singapore8,9. It is still under debate how the vanB transposon facilitates its own transfer, as detailed investigations yielded conflicting results regarding the mechanism of transmission. Launay and co-workers demonstrated that Tn1549 represents a genuine transposon capable of forming a circular intermediate in order to be transferred and integrated into the target bacterial chromosome in the gut of gnotobiotic mice10. Contradicting these results, in vitro experiments performed by Quintiliani, Carias, and others show the movement of large chromosomal elements which would favor a mechanism related to homologous or illegitimate recombination11,12,13,14. Interestingly, Tn1549-vanB is not restricted to enterococci but was also detected among anaerobic bacilli such as Clostridium spp. or Eggerthella lenta10,15,16,17,18. It has been hypothesized that the myriad of reservoirs constitute a source for frequent vanB transposition events and thus acquisition of resistance from gut commensals by susceptible Enterococcus recipients19.
As vanB-positive E. faecium ST192 led to a marked increase of VRE outbreaks and became a highly prevalent lineage of bloodstream infections in Germany in recent years, the present study was conducted to investigate VRE of this particular sequence type with respect to i) population structure, ii) acquisition of the vanB resistance determinant and iii) transfer of the resistance locus.
Results
Description of isolates
The vast majority of the 49 E. faecium strains used in this study was isolated from invasive infections between 2004 and 2014 and sent to our National Reference Centre for Staphylococci and Enterococci from hospitals all across Germany (Table 1). As strains of ST192 have been most prevalent in recent years and have been associated with hospital-related outbreaks in German clinics7, we set out to analyze E. faecium ST192 in more detail with respect to strain relatedness and acquisition of the composite vanB transposon Tn1549. Thus, 39 of the 49 isolates from this study belonged to ST192 whilst the remaining strains represent further prevalent German STs such as ST117, ST17, ST78 or ST203 and were included for comparative reasons. First, strains were analyzed phenotypically, hence displaying variable levels of vancomycin MICs from as low as 2 μg/ml up to 512 μg/ml for vanB-positive isolates, respectively (Table 1). As expected, vanB-negative strains were fully susceptible to vancomycin (≤1 μg/ml) with one exception of UW11625 which harbored the vanA gene cluster but was negative in vanB-specific PCR experiments (data not shown).
Population structure of ST192 and non-ST192 E. faecium in Germany
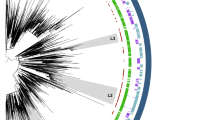
Illumina sequencing was carried out for all E. faecium ST192 strains to reveal relatedness within and between vanB-positive and vanB-negative isolates. The available genomic nucleotide sequence of E. faecium DO/TX16 served as a reference for subsequent mapping of obtained reads. Due to multiple ambiguities after mapping above a set threshold of 10%, 4 ST192 isolates were omitted from the core genome analyses (Table 1). Called variants were condensed to 8,876 single nucleotide polymorphisms (SNPs), thereby excluding every nucleotide variant which most likely resulted from a recombination event (see material and methods). The inferred maximum likelihood tree groups all but one ST192, UW11625 (vanA-positive, but vanB-negative), into one of 4 main clades or subclades (Fig. 1a). The proportional transformed branch diagram was chosen, as it explicitly charts the different clades rather than displays the number of sites altered between individual strains (for nucleotide substitutions per site see Supplementary Fig. S1). It is apparent that the German E. faecium ST192 population is divided into 2 vanB-positive clades or subclades (CIa + b, CII) and 2 vanB-negative clades (CIII and CIV), the latter infrequently exhibiting vanB-positive and vancomycin-resistant isolates (UW5267, UW8130, UW6376, UW 8173). Clade I is represented by 2 subclades which separates the strains UW8084, UW9284 and UW8260 (CIb) and UW8030 from the bigger branch containing 13 isolates (Fig. 1a). Further, clade II is clearly to be distinguished from CI/III/IV, which is congruent with the strains being isolated from a recent outbreak with E. faecium ST192 (vanB-positive) on a neonatal care unit7. Interestingly, there is no apparent correlation based on the year or the region (federal state) the strains originated from, as for example vanB-positive isolates of year 2009 were found both in the vanB-positive clade CIa or the “primarily” vanB-negative clade CIII (Fig. 1a). Together, the data suggest the dissemination of two main subpopulations of E. faecium ST192 in German clinics; one which is represented by the vanB-negative and occasionally vanB-acquiring clades CIII/CVI; and the second which is composed of an epidemic vanB-positive subgroup circulating among hospitals with a geographical preference to the midst and northern parts of Germany (North Rhine-Westphalia, Hamburg, Western Pomerania, Table 1).

Phylogenetic analysis of the core genome of German E. faecium ST192 (a) and non-ST192 (b) clinical isolates. (a) The proportional transformed branch diagram revealed 4 different clades or subclades of ST192 isolates. (b) ST192 and non-ST192 were found to cluster according to their respective MLST type with the exception of UW7027-ST78 falling within clade III of ST192 isolates. Branch labels represent a bootstrap with 1000 permutations. Color assignment was done to visually differentiate ST192 clades as defined in this study (green: VRE clade CIa; red: VRE clade CIb; salmon: VRE clade CII; light blue: VSE/VRE clade CIII and grey VSE/VRE clade IV). Outlying brackets frame the year of isolation of the strains belonging to the respective clade. All isolates are ST192 unless stated next to the strain name. Efm_DO represents the reference E. faecium DO/TX16 (CP003583). “vanB-“ refers to vanB-negative isolates.
A set of non-ST192 were subjected to whole genome sequencing accordingly. Based on 14,177 SNP positions in the core genome, these isolates are only distantly related to the ST192 population (Fig. 1b and Supplementary Fig. S1). Strain E. faecium UW7027 (ST78) represents the sole exception as it groups with cluster CIII of vanB-negative E. faecium ST192 strains (Fig. 1a,b and Supplementary Fig. S1). Further, isolate UW8914 (ST927) is more closely related to the ST192 population of both, vanB-positive and vanB-negative clades, as is UW11625, an ST192 strain which constitutes an outliner in the ST192 core tree and clusters with an ST203 isolate according to the core genome data (Fig. 1a,b). As demonstrated by confidence intervals, the results obtained underscore the importance of whole genome sequencing (WGS) in analyzing population structure in more depth than conventional multi locus sequence typing of only seven housekeeping genes. However, other sequence types analyzed in this study were either represented by their own clade or as singlets (Fig. 1b and Supplementary Fig. S1).
Determination of Tn1549 insertion sites
Insertion sites of the composite vanB transposon Tn1549 were determined for 38 vanB-positive isolates. These included 5 strains which were omitted for core genome analysis (Table 1). In total, 72% of all strains analyzed had the transposon integrated into a novel chromosomal insertion site within the open reading frame HMPREF0351_10592, encoding a hypothetical protein present in E. faecium DO only (BlastN search, data not shown) (Table 1). It is worth noting that the three strains UW7606, UW7652 and UW7816 (all ST192) were counted as one in these analyses, as they were part of a suspected outbreak and do not represent independent samples. Except for the three outbreak strains, which exhibit Tn1549 insertion next to a pathogenicity island (Table 1), all ST192 isolates, as well as the ST78, ST927 and ST202 strains, displayed identical insertion sites with the most frequent locus HMPREF0351_10592. Interestingly, they also comprised the very same coupling sequence of 5 nucleotides (-ATAGAA-) at the 3′- end of Tn1549 (Fig. 2). Four strains of ST16, ST17 and ST208 showed identical insertion sites into the intergenic region of HMPREF0351_10636 and HMPREF0351_10635. However, the sequence types differed in their coupling sequence and orientation of Tn1549 integration (Supplementary Fig. S2). Four isolates of different STs displayed single insertions into genes or intergenic regions widely distributed over the reference genome E. faecium DO (Table 1 and Supplementary Fig. S2). We were unable to resolve the insertion site for strain UW8173 (ST192). In summary, all German vancomycin resistant isolates of E. faecium analyzed in this study showed novel chromosomal, so far undescribed loci for Tn1549 integration.

Insertion and orientation of Tn1549 into HMPREF0351_10592 is depicted alongside six nucleotides framing the specific insertion site. Enumeration of nucleotides is according to the coding sequence of HMPREF0351_10592. The coupling sequence represents nucleotides which were co-transferred from the initial donor strain.
Phylogeny of Tn1549
In order to examine whether the phylogeny of the core genomes correlates with the relatedness of the vanB transposon, reads were mapped to the reference sequence Tn1549 of strain WCF-TC1 (CP013009) and a maximum likelihood analysis was calculated from 276 SNP positions. Clustering of the Tn1549 sequences did not match the clades as described for the core genome (Figs 1b and 3). On the contrary, Tn1549 segments were strictly assorted to independent groups based on their specific site of insertion (Fig. 3). This included strains of different STs which were distantly related according to their core genome data, e.g. UW6293 (ST202) and UW8452 of the ST192 population, but exhibited almost identical vanB-containing mobile genetic elements (Figs 1b and 3).

The proportional transformed branch diagram revealed an insertion site-specific clustering across all sequence types. Differentiation of the various insertion sites was further validated by bootstrap analysis with 1000 permutations and is indicated by branch labeling. For consistency, color coding represents the different ST192 clades as represented in Fig. 1a,b (green: VRE clade CIa; red: VRE clade CIb; salmon: VRE clade CII; light blue: VSE/VRE clade CIII and grey VSE/VRE clade IV). Unless indicated by specific ST enumeration, all isolates belonged to ST192. Insertion sites are depicted as locus_tag numbering according to the reference genome E. faecium DO (CP003583). Tn1549 represents the reference sequence used for mapping (CP013009). Insertion site “PAI” represents the reference locus_tag EFAU085_02779, as it is not present in E. faecium DO, and due to the proximity to a pathogenicity island (PAI) was termed “PAI” in the following. unk insertion site unknown.
It is worth mentioning that isolate UW8173, located within clade CIII of vanB-negative strains, comprised a Tn1549 element which seemed to be completely different to all other VRE isolates investigated and for which the site of insertion could not be resolved (Figs 1a and 3).
Moreover, the afore-mentioned isolates of ST16, ST17 and ST208 showing identical insertion sites but altered coupling sequences, formed 2 separate clusters in the Tn1549 analysis (Fig. 3).
Altogether, Tn1549 elements of German clinical E. faecium isolates generally displayed a high degree of identity; however, refined distinction allows clustering of the Tn1549 sequences based on their site of chromosomal integration, a clustering which is different to their entire core genome background.
Transfer of Tn1549 and determination of transferred fragment sizes
The mechanism by which Tn1549 is transferred from one bacterium to the other is currently under debate as both classical transposition as well as transfer of large chromosomal fragments have been reported to date10,12,13. Our phylogenetic analyses of the transposon and most importantly of the coupling sequences and flanking regions indicate that acquisition of the resistance determinant might rather be due to the transfer of genomic segments than targeted insertion of the sole Tn1549 element from independent origins. To put the hypothesis to proof, a series of in vitro conjugation experiments was conducted with combinations of donor and recipient strains as summarized in Table 2. Generally, Tn1549 was transferred at variable but low frequencies from 3.3 × 10−9 to 1.2 × 10−6 transconjugants (TCs) per recipient cells (Table 2). Two E. faecium clinical isolates (UW5267, UW8030) failed to transfer Tn1549 to E. faecium 64/3 or 64SS, respectively (Table 2).
We further conducted conjugation experiments using E. faecalis OG1RF and JH2-2 as recipient bacteria. No single TC was obtained in filter-mating experiments when E. faecalis served as a recipient, even though an appropriate E. faecium counterpart such as 64/3 or AK-EM40RF received Tn1549 and the vancomycin resistance cassette from donors E. faecium UW7184 and E. faecium UW7606 (Table 2).
A selection of TCs obtained from the 1st round of transfer experiments were subsequently used for further filter-mating analysis. All but one donor TC transferred Tn1549 to the respective recipients and thus produced a set of 2nd generation and vancomycin-resistant TCs which were included in subsequent analyses of transferred fragment sizes.
In order to determine the size of the transferred elements, one or two TCs per experimental setup were subjected to next generation sequencing. Bioinformatics analyses were based on mapping of contigs obtained by de novo assembly to Tn1549 followed by Mauve alignments of the donor and the respective TC sequences. The accumulation of multiple SNPs in close proximity was defined as the region representing non-transferred DNA and thus set to frame the transferred element. It must be noted that the sizes represent approximations and in some cases could not be resolved in detail due to limited contig lengths. However, as a result, none of the analyzed TCs obtained the sole Tn1549 transposon by classical transposition, but rather acquired a larger chromosomal fragment of variable size (Table 2). Notably, even 2 TCs isolated from the very same conjugation experiment showed transfer of differently sized genomic fragments. For instance, 7606 × 6711RF × BM4105SS TC1 revealed the acquisition of a >220 kb element whilst the second TC analyzed, 7606 × 6711RF × BM4105SS TC2, received 140 kb only (Table 2).
The MICs to vancomycin of the transconjugants generally resembled those of the donor strains, as for example both above mentioned 2nd generation TCs exhibited the same MIC of 64 μg/ml as their donor 7606 × 6711RF (Table 2).
Furthermore, no association determining the size of the Tn1549-containing fragment could be deduced from the combination of donor or recipient strain used. For instance, transconjugant 7184 × 64/3 TC1 acquired a >123 kb fragment, while using the very same strain as a donor in subsequent 2nd rounds of conjugations, the resulting TC (7184 × 64/3 × BM4105SS TC1) obtained 81 kb only (Table 2).
Since Tn1549 transmission permits carryover of flanking chromosomal regions, TCs which were obtained by using E. faecium UW7606 as a donor co-transferred important virulence determinants such as a pathogenicity island alongside the resistance cassette.
Together, our data suggests a Tn1549-related transfer different from the supposed mechanism of transposition.
Characterization of plasmid content of donor, recipient and transconjugant bacteria
The mechanism and putative factors which trigger or support transfer of large Tn1549-containing fragments to recipient strains still await elucidation. It was hypothesized by Dahl and colleagues that certain plasmids get transferred simultaneously with Tn1549 in an undetermined manner13, while others did not observe co-transfer of episomal DNA14. In order to analyze the putative involvement of a plasmid co-transfer in our conjugation experiments, we first screened a set of donor, recipient and TC strains by PCR using oligonucleotides specifically amplifying replication initiation genes (rep genes) of the most abundant enterococcal plasmids as well as the toxin/antitoxin system axe-txe of pRUM of E. faecium U37 (AF507977.1) or of plasmid pDO2 from E. faecium DO (CP003585). Table 3 provides a summary of the rep-PCR results, hence indicating the co-transfer of a putative vector carrying rep17/repA of pRUM and/or the axe-txe system to almost every transconjugant of the 1st and 2nd generation. The only exception was represented by 7606 × AK-EM40RF and 7606 × BM4105RF where either the rep17- or the axe-txe-specific primers produced a positive PCR product (Table 3).
Sequencing of the rep gene of the TCs revealed that it was more related to the rep sequence of plasmid 2 of AUS0085 than to that of pRUM. The sequence of the axe-txe system of p7606 × 64/3 TC1 (named pWCF-TC1) is identical to both pRUM and plasmid p1 of the Australian isolate AUS0085 (not shown). Thus, these findings suggest the presence of a novel hybrid plasmid containing a rep17/repA gene as well as the toxin-antitoxin system or 2 separate plasmids that were co-transferred with Tn1549.
Enterococcal plasmids are known to exhibit a high degree of modular dissociability20. Thus, for in-depth analysis of whole plasmid content, bacterial cells were treated with S1-nuclease and separated by PFGE. As depicted in Fig. 4, donor strains generally contained multiple plasmids. Explicitly prominent, however, was an approx. 40 kb plasmid which was present in almost all German clinical isolates investigated (UW7606, UW6711RF, UW7184, UW6293; Fig. 4). Moreover, a plasmid with the estimated size of 70 kb was observed in all donor strains which produced transconjugants, but absent in the one which failed to transfer Tn1549 to a receptive host (7606 × BM4105RF TC1; Table 3). This plasmid was the only one which was co-transferred alongside the Tn1549-containing chromosomal element.

Separation in PFGE disclosed the plasmid content of the strains analyzed. The red rectangle indicates the putative novel plasmid pWCF-TC1 of 66.5 kb. The red asterisks indicate strains that do not possess the respective plasmid. M marker strain S. aureus NCTC8325.
Determination of plasmid sequence pWCF-TC1
In this study, we determined the genome sequence of transconjugant E. faecium WCF-TC1 (CP013009). PacBio sequencing revealed the presence of one putative plasmid which was co-transferred during filter-mating experiments from the clinical donor isolate E. faecium UW7606 to recipient strain E. faecium 64/3. The obtained contig was further analyzed to deplete of overlapping sequences and verified for closed circle confirmation by PCR (data not shown). The final plasmid, termed pWCF-TC1, exhibits a length of 66,496 bp and thus is comparable in size to the co-transferred plasmid as estimated by S1-PFGE (Fig. 4). Subsequent annotation by NCBI revealed 76 coding sequences (CDS), amongst them pre-eminently open reading frames for hypothetical proteins, Rep17/RepA and the toxin-antitoxin system as described above (CP013010). Interestingly, pWCF-TC1 exhibits a CDS (locus_tag AQ614_13110) encoding a coupling protein of the transfer machinery (TraG), which might represent a supportive factor for Tn1549 co-transfer. In order to analyze whether the novel plasmid is present in TC-producing strains and TC-non-producers and thus in general might exert co-transfer capabilities on Tn1549, sequencing data were mapped to pWCF-TC1 and the amount of ambiguous sites (AS) was used as a rough measure for plasmid/gene existence. Expectantly, strains such as E. faecium 64/3 which were plasmid-negative in S1-PFGE analyses displayed no plasmid coverage in mapping attempts (AS 93.7%; Supplementary Table S1). In contrast, 13 of 21 strains analyzed exhibited a substantial coverage with AS in the range of 1.8–7.3%. However, conflicting results were obtained for some isolates which were plasmid-positive (S1-PFGE), but showed poor coverage of the entire novel plasmid sequence, e.g. 7606 × 6711RF × BM4105SS TC1 (AS 94.3%), or did not harbor episomal DNA according to PFGE but displayed a low percentage of ambiguities after mapping analyses (e.g. 7606 × BM4105RF TC1 showing AS of 16.8%; Supplementary Table S1). Further, putative existence of plasmid pWCF-TC1 as inferred from read mappings was not associated with successful transfer of Tn1549, as isolates UW8030 or UW8260 did not produce vanB-positive TCs but showed a decent coverage of plasmid pWCF1 (AS 7.3% and 2.1%, respectively, Supplementary Table S1).
Discussion
E. faecium ST192 has been recognized as one of three highly prevalent sequence types responsible for causing VRE outbreaks and hospital-associated (HA) bloodstream infections in German hospital patients in recent years7. Moreover, emergence of E. faecium ST192 as a dominant clone and belonging to a cluster of HA-strains formerly known as clonal complex 17 (CC17), was reported from Sweden and Korea where outbreaks with these strains were noted21,22,23. Interestingly, and opposed to the data of the present study, vancomycin resistance in these strains was mediated by the acquisition of either vanA, as part of the plasmid-associated transposon Tn1546, or the vanB-containing Tn1549, also of plasmid origin. Sivertsen and colleagues demonstrated that the vanB2-Tn1549 mobile element inserted into a 40 kb rep17/pRUM plasmid which pre-existed in vancomycin-sensitive enterococci (VSE)22. Hence, generation of vancomycin-resistant strains was followed by clonal spread of the resistant population. We herein describe the population structure of German VRE ST192 in comparison with further ST lineages, all exhibiting a chromosomally encoded Tn1549-vanB. However, localization of Tn1549 in enterococcal chromosomes is no unique feature of German E. faecium isolates and, as a matter of fact, had been reported from many countries across the globe11,13,24,25.
Based on the core genome data, VRE and VSE of the German ST192 population can be divided into at least two distinct clades; the VRE only clade CIa and the VSE clades CIII/CIV which occasionally acquired the resistance determinant (Fig. 1a). This would suggest the existence of a quite diverse E. faecium ST192 population, endemic all across Germany with the capabilities of both, clonal spread of VRE strains as well as de novo acquisition of vanB-Tn1549 by susceptible progenitors. In support of this, we recently demonstrated by PFGE and DiversiLab® and by using an expanded set of clinical isolates that German VRE exhibit a versatile population structure across all endemic lineages26. Whether the still susceptible population is comprised of pre-existing, HA- and circulating high risk-VSE strains or represent gut commensals highly prevalent in the human community, remains to be determined. This is of major importance in order to implement countermeasures hence preventing transmission of even a certain VSE subpopulation prone to acquire vancomycin resistance.
The hypothesis of de novo generation rather than clonal spread of VRE was also presumed by Howden et al. in their recent description of 61 E. faecium clinical isolates from Australia19. The study included 36 VRE strains exhibiting a chromosomally encoded vanB-Tn1549. The authors compared the transposon sequence, insertion site and orientation of Tn1549 insertion, as well as the coupling sequences present in clinical E. faecium isolates and in anaerobic gut commensals. In accordance with our data, highly similar Tn1549 were associated with identical coupling sequences and dispersed throughout a phylogenetic tree covering different sequence types19. Moreover, Tn1549 localization revealed seven unique insertion sites. Interestingly, these sites differ from the seven loci for Tn1549 insertion described in the present study. Also, German and Australian isolates exhibited distinct coupling sequences which provide evidence for independent origins of acquisition and random insertion of Tn1549 based on classical transposition events at first. Notably, a Tn1549 sequence obtained from Clostridium spp. of another Australian patient was identical to Tn1549 of a subset of VRE strains, thus indicating that certain gut commensals could act as a reservoir for the vanB resistance determinant. Indeed, extensive PCR screenings have previously demonstrated that vanB is highly prevalent in non-enterococcal anaerobic bacteria of healthy individuals16,27. However, other Tn1549 sequences from anaerobic commensals were shown to be only distantly related to the transposon isolated from VRE strains19. This assumes a more frequent transfer among enterococci (VRE and VSE) rather than de novo acquisition of Tn1549 from the gut microbiota.
In accordance with this hypothesis, our study revealed a highly specific clustering of the transposon according to the respective insertion sites (Fig. 3). Due to identical coupling sequences as well as distribution of homologous Tn1549 elements across the entire phylogenetic tree of the core genome (Fig. 2 and Supplementary Fig. S2), independent acquisition by transposition followed by clonal dissemination of the generated VRE can be excluded. The data are rather indicative of transfer of Tn1549 based on the mechanism of illegitimate and/or homologous recombination, given the presence of a specific target region in the recipient strain. Consistent with this assumption, filter-mating experiments conducted in this study clearly demonstrated that Tn1549 exhibits the capability to transfer as part of a large chromosomal fragment. Transfer was also observed to recipient strains which lacked the apparent hot spot for insertion, but subsequently acquired the entire fragment including insertion sites and flanking regions of variable sizes (Tables 1 and 2). However, a certain restriction must be imposed by relatedness of the core genome, especially of the Tn1549 flanking regions, as no transfer was achieved to the species E. faecalis.
It has been reported by a number of studies that the vanB-containing mobile genetic element is capable of co-transferring adjacent genomic fragments10,12,13,14,28,29. In one case, concomitant transfer led to the development of strains with increased tolerance to antibiotics11. Likewise, we observed co-transfer of a pathogenicity island located in close proximity to the resistance cassette. This is additionally worrisome, as increased prescription and usage of certain antibiotics could trigger the generation and emergence of a bacterial subpopulation with enhanced pathogenicity and/or antibiotic resistances.
It still remains unclear which factors are involved or might trigger the transfer of vanB-Tn1549, either with or without flanking regions. Our study demonstrated the co-transfer of a novel plasmid, termed pWCF-TC1. Whether it has the capability to mobilize Tn1549 or is itself dependent on mobilization remains to be determined; however, transfer of vancomycin resistance to recipient bacteria only occurred from donor strains which were shown to be plasmid-positive. Analysis of the gene content revealed the presence of traG (locus_tag AQ614_13110) encoding a coupling protein of the transfer machinery. However, presence and/or detailed investigation of the nucleotide sequence of the traG open reading frame showed no correlation with the ability of the donor to transfer Tn1549 and/or the novel plasmid pWCF-TC1 (Supplementary Table S1). As an example, clinical isolate UW8260 was carrying pWCF-TC1 and encodes an intact ORF for traG but failed to produce Tn1549-vanB-positive transconjugants. Collectively, our data suggest the presence and simultaneous transfer of certain plasmids together with the vancomycin resistance transposon Tn1549. As enterococcal plasmids are known for their diverse modular structure assignment of transfer capabilities to certain coding sequences cannot be deduced from current data.
In summary, we hypothesize that initial acquisition of Tn1549 by a receptive Enterococcus might occur via transposition from the gut microbiota. However, dissemination of the vanB resistance locus in German clinical isolates is presumably due to the exchange of genomic material between VRE and VSE. This might or might not involve the action of co-mobilizing plasmids. It is important to keep in mind the possibility of a circulating high risk VSE lineage in order to prevent de novo generation and further spread of dominant VRE HA-populations.
Methods
Strain collection
An overview of genotypic and phenotypic features of 49 ST192 and non-ST192 E. faecium clinical isolates analyzed in this study and isolated between 2004 and 2014 is given in Table 1. For comparative reasons 11 vanB-negative E. faecium ST192 strains were included in the strain collection. The strains originated from hospital or diagnostic microbiological laboratories all across Germany and were sent to the National Reference Centre for Staphylococci and Enterococci for resistance determination and further molecular characterization. The majority of these strains were derived from bloodstream infections. Three isolates (UW7606, UW7625, UW7816) belonged to a previously reported outbreak7.
Antimicrobial susceptibility testing
Susceptibility to vancomycin was determined by using the broth microdilution method according to DIN58940 and applying EUCAST breakpoints and, in case of antibiotics with no EUCAST breakpoints, using epidemiological cut-off values (ECOFFs) for interpretation of the results (www.eucast.org). Donor, recipient (Table 1) and transconjugant strains (this study) obtained from filter-mating experiments were analyzed for resistance to vancomycin, fusidic acid, rifampicin, streptomycin and/or spectinomycin, respectively.
Filter-mating experiments
Transfer capabilities of Tn1549 were analyzed by filter-mating experiments and by altering donor and recipient strains as stated in Table 2. To discriminate the recipient from the donor strain rifampicin/fusidic acid (RF)- or streptomycin/spectinomycin (SS)-resistant E. faecium strains were used (Table 1). Likewise, E. faecalis OG1RF or JH2-2 served as recipient bacteria. Putative transconjugants of filter-mating experiments were selected on agar plates containing 5 μg/ml vancomycin, 30 μg/ml rifampicin and 20 μg/ml fusidic acid or 150 μg/ml streptomycin and 150 μg/ml spectinomycin, respectively. Further, transconjugants were routinely screened for acquisition of the vanB gene cluster by PCR using oligonucleotides as published previously7. Additional evaluation of the transconjugants was carried out by determining susceptibility to vancomycin using broth microdilution assays.
DNA extraction
Strains were cultivated overnight in brain heart infusion broth at 37 °C. DNA used for PCR or library preparation for Illumina sequencing was extracted by utilizing the DNeasy Blood and Tissue kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). DNA subjected to sequencing by means of Pacific Bioscience SMRT technology was extracted by using the Genomic-tip 100/G kit (Qiagen, Hilden, Germany).
Polymerase chain reaction (PCR)
To specifically amplify the ORFs of replicase genes with oligonucleotides rep-p-hylF (5′-TGAGCCCCAAGGGATTCAGGGT-3′) and rep-p-hylR (5′-CGCAATCAGCAAACGGCAAATCG-3′) for pLG1 or primers as published elsewhere30,31,32,33, a PCR was carried out as follows: for each sample 12.5 μl of a 2x concentrated DreamTaq Green-Mastermix (Thermo Scientific), as well as forward and reverse primer (final concentration of 0.1 μM) and 0.5 μl of the designated DNA-sample were mixed with DEPC H2O to a final volume of 25 μl. Subsequently, DNA amplification was carried out at 95 °C for 120 sec, followed by 30 cycles at 95 °C for 30 sec, annealing temperature of 50 °C (axe-txe oligonucleotides only) or 55 °C for 30 sec, elongation at 72 °C for 30 sec and a final extension for 7 min at 72 °C. The same protocol was applied for verification of closed circle conformation of plasmid pWCF-TC1. Oligonucleotides pTC1_cc_fw (5′-CTTAAAGGATGTGTGGATTTAT-3′) and pTC1_cc_rv (5′-CGCTCCGTTTACAGTAATAT-3′) were used at a final concentration of 0.1 μM and an annealing temperature of 55 °C.
S1-nuclease macrorestriction
Examination of plasmid content of various isolates was conducted by S1-nuclease treatment prior to separation of the genetic content in pulsed-field gel electrophoresis as published previously32.
Single molecule real-time (SMRT) sequencing
A transconjugant strain of E. faecium UW7606 (donor) and E. faecium 64/3 (recipient), 7606 × 64/3 and termed WCF- TC1 in the following, was sent to GATC (Konstanz, Germany) for whole genome sequencing by means of SMRT technology. The retrieved chromosomal and plasmid contigs were manually trimmed of overlapping sequences utilizing the Geneious software v7.1.4 (Biomatters Ltd.), hence yielding 2 fragments of 2.686.859 bp and 66.496 bp, respectively. Annotation was performed by NCBI.
Illumina whole genome sequencing and bioinformatic analyses
A total of 1 ng of extracted DNA was used for library generation by utilizing the Nextera XT DNA Library Prep Kit according to the manufacturer’s recommendations (Illumina). Sequencing was carried out on a MiSeq benchtop instrument and performed in paired-end mode using a v3 chemistry-based cartridge 600 (600-Cycle Reagent Kit, Illumina). Obtained reads were mapped to a designated reference sequence by utilizing a pipeline based on BWA version: 0.7.12-r1039 (BWA-SW)34 and VarScan v2.3 for variant calling35. As enterococci are highly recombinant, description of the core genome requires depletion of SNPs which might result from recombination events. Thus, reads mapped to reference strain E. faecium DO/TX16 (CP003583; DO in the following) were subjected to a custom-made script thereby excluding all SNPs falling within 300 bp or less of distance from each other. Retained SNPs served as a basis for phylogenetic analyses by using the graphical user interface of Seaview36 in combination with the program PhyML 3.037. Bootstrap confidence intervals are based on 1000 permutations. For visualization purposes, trees were processed with FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/) and/or Adobe Illustrator (Adobe Systems). (Sequence read data are available from the SRA database under accession SRP069166).
In order to determine Tn1549 insertion sites, reads were mapped to the transposon sequence of WCF-TC1 (33.811 bp) (CP013009) and the consensus of read pile ups at both 5′-and 3′-ends of Tn1549 was extracted to search for homologous regions using BlastN (http://www.ncbi.nlm.nih.gov/). For examination of transferred fragments sizes after conjugation experiments, genomic reads were subjected to de novo assembly by utilizing the open source pipeline a5-miseq38. The resulting contigs were mapped to Tn1549 of WCF-TC1 and a consensus sequences including flanking regions was extracted. Subsequently, consensus sequences of transconjugant strains were aligned to the consensus derived from the respective donor strain by using the Mauve plugin in Geneious.
Additional Information
Accession codes: The nucleotide sequences of WCF-TC1 and pWCF-TC1 are available from the GenBank database under the following accession numbers: CP013009; CP013010. http://www.nature.com/srep
How to cite this article: Bender, J. K. et al. Population structure and acquisition of the vanB resistance determinant in German clinical isolates of Enterococcus faecium ST192. Sci. Rep. 6, 21847; doi: 10.1038/srep21847 (2016).
References
Centers for Disease, C. & Prevention. Nosocomial enterococci resistant to vancomycin–United States, 1989–1993. MMWR Morb Mortal Wkly Rep 42, 597–599 (1993).
Schouten, M. A., Hoogkamp-Korstanje, J. A., Meis, J. F. & Voss, A. & European, V. R. E. S. G. Prevalence of vancomycin-resistant enterococci in Europe. Eur J Clin Microbiol Infect Dis 19, 816–822 (2000).
Sujatha, S. & Praharaj, I. Glycopeptide resistance in gram-positive cocci: a review. Interdiscip Perspect Infect Dis 2012, 781679, doi: 10.1155/2012/781679 (2012).
Arthur, M., Molinas, C., Depardieu, F. & Courvalin, P. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol 175, 117–127 (1993).
Garnier, F., Taourit, S., Glaser, P., Courvalin, P. & Galimand, M. Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 146 (Pt 6), 1481–1489 (2000).
Deshpande, L. M., Fritsche, T. R., Moet, G. J., Biedenbach, D. J. & Jones, R. N. Antimicrobial resistance and molecular epidemiology of vancomycin-resistant enterococci from North America and Europe: a report from the SENTRY antimicrobial surveillance program. Diagn Microbiol Infect Dis 58, 163–170, doi: 10.1016/j.diagmicrobio.2006.12.022 (2007).
Werner, G. et al. Vancomycin-resistant vanB-type Enterococcus faecium isolates expressing varying levels of vancomycin resistance and being highly prevalent among neonatal patients in a single ICU. Antimicrob Resist Infect Control 1, 21, doi: 10.1186/2047-2994-1-21 (2012).
Bjorkeng, E. et al. Clustering of polyclonal VanB-type vancomycin-resistant Enterococcus faecium in a low-endemic area was associated with CC17-genogroup strains harbouring transferable vanB2-Tn5382 and pRUM-like repA containing plasmids with axe-txe plasmid addiction systems. APMIS 119, 247–258, doi: 10.1111/j.1600-0463.2011.02724.x (2011).
Freitas, A. R. et al. In 22nd European Congress of Clinical Microbiology and Infectious Diseases (ECCMID) (2012).
Launay, A., Ballard, S. A., Johnson, P. D., Grayson, M. L. & Lambert, T. Transfer of vancomycin resistance transposon Tn1549 from Clostridium symbiosum to Enterococcus spp. in the gut of gnotobiotic mice. Antimicrob Agents Chemother 50, 1054–1062, doi: 10.1128/AAC.50.3.1054-1062.2006 (2006).
Carias, L. L., Rudin, S. D., Donskey, C. J. & Rice, L. B. Genetic linkage and cotransfer of a novel, vanB-containing transposon (Tn5382) and a low-affinity penicillin-binding protein 5 gene in a clinical vancomycin-resistant Enterococcus faecium isolate. J Bacteriol 180, 4426–4434 (1998).
Quintiliani, R., Jr. & Courvalin, P. Conjugal transfer of the vancomycin resistance determinant vanB between enterococci involves the movement of large genetic elements from chromosome to chromosome. FEMS Microbiol Lett 119, 359–363 (1994).
Dahl, K. H., Rokenes, T. P., Lundblad, E. W. & Sundsfjord, A. Nonconjugative transposition of the vanB-containing Tn5382-like element in Enterococcus faecium . Antimicrob Agents Chemother 47, 786–789 (2003).
Poyart, C. et al. Emergence of vancomycin resistance in the genus Streptococcus: characterization of a vanB transferable determinant in Streptococcus bovis . Antimicrob Agents Chemother 41, 24–29 (1997).
Domingo, M. C. et al. Characterization of a Tn5382-like transposon containing the vanB2 gene cluster in a Clostridium strain isolated from human faeces. J Antimicrob Chemother 55, 466–474, doi: 10.1093/jac/dki029 (2005).
Stinear, T. P., Olden, D. C., Johnson, P. D., Davies, J. K. & Grayson, M. L. Enterococcal vanB resistance locus in anaerobic bacteria in human faeces. Lancet 357, 855–856, doi: 10.1016/S0140-6736(00)04206-9 (2001).
Ballard, S. A., Grabsch, E. A., Johnson, P. D. & Grayson, M. L. Comparison of three PCR primer sets for identification of vanB gene carriage in feces and correlation with carriage of vancomycin-resistant enterococci: interference by vanB-containing anaerobic bacilli. Antimicrob Agents Chemother 49, 77–81, doi: 10.1128/AAC.49.1.77-81.2005 (2005).
Ballard, S. A., Pertile, K. K., Lim, M., Johnson, P. D. & Grayson, M. L. Molecular characterization of vanB elements in naturally occurring gut anaerobes. Antimicrob Agents Chemother 49, 1688–1694, doi: 10.1128/AAC.49.5.1688-1694.2005 (2005).
Howden, B. P. et al. Genomic insights to control the emergence of vancomycin-resistant enterococci. MBio 4, doi: 10.1128/mBio.00412-13 (2013).
Freitas, A. R. et al. Microevolutionary events involving narrow host plasmids influences local fixation of vancomycin-resistance in Enterococcus populations. PLoS One 8, e60589, doi: 10.1371/journal.pone.0060589 (2013).
Wardal, E. et al. Molecular analysis of vanA outbreak of Enterococcus faecium in two Warsaw hospitals: the importance of mobile genetic elements. Biomed Res Int 2014, 575367, doi: 10.1155/2014/575367 (2014).
Sivertsen, A. et al. A multicentre hospital outbreak in Sweden caused by introduction of a vanB2 transposon into a stably maintained pRUM-plasmid in an Enterococcus faecium ST192 clone. PLoS One 9, e103274, doi: 10.1371/journal.pone.0103274 (2014).
Lee, W. G., Ahn, S. H., Jung, M. K., Jin, H. Y. & Park, I. J. Characterization of a Vancomycin-resistant Enterococcus faecium Outbreak Caused by 2 Genetically Different Clones at a Neonatal Intensive Care Unit. Ann Lab Med 32, 82–86, doi: 10.3343/alm.2012.32.1.82 (2012).
Lam, M. M. et al. Comparative analysis of the first complete Enterococcus faecium genome. J Bacteriol 194, 2334–2341, doi: 10.1128/JB.00259-12 (2012).
Lam, M. M. et al. Comparative analysis of the complete genome of an epidemic hospital sequence type 203 clone of vancomycin-resistant Enterococcus faecium . BMC Genomics 14, 595, doi: 10.1186/1471-2164-14-595 (2013).
Werner, G. et al. Evaluation of DiversiLab(R), MLST and PFGE typing for discriminating clinical Enterococcus faecium isolates. J Microbiol Methods 118, 81–84, doi: 10.1016/j.mimet.2015.08.019 (2015).
Graham, M., Ballard, S. A., Grabsch, E. A., Johnson, P. D. & Grayson, M. L. High rates of fecal carriage of nonenterococcal vanB in both children and adults. Antimicrob Agents Chemother 52, 1195–1197, doi: 10.1128/AAC.00531-07 (2008).
Dahl, K. H., Lundblad, E. W., Rokenes, T. P., Olsvik, O. & Sundsfjord, A. Genetic linkage of the vanB2 gene cluster to Tn5382 in vancomycin-resistant enterococci and characterization of two novel insertion sequences. Microbiology 146 (Pt 6), 1469–1479 (2000).
Dahl, K. H. & Sundsfjord, A. Transferable vanB2 Tn5382-containing elements in fecal streptococcal strains from veal calves. Antimicrob Agents Chemother 47, 2579–2583 (2003).
Garcia-Migura, L., Liebana, E. & Jensen, L. B. Transposon characterization of vancomycin-resistant Enterococcus faecium (VREF) and dissemination of resistance associated with transferable plasmids. J Antimicrob Chemother 60, 263–268, doi: 10.1093/jac/dkm186 (2007).
Sletvold, H. et al. Comparative DNA analysis of two vanA plasmids from Enterococcus faecium strains isolated from poultry and a poultry farmer in Norway. Antimicrob Agents Chemother 51, 736–739, doi: 10.1128/AAC.00557-06 (2007).
Laverde Gomez, J. A. et al. A multiresistance megaplasmid pLG1 bearing a hylEfm genomic island in hospital Enterococcus faecium isolates. Int J Med Microbiol 301, 165–175, doi: 10.1016/j.ijmm.2010.08.015 (2011).
Moritz, E. M. & Hergenrother, P. J. Toxin-antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc Natl Acad Sci USA 104, 311–316, doi: 10.1073/pnas.0601168104 (2007).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, doi: 10.1093/bioinformatics/btp698 (2010).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576, doi: 10.1101/gr.129684.111 (2012).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27, 221–224, doi: 10.1093/molbev/msp259 (2010).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321, doi: 10.1093/sysbio/syq010 (2010).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589, doi: 10.1093/bioinformatics/btu661 (2015).
Werner, G. et al. Host range of enterococcal vanA plasmids among Gram-positive intestinal bacteria. J Antimicrob Chemother 66, 273–282, doi: 10.1093/jac/dkq455 (2011).
Qin, X. et al. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol 12, 135, doi: 10.1186/1471-2180-12-135 (2012).
Dunny, G. M., Brown, B. L. & Clewell, D. B. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci USA 75, 3479–3483 (1978).
Jacob, A. E. & Hobbs, S. J. Conjugal transfer of plasmid-borne multiple antibiotic resistance in Streptococcus faecalis var. zymogenes . J Bacteriol 117, 360–372 (1974).
Acknowledgements
We would like to thank all laboratories which provided us with strains and Dr. Roman Gerlach for critical reading of the manuscript. We would further like to acknowledge the contribution of Dr. Ralph Vogelsang and Dr. Gerrit Kuhn to the de novo assembly of WCF-TC1 (CP013009).
Author information
Authors and Affiliations
Contributions
J.K.B., I.K. and G.W. conceived the experiments, J.K.B, A.K. and C.F. conducted the experiments, J.K.B., A.K., I.K., S.F. and G.W. analyzed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bender, J., Kalmbach, A., Fleige, C. et al. Population structure and acquisition of the vanB resistance determinant in German clinical isolates of Enterococcus faecium ST192. Sci Rep 6, 21847 (2016). https://doi.org/10.1038/srep21847
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21847
This article is cited by
-
An Economic Evaluation Estimating the Clinical and Economic Burden of Increased Vancomycin-Resistant Enterococcus faecium Infection Incidence in Japan
Infectious Diseases and Therapy (2023)
-
The global dissemination of hospital clones of Enterococcus faecium
Genome Medicine (2021)
-
Increase of vancomycin-resistant Enterococcus faecium strain type ST117 CT71 at Charité - Universitätsmedizin Berlin, 2008 to 2018
Antimicrobial Resistance & Infection Control (2020)
-
Enterococcus faecium: from microbiological insights to practical recommendations for infection control and diagnostics
Antimicrobial Resistance & Infection Control (2020)
-
Near-ubiquitous presence of a vancomycin-resistant Enterococcus faecium ST117/CT71/vanB –clone in the Rhine-Main metropolitan area of Germany
Antimicrobial Resistance & Infection Control (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
