
Abstract
Exaggerated pulmonary pressor response to hypoxia is a pathgonomic feature observed in high altitude pulmonary edema (HAPE) susceptible mountaineers. It was investigated whether measurement of basal pulmonary artery pressure (Ppa) and brain natriuretic peptide (BNP) could improve identification of HAPE susceptible subjects in a non-mountaineer population. We studied BNP levels, baseline hemodynamics and the response to hypoxia (FIo2 = 0.12 for 30 min duration at sea level) in 11 HAPE resistant (no past history of HAPE, Control) and 11 HAPE susceptible (past history of HAPE, HAPE-S) subjects. Baseline Ppa (19.31 ± 3.63 vs 15.68 ± 2.79 mm Hg, p < 0.05) and plasma BNP levels (52.39 ± 32.9 vs 15.05 ± 9.6 pg/ml, p < 0.05) were high and stroke volume was less (p < 0.05) in HAPE-S subjects compared to control. Acute hypoxia produced an exaggerated increase in heart rate (p < 0.05), mean arterial pressure (p < 0.05) and Ppa (28.2 ± 5.8 vs 19.33 ± 3.74 mm Hg, p < 0.05) and fall in peripheral oxygen saturation (p < 0.05) in HAPE-S compared to control. Receiver operating characteristic (ROC) curves showed that Ppa response to acute hypoxia was the best variable to identify HAPE susceptibility (AUC 0.92) but BNP levels provided comparable information (AUC 0.85). BNP levels are easy to determine and may represent an important marker for the determination of HAPE susceptibility.
Similar content being viewed by others

Introduction
HAPE is hydrostatic edema which occurs in predisposed subjects at altitude above 2500 m1,2,3. The exact etiology of HAPE is not known, however, rapid ascent and individual susceptibility are important factors in the occurrence of HAPE4.
It is important to consider that HAPE represents an extreme pathological version of an otherwise physiological response to hypoxia, with various studies showing that exposure to chronic hypoxia enhances pulmonary artery pressures and vascular reactivity to acute hypoxia5,6. During ascent to high altitude there is a normal increase in pulmonary artery pressure that is initially reversible by oxygen administration, but HAPE susceptible (HAPE-S) subjects are prone to develop more severe pulmonary hypertension (PH) unresponsive to acute oxygen breathing7. It has been suggested that HAPE-S individuals may have residual structural changes in pulmonary vasculature that explain the fixed high pulmonary artery pressure8. It is hypothesized that there is a constitutional abnormality in HAPE-S individuals which causes such changes. Reduced vascular capacity, sub-clinical PH or ventricular dysfunction are such possibilities9,10,11,12. Previous studies on susceptibility to HAPE were done on mountaineers and experimental protocol involved rapid ascent to high altitude. It is well known that physical exertion associated with rapid ascent is an independent factor for occurrence of HAPE and this along with selection bias may account for negative findings regarding baseline differences in pulmonary vascular function13,14. In contrast, Indian armed forces personnel are routinely deployed at high altitude location in the Himalaya Mountains, without any previous mountaineering experience, but following a carefully controlled acclimatization schedule that lowers the risk of HAPE. Yet, despite these precautions during deployment, some of these personnel develop HAPE, presumably reflecting an underlying HAPE-S defect. Identification of a biomarker to screen HAPE-S subjects is a vital need in this context. In this study acute normobaric hypoxic stress was given to both HAPE-S and control subjects at sea level and their pulmonary and systemic hemodynamic responses were compared before and after hypoxia.
B type natriuretic peptide (BNP) is up regulated in pulmonary artery hypertension disease15. Studies in patients with pulmonary hypertension (PH) have demonstrated that plasma BNP levels are raised proportionally to the extent of right ventricular dysfunction, Ppa and PVR16. It is not known whether BNP may serve as a marker of sub-clinical pathology and HAPE susceptibility. Thus, an additional aim of this study was to examine whether HAPE-S subjects can be identified by elevated BNP levels.
Methods
Study population
We studied 11 Control and 11 HAPE-S who were categorized based upon their resistance or susceptibility to HAPE and were radiologically documented during their previous stay at high altitude. Our study population consists of Army troops who stayed at high altitude as a part of their tenure posting. All control subjects stayed at high altitude more than 2500 m for duration of 2 years of which 3 month stay was at altitude more than 4500 m. Both the groups were air inducted from sea level and had repeated exposures to high altitude. HAPE-S subject also stayed at high altitude more than 2500 m. The altitude of induction was 3500 m and everyone had to follow six day acclimatization schedule which includes complete rest for first two days followed by graded increase in physical activity for next four days of induction to high altitude. The HAPE-S participants suffered the illness during induction at 3500 m which was confirmed radiologically in spite of observing acclimatization schedule. All volunteers were nonsmoking natives of low altitude free of airway infection and receiving no medication when data was collected. None of the subject had resided at high altitude within last 6 months before the baseline measurements were carried out in Delhi, India at an altitude 293 m above sea level. The group of HAPE-S subjects consisted of individuals with mean age, 31.91 ± 4.74 y; wt, 69.7 ± 6.29 Kg; height, 168.6 ± 6.24 cm who had developed at least one case of radiologically documented HAPE within last 5 years. Control subjects consisted of individuals with mean age, 29.82 ± 2.68 y; wt, 69.73 ± 7.88 Kg; height, 171 ± 3.38 cm who did not develop HAPE during their stay at high altitude. All experimental protocols were approved by Defence Institute of Physiology and Allied Sciences Ethics Committee for scientific experiments. Informed written consent was obtained from all participants before enrolment in the study. All methods were carried out in accordance with the approved guidelines and regulations.
General Procedures
The subjects were investigated in the supine position while breathing synthetic gas mixtures consisting of 21 or 12% oxygen (FiO2 = 0.12) mixed in nitrogen. The hypoxic gas mixture corresponded to an altitude of 4500 m. Inhalation was performed via a tight fit face mask. Systolic blood pressure (SBP), Diastolic blood pressure (DBP), Mean blood pressure (MBP), heart rate (HR) and peripheral oxygen saturation (Spo2) were recorded before and at the end of 30 min. of hypoxic stress by using Multi parameter monitor, BPL, India.
Determination of pulmonary artery systolic pressure17
Pulmonary artery hemodynamics was measured noninvasively using echocardiography. Echocardiography studies were performed with My Lab 30 Gold Line ultrasonograph (Esaote India). Standard parasternal and apical two dimensional views were obtained and color flow directed pulse wave Doppler measurements of transvalvular flows and continuous wave Doppler measurements of tricuspid regurgitant flow were obtained. A single lead electrocardiogram was recorded on the ultrasonograph. Measures obtained using this noninvasive technique correlates closely with those obtained using cardiac catheterization. Pulmonary artery systolic pressure (sPpa) was calculated as follows

Where TRvel is tricuspid regurgitation jet velocity and RAP is the estimated right atrial pressure based on respiration variation in inferior vena cava size.
Calculation of Mean pulmonary artery pressure18

Cardiac output (Q) was calculated as follows

Where P(D/2)2 is the cross sectional area of blood flow into the aorta, Aovti is the velocity time integral through the aorta and HR is the heart rate.
Systemic vascular resistance (SVR) was measured as follows19

Pulmonary vascular resistance (PVR) was measured as follows20

Measurements of sPpa, Q, SVR, PVR were obtained before and at the end of 30 min. of hypoxic stress.
*This is a simplified estimate, since the full expression is [(Mean pulmonary artery pressure–Pulmonary capillary wedge pressure)/Cardiac output] × 80 dynes. sec. cm−5.
Pulmonary function measurement
Basal pulmonary function data was measured using dry, rolling seal spirometer (P.K. Morgan, Kent, UK). In addition to lung volumes and flow measurements, Functional residual capacity (FRC) was measured by the closed circuit Helium Dilution test. Diffusion capacity of the lung (DLCO) was also determined. The best value from three attempts were recorded as both absolute values and as percentage of the predicted values, based on age and body weight.
Biochemical parameters
Baseline venous blood samples were collected in heparin tube, centrifuged and plasma stored at −80 °C until analyzed.
Plasma BNP levels (pg/ml) was measured by enzyme-immuno assay method (Elabscience; E-EL-H0598). The sensitivity of assay was 18.75 pg/ml. The coefficient of variation was <10%.
Statistics
Corrected p values were used for multiple comparisons within and between the groups for normally distributed data. Non-parametric statistical tests were used for nonnormally distributed data. Wilcoxon signed rank test for paired and Wilcoxon rank sum test for comparison in between the two groups. ROC analysis was done for Ppa and BNP and cutoff values for all the variables, their sensitivities and specificities were calculated and compared. All the analysis was done using R statistical programming language.
All data are presented as means ± SD. P < 0.05 is considered significant.
Results
Baseline anthropometry and pulmonary function
The HAPE-S and control subjects were matched for age and lung function. Table 1 confirms that there were no significant differences in age, height and weight between two groups. There were no significant difference in forced vital capacity (FVC), Forced expiratory volume in 1 sec (FEV1), FEV1/FVC, Total lung capacity (TLC), Functional residual capacity (FRC), pulmonary diffusion capacity for carbon monoxide (DLCO) and Alveolar ventilation (VA), DLCO/VA between two groups.
Hemodynamics before and after hypoxia
HAPE-S subjects had baseline higher Ppa and reduced stroke volume (SV) as compared to control. HAPE-S subject showed higher baseline levels of BNP compared to control. Hypoxia produced a significant fall in SVR (formula No. 3) in HAPE-S compared to control. An exaggerated increase in heart rate (HR), mean arterial pressure (MAP) and Ppa (formula No. 1) was seen in HAPE-S subjects in response to hypoxia compared to control. Hypoxia produced a fall in peripheral oxygen saturation (SpO2) in both the groups however HAPE-S showed exaggerated fall compared to control.
Systemic and pulmonary hemodynamic parameters during normoxia and hypoxia
Heart rate (HR): There was no significant difference between two groups during normoxia. HAPE-S subjects showed an exaggerated increase in HR (p < 0.05) compared to control (23% in HAPE-S vs 15% in control) during hypoxia.
Blood pressure: There was no significant difference in systolic blood pressure (SBP), diastolic blood pressure (DBP) and MAP during normoxia. Hypoxia led to a significant increase in DBP and MAP in HAPE-S subjects, when compared to control.
Systemic vascular resistance (SVR): No significant difference in SVR was found during normoxia between the two groups. Hypoxia led to a significant fall in SVR in HAPE-S subjects, when compared to normoxia.
Cardiac output (Q): There was no significant difference in Q (formula No. 2) during normoxia between the two groups. Hypoxia led to a significant increase in cardiac output in HAPE-S (26% vs 14% in control).
Stroke volume (SV): HAPE-S subjects showed a significantly lower SV (24% less than control) during normoxia. No significant difference was observed during hypoxia between two groups.
Pulmonary vascular resistance (PVR): HAPE-S subjects showed a significantly higher PVR (31% greater than control) during normoxia. Hypoxia led to an exaggerated increase (p < 0.05) in PVR (formula No. 4) in HAPE-S (30% higher than control). All these findings are summarized in Table 2.
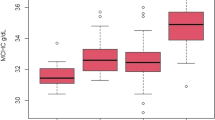
Pulmonary artery pressure under normoxic and hypoxic condition and baseline brain natriuretic peptide levels in HAPE susceptible subjects
Figures 1 and 2 Pulmonary artery pressure (Ppa): HAPE-S subjects showed a significantly higher (p < 0.05) Ppa (25% higher than control) during normoxia. Hypoxia produced an exaggerated increase (p < 0.01) in Ppa in HAPE-S (43% vs 23% increase) compared to control.

Mean pulmonary arterial pressure (Ppa) levels in normoxic and hypoxic conditions in controls (A) and in HAPE-S (B) individuals. To illustrate paired nature of data values of same individual are connected by a line. P values shown are obtained from paired t-test.

Boxplots showing comparison between control and HAPE-S individuals of mean pulmonary arterial pressure (Ppa) in normoxia (A) acute hypoxia (B), response to acute hypoxia (C) and baseline Brain natriuretic peptide (BNP) levels (D).
HAPE-S subject showed significantly higher (p < 0.05) baseline levels of BNP (52.39 ± 32.9 vs 15.05 ± 9.6 pg/ml) compared to control. The value of ROC area under the curve (AUC) for basal BNP value was in the diagnostically suitable range and comparable to Ppa in hypoxic condition (0.85 vs. 0.92).
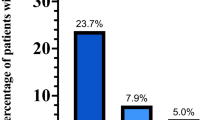
ROC curves and values of area under the curve for pulmonary artery pressure under normoxic and hypoxic condition and baseline brain natriuretic peptide levels in HAPE-S subjects is summarized in (Fig. 3). The thresholds associated with points on the ROC curve can be read from the color coded secondary Y-axis.

Receiver operator curves indicating the AUC for Pulmonary arterial pressure (Ppa) in normoxic (A) and hypoxic conditions (B), Ppa response to hypoxia (C) and baseline Brain natriuretic peptide (BNP) levels (D). Curves are color coded based on cutoff values which are shown as the second Y-axis.
Discussion
Exaggerated increase in Ppa in response to acute hypoxia is a pathogonomic feature of HAPE susceptiblity in mountaineers and trackers21,22. In this study, we show that non-mountaineers who developed HAPE, have not only a similar exaggerated Ppa increase but also increased sub-clinical baseline Ppa. To the best of our knowledge, this is the first such study in, a non-mountaineer population that followed acclimatization schedule on arrival at high altitude. This increases the likelihood that HAPE-S subjects from this population are representative of a true HAPE susceptible population with underlying abnormalities that predispose to exaggerated responses to hypoxia. Additionally, our study shows for the first time that elevation of BNP is comparable to Ppa response to acute hypoxia in identifying subjects who are HAPE susceptible.
The major limitation of our study is that subjects were studied after the development of HAPE. In our soldier population that undergoes acclimatization, the incidence of HAPE is very low and prospective studies were not feasible. The possibility that the observed differences are a consequence of HAPE, rather than risk markers, appears unlikely for the following reasons. First, the possibility of residual effects of high altitude stay as a cause of PH at sea level in HAPE-S subjects can be ruled out since none of the subject had resided at high altitude within the preceding 6 months from baseline data collection. Second, DLCO/Va was not different between two groups. This shows that no persistent pulmonary membrane defect existed in subjects with previous history of HAPE. Similar results were shown in a larger set of subjects studied earlier23. Further, the well known noninflammatory nature of HAPE and its quick resolution with descent and/or oxygen speaks against residual traces of previous episode24. While aging itself causes an increase in Ppa9, subjects with lower age were included in the present study compared to earlier studies14. Other limitations include absence of pulmonary capillary wedge pressure measurements for more precise estimation of PVR, a short duration of hypoxia (30 min at 12% Fio2) and measurement of BNP at baseline but not post-hypoxia. In this group of non-mountaineers, 30 minutes of hypoxia led to substantial declines in oxygenation, as shown in Table 2. Further, our intent was to identify the utility of BNP as a rapid screening biomarker for HAPE susceptibility in our non-mountaineer population. Therefore, it was measured only at baseline. However, additional information provided by post-hypoxia measurement could have been useful for biological understanding.
Our findings are generally comparable to other reports, but with some important differences. Control subjects had resting normoxic Ppa and showed hypoxic pulmonary vascular response (HPVR) to acute normobaric hypoxia (FIO2 = 0.12) similar to that reported previously21,25,26. We found higher basal Ppa and PVR in HAPE-S subjects compared to control. This is in contrast to studies on mountaineers, which showed no difference in basal Ppa between two groups13,14,24,26. Studies showed that hypoxic pulmonary vasoconstriction caused by high altitude is comparable to breathing 4 hr of 12% oxygen at sea level. Normobaric hypoxia (12% oxygen) when given for 4 hr in mountaineer population showed Spo2 falls to 82 ± 2% in HAPE-S and 87 ± 2% in control13. In our population, Spo2 fell to 68 ± 11.8% in HAPE-S and 78.7 ± 3.8% in control after 30 min. of 12% hypoxia. Therefore, a strictly comparable frame of reference may not be available. Exposure to acute hypoxia resulted in abnormally high HPVR in HAPE-S which is in agreement with reports of other investigators21,22. Some investigators have not reported exaggerated HPVR to hypoxia27,28. The reason for this difference in HPVR is not clear but may be related to inter-individual variations in HPVR and HAPE susceptibility, different experimental design and different pathology like reentry HAPE8. Greater desaturation in HAPE-S subjects in the present study could be explained based on blunted ventilatory response to hypoxia, as has been reported in some studies, leading to lower levels of alveolar Po223. However, reduced ventilatory response is an inconsistent observation and HAPE can occur in susceptible subjects despite the presence of a normal or high ventilatory response to hypoxia29.
Genetic variations in EGLN1 (HIF-prolyl hydroxylase 2), which normally hydroxylates HIF1α and marks it for degradation during normoxia, have been associated with HAPE30. EGLN1 expression was upregulated in larger set of HAPE patients (n = 250) and inversely correlated to SpO2 levels. Chronic hypoxia is closely associated with reduced expression of endothelial nitric oxide synthase which may explain low nitric oxide levels in pulmonary and systemic vasculature as also previously observed in HAPE-S13,31,32,33. Chronic hypoxia is known to cause vascular remodeling which explains baseline elevated Ppa and exaggerated pulmonary pressor response to acute hypoxia in the present study. Fall in SVR in response to acute hypoxia in HAPE-S is an exaggeration of a normal response since healthy individuals also show such hemodynamic response with increased severity and duration of hypoxia34.
Several reports have documented that HAPE resistant subjects have roughly 10% greater vital capacity that might indicate a smaller vascular bed in subjects susceptible to HAPE8,12. HAPE-R subjects have shown higher vascular capacity (Q and SV) during exercise compared to HAPE-S12. Since there was no ventilation perfusion mismatch at rest in between two groups therefore it was suggested that reduced vascular capacity could be a factor contributing to HAPE susceptibility35,36,37. Normal DLCO values in our HAPE-S subjects imply absence of any clinically significant reductions in baseline vascular capacity, but the possibility of sub-clinical differences that manifest during hypoxic stress cannot be excluded.
Dyspnoea on exertion (DOE) is a common symptom in both HAPE-S and pulmonary hypertension (PH) and associated with simultaneous rise in Ppa and pulmonary capillary wedge pressure (PCWP)8,38. Reduction of high PCWP in PH caused significant relief in DOE38. Acute hypoxia in HAPE-S induces increase in Ppa, as well as a possible rise in pulmonary capillary wedge pressure due to pulmonary venoconstriction in accordance to previous study26,39. This may cause pulmonary vascular congestion and arterial desaturation consistent with exaggerated fall in Spo2 in HAPE-S subjects in the present and past study20.
In the present study we have also found that the plasma BNP levels in HAPE susceptible subjects with no pulmonary disease were higher than healthy control subjects. It has been reported that an increase in BNP concentration is a risk factor for death independent of chronic lung disease40. These results were unchanged when factors related to age and gender were eliminated. Patients with HAPE showed increase in pulmonary artery pressure with normal left atrial pressures41. Pulmonary hypertension leading to shift of interventricular septum towards left was observed in 5 of 9 subjects with HAPE within first 24 hr of ascent to 4559 m42. BNP is transcribed in cardiac myocytes during mechanical strain, which supports our interpretation of the observed Ppa elevation being clinically relevant. Assuming that the observed increase in Ppa and BNP are related, as discussed above, it is useful to briefly consider the molecular pathways that intersect between PH and HAPE-S, since they would best describe the observed phenotype in our study like low levels of nitric oxide in pulmonary and systemic vasculature have been noted in both PH and HAPE-S13,31,43. Hypoxic response signaling has also been directly or indirectly related to both HAPE and PH and there is a strong rationale to further investigate variations in hypoxic response as a cause of the observed HAPE-S phenotype. The role of mitochondria in this context is particularly intriguing in view of their important role in oxygen sensing and free radical generation as mitochondrial redox distress has an association with HAPE susceptibility44.
Pulmonary hypertensive response to hypoxia and exercise has been found in relatives of patient with idiopathic and familial pulmonary artery hypertension45. The present study is first to associate elevated Ppa and baseline elevation of BNP with HAPE susceptibility. The potential use of BNP as a marker for HAPE susceptibility is attractive because of ease of testing. However, this is a very small dataset and larger independent studies are necessary before BNP can be considered to be a feasible screening approach, using consistent well-defined thresholds.
Additional Information
How to cite this article: Gupta, R. K. et al. Elevated pulmonary artery pressure and brain natriuretic peptide in high altitude pulmonary edema susceptible non-mountaineers. Sci. Rep. 6, 21357; doi: 10.1038/srep21357 (2016).
References
Swenson, E. R. & Bärtsch, P. High‐Altitude Pulmonary Edema. Compr Physiol 2, 2753‐2773 (2012).
Hultgren, H. N., Lopez, C. E., Lundberg, E. & Miller, H. Physiological studies of pulmonary edema at high altitude. Circulation. 29, 393–408 (1964).
Roy, S. B. et al. Haemodynamic studies in high altitude pulmonary edema. Br. Heart J. 31, 52–58 (1969).
Bärtsch, P. & Swenson, E. R. Acute High-Altitude Illnesses. N Engl J Med 368, 2294–2302 (2013).
Grover, R. F. et al. Pulmonary hypertension and pulmonary vascular reactivity in beagles at high altitude. Journal of Applied Physiology. 65(6), 2632–2640 (1988).
Penaloza, D., Sime, I., Banchero, N. & Gamboa, R. Pulmonary hypertension in healthy men born and living at high altitude. Am J. Cardiology. 11: 150–157 (1963).
Hultgren, H. N., Grover, R. F. & Hartley, L. H. Abnormal Circulatory Responses to High Altitude in Subjects with a Previous History of High-Altitude Pulmonary Edema. Circulation. 44, 759–770 (1971).
Eldridge, M. W. et al. Pulmonary hemodynamic response to exercise in subjects with prior high-altitude pulmonary edema. Journal of Applied Physiology 81(2), 911–921 (1996).
Emmanuel Weitzenblum. Chronic cor pulmonale. Heart 89, 225–230 (2003).
Shena, M. et al. Echo-doppler evaluation of left ventricular impairment in chronic cor pulmonale. Chest 109, 1446–1451 (1996).
Jessup, M. et al. The effect of chronic pulmonary hypertension on left ventricular size, function and interventricular septal motion. Am Heart J 113, 1114–1122 (1987).
Steinacker, J. M. et al. Lung diffusion capacity and exercise in subjects with previous high altitude pulmonary edema. Eur Respir J 11, 643- 650 (1998).
Busch, T. et al. Hypoxia decreased exhaled nitric oxide in mountaineer susceptible to high altitude pulmonary edema. Am J Respir Crit care Med 163, 368–373 (2001).
Gru¨nig, E. et al. Stress Doppler Echocardiography for Identification of Susceptibility to High Altitude Pulmonary Edema. J Am Coll Cardiol 35, 980–7 (2000).
Hargett, C. W. & Tapson, V. F. Brain Natriuretic Peptide: Diagnostic and Therapeutic Implications in Pulmonary Arterial Hypertension. Seminars in respiratory and critical care medicine 26 (4), 385–393 (2005).
Nagaya, N. et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol 31(1), 202–208 (1998).
Yock, P. G. & Popp, R. L. Non invasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation 70, 657–62, (1984).
Hoit, B. D. et al. Nitric oxide and cardiopulmonary hemodynamics in Tibetan highlanders. J Appl Physiol 99, 1796–1801 (2005).
Fahim, M. & Pressman, B. C. Improvement of cardiac performance by carboxylic ionophore monensin in greyhound and mongrel dogs. Naunyn-Schmiedeberg’s Arch Pharmacol 333, 412–20 (1986).
Duplain, H. et al. Augmented Sympathetic Activation During Short-Term Hypoxia and High-Altitude Exposure in Subjects Susceptible to High-Altitude Pulmonary Edema. Circulation 99(13), 1713–18 (1999).
Kawashima, A., Kubo, K., Kobayashi, T. & Sekiguchi, M. Hemodynamic responses to acute hypoxia, hypobaria and exercise in subjects susceptible to high altitude pulmonary edema. J Appl Physiol 67(5), 1982–1989 (1989).
Fasules, Wiggins J. J. & Wolfe, R. Increased lung vasoreactivity in children from Leadville, Colorado, after recovery from high altitude pulmonary edema. Circulation. 72, 957–962 (1985).
Hohenhaus, E., Paul, A., McCullough, R. E., Kucherer, H. & Bartsch, P. Ventilatory and pulmonary vascular response to hypoxia and susceptibility to high altitude pulmonary edema. Eur Respir J 8, 1825–1833 (1995).
Swenson, E. R. et al. Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. JAMA 287(17), 2228–35 (2002).
Donoyle, J., Wilson, J. & Warren, J. The pulmonary vascular response to short term hypoxia in human subjects. Circulation 5, 256–270 (1952).
Maggiorini, M. et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 103(16), 2078–83 (2001).
Viswanathan, R. et al. Pulmonary edema of high altitude II: clinical, aerohemodynamic and studies in a group with history of pulmonary edema of high altitude. Am Rev RespirDis 100, 334–341 (1969).
Hultgren, H. Pulmonary hypertension and pulmonary edema. In: Oxygen transport to human tissues, edited by J. Leoppky & M. Riedesel. New York: Elsevier, p. 253–254 (1982).
Selland, M. A. et al. Pulmonary function and hypoxic ventilatory response in subjects susceptible to high-altitude pulmonary edema Chest. 103(1), 111–6 (1993).
Mishra, A., Mohammad, G., Thinlas, T. & Pasha, M. A. EGLN1 variants influence expression and SaO2 levels to associate with high-altitude pulmonary oedema and adaptation. Clinical Sciences 124, 479–489 (2013).
Berger, M. M. et al. Hypoxia impairs systemic endothelial function in individuals prone to high altitude pulmonary edema. Am J Respir Crit Care Med 172, 763–767 (2005).
Fike, C. D., Kaplowitz, M. R., Thomas, C. J. & Leif, D. N. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am. J. Physiol. 274 (Lung Cell. Mol. Physiol. 18) L517–L526 (1998).
Mourad, T. et al. Downregulation of endothelial nitric oxide synthase in rat aorta after prolonged hypoxia in vivo. Circ Res. 86, 671–675 (2000).
Boos, C. J., Hodkinson, P., Mellor, A., Green, N. P. & Woods, D. R. The Effects of Acute Hypobaric Hypoxia on Arterial Stiffness and Endothelial Function and its Relationship to Changes in Pulmonary Artery Pressure and Left Ventricular Diastolic Function. High Altitude Medicine & Biology 13(2), 105–111 (2012).
Podolsky, A. et al. Exercise-induced VA/Q inequality in subjects with prior high-altitude pulmonary edema. J Appl Physiol 81(2), 922–932 (1996).
Wagner, P. D. et al. Pulmonary gas exchange in humans exercising at sealevel and simulated altitude. J Appl Physiol 61, 260–270 (1986).
Hopkin, S. R., McKenzie, D. C., Schoene, R. B., Glenny, R. W. & Robertson, H. T. Pulmonary gas exchange during exercise in atheletes I. Ventilation- perfusion mismatch and diffusion limitation. J Appl Physiol 77, 912–917 (1994).
Anand, A. et al. Early-symptom relief after valvulotomy in mitral stenosis indicates role of lobeline-sensitive intrapulmonary receptors. Respiratory Physiology & Neurobiology 169, 297–302 (2009).
Zhao, Y., Packer, C. S. & Rhoades, R. A. Pulmonary vein contracts in response to hypoxia. Am J Physiol 265, L87–L92 (1993).
Casserly, B. & Klinger, J. R. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Design, Development and Therapy 3, 269–287 (2009).
Kobayashi, T. et al. Clinical features of patients with high altitude pulmonary edema in Japan. Chest 92, 814–821 (1987).
Allemann, Y. et al. Impact of acute hypoxic pulmonary hypertension on LV diastolic function in healthy mountaineers at high altitude. Am J Physiol Heart Circ Physiol. 286, H856–H862 (2004).
Giaid, A. & Saleh, D. Reduced Expression of Endothelial Nitric Oxide Synthase in the Lungs of Patients with Pulmonary Hypertension. N Engl J Med 333, 214–221 (1995).
Adaptability to hypobaric hypoxia is facilitated through mitochondrial bioenergetics: an in vivo study. Chitra L and Boopathy R. British Journal of Pharmacology169, 1035–1047 (2013).
Grünig, E. et al. Stress doppler echocardiography in relatives of patients with idiopathic and familial pulmonary arterial hypertension. Circulation 119, 1747–1757 (2009).
Acknowledgements
Authors are grateful to the commanding officers and the troops of different units for volunteering to participate in the study. We also express our gratitude to Army Medical Corps authorities including DGAFMS, AFMC Pune and SD branch for providing nominal roll of HAPE patients. Due thanks are also expressed to Mr. CGC Sekharan, V. P. Chest Institute for conducting lung functions and diffusion studies. This work was supported by DRDO, Ministry of Defence, Govt. of India.
Author information
Authors and Affiliations
Contributions
R.K.G. has set up the study concept and design, data acquisition, interpretation and drafting of manuscript. G.H. and K.S. were responsible for data acquisition and preparation of manuscript. P.S. performed data acquisition and biochemical analysis, K.D. and A.A. were responsible for drafting of manuscript, statistical analysis and preparation of figures. D.D., D.G., P.K.R. and U.P. have set up normobaric hypoxia protocol, data acquisition and analysis. S.B.S. was responsible for drafting of manuscript, supervision and coordination of study process. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gupta, R., Himashree, G., Singh, K. et al. Elevated pulmonary artery pressure and brain natriuretic peptide in high altitude pulmonary edema susceptible non-mountaineers. Sci Rep 6, 21357 (2016). https://doi.org/10.1038/srep21357
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21357
This article is cited by
-
Biofluids in hypobaric hypoxia: best possible use, investigative strategies and putative markers
Journal of Proteins and Proteomics (2019)
-
Subclinical pulmonary dysfunction contributes to high altitude pulmonary edema susceptibility in healthy non-mountaineers
Scientific Reports (2017)
-
Raised HIF1α during normoxia in high altitude pulmonary edema susceptible non-mountaineers
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
