
Abstract
The global spread of human infectious diseases is of considerable public health and biomedical interest. Little is known about the relationship between the distribution of ancient parasites and that of their human hosts. Schistosoma japonicum is one of the three major species of schistosome blood flukes causing the disease of schistosomiasis in humans. The parasite is prevalent in East and Southeast Asia, including the People’s Republic of China, the Philippines and Indonesia. We studied the co-expansion of S. japonicum and its human definitive host. Phylogenetic reconstruction based on complete mitochondrial genome sequences showed that S. japonicum radiated from the middle and lower reaches of the Yangtze River to the mountainous areas of China, Japan and Southeast Asia. In addition, the parasite experienced two population expansions during the Neolithic agriculture era, coinciding with human migration and population growth. The data indicate that the advent of rice planting likely played a key role in the spread of schistosomiasis in Asia. Moreover, the presence of different subspecies of Oncomelania hupensis intermediate host snails in different localities in Asia allowed S. japonicum to survive in new rice-planting areas and concurrently drove the intraspecies divergence of the parasite.
Similar content being viewed by others

Introduction
The global spread of human infectious diseases is of considerable biomedical interest as this process relies on the dispersal of both host and pathogen. Indeed, the current Ebola outbreaks caused by viruses of the genera Ebolavirus and Marburgvirus resulted from the spread of their human hosts1. Previous studies have also reported geographically structured populations for a number of human pathogens2,3,4,5,6, some of which have been linked to ancient human host migrations3,4,5,6. However, for many parasites, which may have several hosts in their life cycle, there is no information as to whether they migrated with human or other hosts.
Schistosomes are ancient parasites with two distinct hosts during their lifecycle: an intermediate fresh water snail host and a definitive human or other mammalian host. These blood flukes cause schistosomiasis which ranks as the second most serious human parasitic infection globally in terms of burden of disease estimates. There are three major species that infect humans: Schistosoma mansoni, S. haematobium and S. japonicum. The latter schistosome has exploited a wide range of at least 46 species of mammalian definitive hosts, including humans and a variety of wild and domesticated animals, but uses only Oncomelania hupensis as its intermediate host7. S. japonicum is prevalent in the People’s Republic of China, particularly in the marsh and lake regions along the Yangtze River basin (Hunan, Hubei, Jiangxi, Anhui and Jiangsu province) and in mountainous areas (Sichuan and Yunnan province) and in parts of the Philippines and Indonesia8,9. The population genetic structure of S. japonicum distributed in East Asia still remains unclear. Several studies of genetic variation, based on the use of mitochondrial (mt) genes and microsatellite loci as gene markers, have shown that in mainland China worms from the lake regions and from the mountain regions are different10. However, there is limited information regarding the relationship between Chinese mainland S. japonicum and other strains of the parasite in East Asia11. Where did the ancestor of all strains of the S. japonicum group originate? How and when did the different strains diverge during the course of evolutionary history? Why did S. japonicum expand into new areas and hosts? Addressing these questions will provide an understanding of the evolution of this parasite and shed light on its history of disease transmission.
We collected 119 S. japonicum samples from 13 locations endemic for Asiatic schistosomiasis, including Japan, Indonesia, Philippines, Taiwan and mainland China and we investigated the co-expansion of S. japonicum and its human definitive host. Phylogenetic reconstruction based on complete mitochondrial genome sequences showed that S. japonicum radiated from the middle and lower reaches of the Yangtze River to the mountainous areas of China, Japan and Southeast Asia. In addition, the parasite experienced two population expansions during the Neolithic agricultural era, coinciding with human migration and population expansion. The data indicate that the advent of rice planting likely played a key role in the spread of schistosomiasis japonica in Asia.
Results
Haplotype analysis
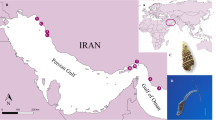
We collected S. japonicum samples from Indonesia, Japan, the Philippines, Chinese Taiwan and nine locations on mainland China, including two populations from mountainous areas and seven from the lake regions (for detailed information, see Fig. 1 and Table S1). In total, 119 complete S. japonicum mitochondrial DNA (mtDNA) genomes (~14-kilobase) were sequenced successfully using Next-Generation Sequencing (NGS) technology, with high quality (the coverage ranged from 571 to 6,593, with the average being 3,588; Fig. S1). For S. japonicum lineages from Indonesia, Japan, the Philippines and Taiwan, only one haplotype was detected per population based on scrutiny of their complete mtDNA genomes (Fig. 2), indicating an extremely low effective population size (Ne) in these groups. However, lower nucleotide diversity was evident in the two S. japonicum populations from the mountainous areas of China compared with those from the lake areas (Table S1 and Fig. 1). In total, 45 haplotypes were distributed among S. japonicum populations from the lake areas of mainland China indicating a very rich level of genetic variation.

Location of the Schistosoma japonicum samples used in analysis.
The arrows indicate the direction of spread of S. japonicum. For abbreviations of each sample, refer to Table S1. The migration times are annotated in the figure (BP: years before present). The Map was created using ArcGIS® version 10.2 software by Esri http://www.esri.com/software/arcgis).

Phylogenetic tree of 119 complete Schistosoma japonicum mt genomes, based on maximum likelihood and Bayesian methods.
The S. japonicum phylogeny was calibrated with the mt genome of S. mekongi, a close relative found exclusively in the Mekong river basin of Laos and Cambodia in South-east Asia. Nodes with high statistical support (>80% approximate likelihood branch support in ML analysis and >0.9 posterior probability in Bayesian analysis, respectively) are highlighted by asterisks. The numbers in brackets indicate the number of identical mt genome sequences obtained. The green circles indicate lake samples, the pink circles indicate samples collected from mountainous areas of mainland China, whereas the coloured triangles represent samples from Chinese Taiwan, Japan, the Philippines and Indonesia. For the abbreviations of each sample, refer to Table S1.
Phylogeny reconstruction
The phylogenetic history of the 119 complete mtDNA sequences obtained for the S. japonicum samples was inferred by a Bayesian model. The topology of the phylogeny was further confirmed by both the maximum likelihood (ML) method and median-joining network (refer to Table S2 and Fig. S2 for details of the mtDNA sequence variations). Important nodes of the topology showed relative high support (posterior probability by the Bayesian method and approximate likelihood support by the maximum likelihood method). In total, 23 S. japonicum haplogroups were determined and their defining variants (Fig. S3) were classified into four major haplogroups (i.e. Haplogroup A, B, C and D) (Fig. 2). First of all, S. japonicum samples from Taiwan (Haplogroup D) diverged out from mainland lineages. In addition, the distance between the Taiwan and the ancestor of non-Taiwan lineages (Haplogroup A'B'C) showed ~6 times the average genetic distance non-Taiwan lineages diverging from their most recent common ancestor. Large divergence indicated a considerable difference in schistosome infection, which coincided with the fact that non-Taiwan S. japonicum can infect humans while Taiwan lineage do not.
S. japonicum from mainland of China was mainly composed of 3 major haplogroups which almost diverged tridently. Haplogroup C split out from Haplogroup A′B′C little earlier than Haplogroup A and B, which separated after additional 3 variants (Fig. S3). Japan and Southeast Asian samples belonged to Haplogroup A and B, respectively. Mountain region lineages showed a definite monophyletic clade, belonging to Haplogroup A. According to the phylogeny, we detected 878 variant sites, 1055 substitutions and 30 indels (Table S2). High nucleotide diversity (π) in the ND5 gene was revealed by the analysis of sliding window of 200 bp (step size = 50 bp) along the entire genome (Fig. S2). A similar plot of distribution on total substitutions showed an unexpected high mutation hot spot at the end of the mitochondrial genome. After inspection, we found that substitutions occurred relatively frequently at 3 sites in the whole phylogeny (13971, 7 times, 13980, 8 times, 13981, 9 times, respectively). Interestingly, variants of these sites were linked 7 times but the mutation mechanisms involved require further research.
Inference of reference sequence
According to the phylogenetic tree (Fig. 2), the lineages of Taiwan S. japonicum, which does not colonize humans12, diverged first from the other lineages that can infect humans (Fig. 2). The extant S. japonicum reference mt genome is 14085 bp (Genbank access: NC_002544). Compared with the extant reference mtDNA genome sequence, we detected a haplotype bearing 43 nucleotide mismatches with two additional insertions (a guanine ‘insertion’ at position 2318 and a thymine ‘insertion’ at position 2450) which all the human-colonizing lineages originated from. The guanine insertion at 2318 encodes COX3, which affects the codon frame and results in an extended 2 amino acids compared with the original protein product. The N-terminal of the corrected COX3 protein subunit was more similar to that of S. mekongi. The thymine insertion at 2450 encodes tRNAGlu. Both insertions were also found in other published S. japonicum mtDNA sequences13. In addition, the original S. japonicum mtDNA reference showed many mismatches in nucleotides compared to the extant sequences. Considering the potential error of the extant reference sequence and the phylogenetic significance, we proposed to use the reconstructed sequence, which was the ancestor of all human-infecting lineages (ancestor of Haplogroup A, B and C, i.e., the defining sequence of Haplogroup A'B'C) and to renumber the reference to 14087 bp (Supporting information). Mismatches to the new reference sequence showed the derived allele in S. japonicum evolution, which would have a practical influence.
Estimation of divergence
For the calibration point, we assumed that the divergence of S. japonicum from the related S. mekongi, found in Laos and Cambodia, occurred 3.8 million years ago (mya) according to a recent study14. Then, we employed two strategies, a ML method via PAML package v4.7 and a Bayesian method via BEAST v1.8, to estimate the ages of the S. japonicum haplogroups (Fig. S4 and Table S3) and the mutation rates (Table S4). In both analyses, whole genome sequences partitioned in six regions were used. For comparison, protein coding sequences were employed in ML analysis considering that gene rearrageenment might cause alignment problem between S. japonicum and S. mekongi. In addition, mtDNA was shaped by selective constraints, which affected the time estimates based on whole genome sequences. However, synonymous mutation was considered free of the pressure. In the phylogeny of S. japonicum, there were 529 variants on the third codon, of which only 27 variants were non-synonymous mutations. Thus, we also used the third position of the protein codon to estimate an approximately ‘neutral’ rate in ML analysis.
Generally, in total, four age estimates via ML and Bayesian strategies were obtained for each haplogroup and these did not show large discrepancies (Fig. S4 and Table S3). Haplogroup D exhibited a large genetic distance compared with the others and diverged into Taiwanese S. japonicum isolates ~75 thousand years ago (kya), whereas Haplogroups A, B and C separated much more recently and almost simultaneously, about 22 kya. Moreover, Haplogroup C split out a little earlier than Haplogroup A or B and the Haplogroups A'B and A'B'C were only differentiated in several hundred years. Two star-like lineages (Haplogroup B and A1) took place ~10 kya, showing a great expansion in S. japonicum. In Haplogroup B, Japan lineages split from mainland China ~7–9 kya. In Haplogroup A1a, S. japonicum migrated to Southeast Asia ~3–4 kya and to mountain regions ~5 kya. Two star-like lineages (A1b1 and A1a1a) were within ~5 kya and indicated recent expansions in the agriculture era. Mutation rate estimates showed that the non-coding region evolved fastest for free of selective constraints while rRNA and tRNA were relatively conservative (Table S4).
Inference of population expansions
Given the information we obtained on the phylogeny and haplogroup age of S. japonicum, we further reconstructed its demographic history using BSP via mtDNA coding regions (Fig. 3 and Table S5). This showed that the effective population size (Ne) of S. japonicum expanded from 15,000 to 50,000 at ~11 to 7 kya, assuming half a year as the length of generation time. This expansion corresponded with two star-like lineages (Haplogroup B and A1; Fig. 2) both coalescing at ~10 kya. Interestingly, S. japonicum showed an additional growth peak ~5–2 kya in the agricultural era, with Ne ranging from 70,000 to 300,000, which might correlate with an expansion in the lineages A1b1, A1a2, A1a1b and A1a1a (Fig. S3). For comparison, we also employed three human mtDNA lineages (B5, M7 and F) to construct human BSP for comparison. Haplogroup B5, M7 and F were major haplogroups in the Southern part of East Asia and then some sub-lineages migrated to Southeast Asia, probably corresponding to the human expansion due to rice agriculture in Southern China (Fig. 3 and Table S5). Human BSP also showed two peaks of rapid Ne growth. The former was a ten-fold increase ~12–8 kya, followed by a subsequent three-fold expansion ~5–3 kya in the agriculture era. Human BSP correlated well with S. japonicum BSP (Fig. 3), indicating that S. japonicum expanded accompanying human activity, especially the advent of the agricultural era. The latter expansion of S. japonicum appeared much greater than the former and corresponded to more human migration events.

Demographic analysis of Schistosoma japonicum and Homo sapiens.
(A) Bayesian skyline plots of Schistosoma japonicum (blue) and three lineages of Homo sapiens (red). The x-axis is the time from present in units of years and the y-axis is the product of maternal effective size and generation time. The solid line is the median estimate and the dashed lines show the 95% highest posterior density limits. (B) Correlations of effective population size between Homo sapiens and S. japonicum. The linear regression line and coefficient were denoted.
Discussion
In this work, we constructed the phylogeny tree based on whole mitochondrial genome sequences and four major haplogroups (i.e. Haplogroup A, B, C and D; Fig. 2) were determined for S. japonicum. All the Taiwanese lineages were placed in Haplogroup D, the Japanese lineages were placed in Haplogroup B and the lineages from Southeast Asia were represented in Haplogroup A. In addition, the lineages from the Chinese lake regions had the highest diversity, spreading out into all the lineages of Haplogroup C and some sub-haplogroups of A and B. The lineages from the mountainous regions of China were clearly clustered, presenting as a specific sub-haplogroup of A, which we termed Haplogroup A1a1 (Figs 1 and 2). The reason that the lake worms showed high diversity are gene flow happened in this area because of the rich waternet, while the lineages from the mountainous regions of China and the island strain showed low diversity as result of their geographic isolation. It was very clear that the Taiwan samples generated an independent clade which was separated from the other human colonized worms. This result may coincide with its unique infection feature. Therefore, the 14,087 bp mtDNA reference genome (see Supporting information) of S. japonicum we constructed represents the ancestor of all human-colonizing lineages. This new reference sequence will have practical and phylogenetic importance in future research as the potential mismatches could help in the determination of derived alleles in S. japonicum evolution.
Furthermore, the ages of the S. japonicum haplogroups were estimated; the Haplogroup D exhibited a large genetic distance compared with the others and diverged into Taiwanese S. japonicum isolates ~75 thousand years ago (kya), whereas Haplogroups A, B and C separated much more recently and almost simultaneously, about 22 kya. Moreover, Haplogroup C split out a little earlier than Haplogroup A or B and the Sub-haplogroups A'B and A'B'C were only differentiated in several hundred years. In addition, the Japanese lineages in Haplogroup B split from the Chinese S. japonicum mainland lake area lineages ~7–9 kya and S. japonicum from the lake regions in Haplogroup A1a migrated to Southeast Asia at ~3–4 kya and to the mountainous regions at ~5 kya. The data show that S. japonicum originated in the lake area of China, with the parasite radiating to Japan around 7 kya, to the mountainous region of China about 5 kya and to the Philippines and Indonesia about 4 kya. The Taiwan parasite in this study, as the sister clade of human colonizing S. japonicum, diverged as a relatively independent isolate about 75 kya. This lineage might have lost the capacity to infect or was never able to infect humans during its evolutionary history. Indeed, it has been proposed that the genus Schistosoma arose in Asia from an avian schistosomatid and this was followed by a host shift utilizing ungulates during the mid to late Miocene15. Interestingly, there is a recent report of a strain of S. japonicum from Changhua, Taiwan (KF279410) by Attwood et al.16, which might have migrated to Taiwan from mainland China. Combined with our data, we confirm that this Taiwan strain originated recently from the Lake region of mainland China ~5 kya (5.39 ± 0.81 kya) (Fig. S5). As a result, we assumed that there might be at least two genetically distinct isolates of S. japonicum in Taiwan, one being an ancient form we report here and the other is an isolate which may have arrived recently from mainland China16. However, as there has never been any report of an autochthonous case of S. japonicum from Taiwan, we consider the newly reported “Taiwan isolate” may instead be a “Chinese Lake isolate” which originated from a patient with a S. japonicum infection recently visiting or migrating from mainland China, although there is no detailed information provided for the sample analysed by Attwood et al.16.
Thus, we posed the question how could this parasite have radiated from the Lake area of China to other endemic areas? According to the life cycle of S. japonicum, there are two possibilities: the parasite was dispersed by its intermediate host or definitive hosts. However, almost no other species disperses as widely as Homo sapiens and, therefore, many human pathogens have achieved wide-spread distribution along with their human hosts17. Consequently, the spread of a pathogen is critically dependent on the extent of expansion of its human host. For example, Mycobacterium tuberculosis has experienced strong population expansion as a consequence of the recent human population increase18. Therefore, we hypothesized that there could be a similar correlation in expansion events between S. japonicum and its human host. Here, we investigate the demographic history of human being and S. japonicum. The reconstructed demographic history (Fig. 3 and Table S5) showed two peaks of population expansion, the first one happened at ~11 to 7 kya corresponded with two star-like lineages (Haplogroup B and A1; Fig. 2) and the second one happened ~5–2 kya, which might correlate with an expansion in the lineages A1b1, A1a2, A1a1b and A1a1a (Fig. S3). To investigate human migration and expansion in areas where S. japonicum was endemic during the transition to the agricultural era, we employed three human mtDNA lineages (i.e. B5, M7 and F) to construct BSPs for the human population. B5, M7 and F are three major human Haplogroups in the Southern part of East Asia with some sub-lineages having migrated to Southeast Asia19,20. Human BSPs showed two peaks of rapid Ne growth. One was a ten-fold increase at ~12–8 kya, followed by a subsequent three-fold expansion at ~5–3 kya (Fig. 3 and Table S5). The latter also coincided with the Neolithic dispersal of S. japonicum. To summarize, dispersal of the human-colonizing S. japonicum, including major expansions and migrations, occurred in Neolithic Time.
The transmission cycle of many pathogens can comprise a suite of species, each distributed according to its own ecological needs, thereby constraining the pathogen to the region where the host ranges intersect. Thus, the human BSPs correlate well with the S. japonicum BSPs (Fig. 3), indicating that S. japonicum expansion accompanied human activity, especially during the advent of the agriculture era. The data also imply that the migration of S. japonicum would be promoted by the migration of its human host over the past 10 kya. Indeed, modern humans have been reported to have migrated to South East Asia in the Neolithic period, with representative mtDNA haplogroups including M7b3, M7c3c and Y221. Autosomal data also strongly support large demic movements of Austronesian speaking populations into Indonesia from ~4 kya22. This corresponds very well with the coalescence time (3–4 kya) of the Southeast Asian lineages of S. japonicum.
However, the data we present led us to pose another question. Why did this parasitic worm radiate from a lake region on the mainland of China to other endemic areas? Interestingly, when we reviewed the distribution of S. japonicum, we found that it coincided well with traditional rice-planting areas. This encouraged us to consider a possible relationship between the dawn of rice agriculture and the transmission of schistosomiasis japonica. Beginning at about 12 kya, hunter-gatherer populations in the Fertile Crescent of West Asia began developing horticultural practices and commenced animal domestication23. In East Asia, agriculture started in the Yangtze and Yellow River Basins about 9 kya23. It is widely accepted that the Yangtze River is the original center of rice cultivation in China and other parts of Asia24,25. The planting of rice commenced about 8 kya in the middle and lower reaches of the Yangtze River, covering Zhejiang, Jiangsu, Jiangxi, Hunan and Hubei provinces, which are endemic areas for S. japonicum in the lake region. Rice-planting radiated to the southwest (endemic mountainous area of China), as well as to Southeast Asia (the Philippines and Indonesia) around 4 kya26 and it seems that the spread of S. japonicum followed the track of this agricultural practice. To achieve success in rice planting, people with appropriate skills and requisite tools and a suitable environment are critical. Along with the spread of the practice of planting rice, subjects infected with S. japonicum would transmit the parasite from one location to another when they migrated to look for new cultivation areas. Moreover, the humid and warm environment required for successful rice cultivation is also highly favorable for breeding of Oncomelania hupensis, the intermediate host of S. japonicum, so that the parasite life cycle could be readily established and maintained in a new rice growing area. Considering that O. hupensis would already have been present in the agricultural era, as the divergence of these snails in the lake and mountainous regions of China occurred about 2–6 million years ago27 well before the parasites were introduced, their contribution to the life cycle of S. japonicum would be pivotal. As there are different subspecies of O. hupensis distributed in different endemic areas for schistosomiasis japonica e.g.28,29,30 and there are differences in compatibility between the snails and worms from different geographical locations31, we consider this separation of subspecies might not only have been an important reason for the survival of S. japonicum in new locations, but also led to the divergence of different geographically separated parasite populations.
The agricultural revolution, which involved the transition from hunting and gathering to settled agrarian societies, not only resulted in human migration but also led to a growth in human population size in Europe, Southeast Asia and sub-Saharan Africa over a period of 10 kya32. Our results lend support to the concept of the two population expansions of S. japonicum during the agriculture era, coinciding with human movement and population increase. For S. japonicum, many different species of definitive mammalian hosts and the key intermediate snail host (O. hupensis) characterize its zoonotic transmission cycle. The agriculture era not only resulted in expansion events of H. sapiens33, domestic and wild animals e.g.34,35, but also heralded the advent of rice-planting, increased land use and the introduction of irrigation along with human migration, which would have promoted the spread of O. hupensis and the transmission of schistosomiasis to new areas in East Asia. Indeed, we propose that the introduction of rice planting would have played a key role in promoting the transmission of schistosomiasis in East Asia.
Methods
Sample collection
119 Schistosoma japonicum worm samples in 13 locations were collected from Indonesia (Lindu lake, Sulawesi), Japan (Yamanashi strain maintained in the laboratory of Dr Sugiyama, National Institute of Infectious Diseases of Japan), the Philippines (Leyte), Chinese Taiwan (Changhua) and nine locations in mainland China (Table S1). In mainland China, two samples were collected from mountain regions (i.e. Eryuan County in Yunnan Province and Xichang City in Sichuan Province), while the other seven were obtained from locations in the lake regions (Duchang and Nanchang City in Jiangxi Province, Guichi and Tongling City in Anhui Province, Yueyang and Changde City in Hunan Province and Shashi City in Hubei Province). From each location, 10 individual adult worms were sampled for sequencing as representatives of population diversity, with the exception that two individuals were collected from Japan. The protocol for worm collection is described in a previous study36.
Preparation of genomic libraries and sequencing
Next generation sequencing (NGS) technology was applied to sequence the complete mitochondrial genomes of all the collected samples of S. japonicum. We first amplified the complete mitochondrial DNA (mtDNA) genomes using 13 PCRs, which cover the whole mtDNA genome. These 13 overlapping products were then mixed in roughly equal amounts after determining the concentration of each amplicon. Then, fragment libraries were prepared using the optimized protocol provided by Illumina and published37. Briefly, the complete mtDNA genome of each sample was sheared with DNase I and the sheared fragments were purified and concentrated using a QIAquick PCR purification spin column (QIAgen Inc.). T4 DNA polymerase, T4 phosphonucleotide kinase and the Klenow fragment of Escherichia coli DNA polymerase were used to fill 5′ overhangs and remove 3′ overhangs of sheared fragments and then were added A-residues at the 3′ terminal sides using dATP and Klenow (3′–5′exonulcease). Adaptors containing unique barcode sequences were then ligated to the fragments. We harvested fragments ranging from 200 bp to 250 bp through an agarose electrophoresis platform, the products were isolated using QIAgen MiniElute gel extraction spin columns and then each sample was amplified using standard Illumina primers and running 15 PCR cycles. After these libraries were re-purified, we quantified the DNA concentration of all samples and 30 ng of each were pooled together. The oligonucleotide mix was finally sequenced on an Illumina HiSeq 2000 by BGI, China.
Whole mtDNA sequence assembly
Original sequencing reads were exported to Fastq files and then bwa v0.6.238 was used to align reads to an existing S. japonicum mitochondrial genome reference sequence (Genbank accession: NC_002544) to generate binary sequence alignment/map (BAM) files of the mtDNA genomes39. Duplicate reads were removed by MarkDuplicates, implemented in Picard v1.82 (http://picard.sourceforge.net) and the mtDNA sequences were locally realigned by GATK v1.2.5940. Pileup files were generated by SAMtools v1.0.1839. Consensus sequences were then obtained based on the pileup files and indels were checked manually afterwards. Variations for haploid were called according to the criteria used in Zheng et al.41. All the 119 sequences obtained were deposited in Genbank (KU196299-KU196417).
Phylogenetic analysis and time estimation
As an outgroup to S. japonicum, the mt genome sequence of S. mekongi (Genbank accession: NC_002529) was selected and aligned to S. japonicum sequences considering gene rearrangement15. The phylogeny of 119 S. japonicum mt genome sequences and one S. mekongi mt genome sequence was inferred by Mrbayes v3.2.142 with the HKY+I+G model. 106 generations were performed with 4 chains (i.e. 1 cold chain and 3 hot chains) and the first 7,000 generations were regarded as burn-in. Moreover, the PhyML v3.043 with HKY+G model and Network v4.6.1.1 (http://www.fluxus-engineering.com/sharenet_rn.htm) were applied to generate topologies. Haplogroups were defined according to the topology and assigned to each sample.
For the calibration point, we assumed that the divergent time between S. japonicum and S. mekongi was 3.8 million years ago (MYA), according to a recent study14. The phylogenetic tree was used to examine the assumption of a molecular clock under the HKY+G mutation model. The null hypothesis of a molecular clock cannot be rejected (P = 1.00) using the PAML package v4.744. We employed two strategies to estimate the ages of S. japonicum haplogroups, i.e., the maximum likelihood (ML) method via PAML package v4.7 and the Bayesian method via BEAST v1.845. In ML analysis, three sequence partitioning methods were performed with the HKY+G model. First, we used the complete mtDNA genome sequences, which were partitioned into 6 regions (i.e. the first, second and third positions of the codons, tRNA, rRNA and non-coding regions). Second, we used protein coding regions only (i.e. partitioned in three positions of codons), considering that historical gene re-arrangements between S. japonicum and S. mekongi might cause alignment problems with whole sequences. Third, we used the third position of codons only for the reason that most variants on the third codon were synonymous mutations. Natural selection on mtDNA is thought to have an effect on the mutation rate of the whole genome and time estimation. However, synonymous mutation rates are regarded as neutral and free from negative selection. In Bayesian analysis, whole mitochondrial sequences were employed and partitioned into 6 regions as with the ML analysis. We ran 108 iterations, with samples drawn every 5,000 steps and the first 107 iterations considered burn-in. A strict clock was selected in all analysis.
We reconstructed the historical demographic variation of 109 human-infecting S. japonicum sequences via Bayesian skyline plots (BSPs) implemented in BEAST v1.8. Tracer v1.5 was used to visualize the results and to construct the BSPs.
Human mtDNA data
The 435 complete human mtDNA sequences of Haplogroups B5, M7 and F were used to construct the BSPs for the human population. B5, M7 and F are three major human Haplogroups in the Southern part of East Asia with some sub-lineages having migrated to Southeast Asia19,20. The 435 human mtDNA sequences were from 3 random population data sets, of which 175 sequences were from the 1000 Genomes Project41, 68 from the Human Genome Diversity Cell Line Panel (HGDP-CEPH, Genbank: KJ445738- KJ446778 and KP240908-KP240930) and 253 newly generated sequence data (Genbank: KP240655-KP240907). The sequencing and variant calling method have been described previously46. The parameters for BSP construction were as we have described previously41,47. Population growth rates were calculated from the BSP using the method described in Gignoux et al.32.
Additional Information
How to cite this article: Yin, M. et al. Co-dispersal of the blood fluke Schistosoma japonicum and Homo sapiens in the Neolithic Age. Sci. Rep. 5, 18058; doi: 10.1038/srep18058 (2015).
References
Baize, S. et al. Emergence of Zaire Ebola virus disease in Guinea. New Engl. Med. J. 371, 1418–1425 (2014).
Agostini, H. T., Yanagihara, R., Davis, V., Ryschkewitsch, C. F. & Stoner, G. L. Asian genotypes of JC virus in Native Americans and in a Pacific island population: Markers of viral evolution and human migration. Proc. Natl Acad. Sci USA 94, 14542–14546 (1997).
Falush, D. et al. Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585 (2003).
Monot, M. et al. On the origin of leprosy. Science 308, 1040–1042 (2005).
Moodley, Y. et al. The peopling of the pacific from a bacterial perspective. Science 323, 527–530 (2009).
Comas, I. et al. Out-of-Africa migration and Neolithic co-expansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182 (2013).
McGarvey, S. T., Zhou, X. N., Willingham, A. L., Feng, Z. & Olveda, R. The epidemiology and host-parasite relationships of Schistosoma japonicum in definitive hosts. Parasitol. Today 15, 214–215 (1999).
McManus, D. P. et al. Schistosomiasis in the People's Republic of China: the Era of the Three Gorges Dam. Clin. Microbiol. Rev. 23, 442–466 (2010).
Gryseels, B., Polman, K., Clerinx, J. & Kestens, L. Human schistosomiasis. Lancet 368, 1106–1118 (2006).
Zhao, Q. P., Sen Jiang, M., Dong, H. F. & Nie, P. Diversification of Schistosoma japonicum in mainland China revealed by mitochondrial DNA. Plos Neglect. Trop. Dis. 6 (2012).
Shrivastava, J., Qian, B. Z., McVean, G. & Webster, J. P. An insight into the genetic variation of Schistosoma japonicum in mainland China using DNA microsatellite markers. Mol. Ecol. 14, 839–849 (2005).
Hsu, H. F. & Hsu, S. Y. L. Schistosoma japonicum in formosa - a crtical review. Exp. Parasitol. 12, 459–465 (1962).
Zhao, G.-H. et al. A specific PCR assay for the identification and differentiation of Schistosoma japonicum geographical isolates in mainland China based on analysis of mitochondrial genome sequences. Infect. Genet. Evol. 12, 1027–1036 020 (2012).
Attwood, S. W., Fatih, F. A. & Upatham, E. S. DNA-sequence variation among Schistosoma mekongi populations and related taxa; phylogeography and the current distribution of Asian Schistosomiasis. Plos Neglect. Trop. Dis. 2, (2008).
Lawton, S. P., Hirai, H., Ironside, J. E., Johnston, D. A. & Rollinson, D. Genomes and geography: genomic insights into the evolution and phylogeography of the genus Schistosoma. Parasit Vectors 4, (2011).
Attwood, S. W., Ibaraki, M., Saitoh, Y., Nihei, N. & Janies, D. A. Comparative phylogenetic studies on Schistosoma japonicum and its snail intermediate host Oncomelania hupensis: Origin, dispersal and coevolution. Plos Neglect. Trop. Dis. 9, (2015).
Smith, K. F. & Guegan, J.-F. Changing geographic distributions of human pathogens. Ann. Rev. Ecol. Evol. Systematics 41, 231–250 (2010).
Hershberg, R. et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6, 2658–2671 (2008).
Wen, B. et al. Genetic evidence supports demic diffusion of Han culture. Nature 431, 302–305 (2004).
Xue, F. et al. A spatial analysis of genetic structure of human populations in China reveals distinct difference between maternal and paternal lineages. Eur. Hum. Genet. 16, 705–717 (2008).
Tumonggor, M. K. et al. The Indonesian archipelago: an ancient genetic highway linking Asia and the Pacific. J. Hum. Genet. 58, 165–173 (2013).
Xu, S. et al. Genetic dating indicates that the Asian-Papuan admixture through Eastern Indonesia corresponds to the Austronesian expansion. Proc. Natl Acad. Sci USA 109, 4574–4579 (2012).
Diamond, J. & Bellwood, P. Farmers and their languages: The first expansions. Science 300, 597–603 (2003).
Molina, J. et al. Molecular evidence for a single evolutionary origin of domesticated rice. Proc. Natl Acad. Sci USA 108, 8351–8356 (2011).
Callaway, E. Domestication : The birth of rice. Nature 514, S58–S59 (2014).
He, S. The rice road: the origin and development of rice cultivation in China and Southeast Asia. Orient Forum. 5, (2013).
Zhao, Q. P., Sen Jiang, M., Littlewood, D. T. J. & Nie, P. Distinct genetic diversity of Oncomelania hupensis, intermediate host of Schistosoma japonicum in mainland China as revealed by ITS sequences. Plos Neglect. Trop. Dis. 4, (2010).
Liu, Y., Lou, T., Wang, Y. & Zhang, W. Subspecies difference of oncomelaniid snails. Acta zootaxon sin. 6, 253–267 (1981).
Zhou, X. N. Population genetics morphology and distribution of Oncomelania hupensis, intermediate host of Schistosoma japonicum in mainland China. PhD thesis, University of Copenhagen, (1994).
Saijuntha, W. et al. Genetic structure inferred from mitochondrial 12S ribosomal RNA sequence of Oncomelania quadrasi, the intermediate snail host of Schistosoma japonicum in the Philippines. Am. J. Trop. Med. Hyg. 90, 1140–1145 (2014).
Moose, J. W. & Williams, J. E. Suseptibility of Oncomelania formosana from three different areas of Taiwan to infection with formosan strain of Schistosoma japonicum. J. Parasitol. 49, 702–703 (1963).
Gignoux, C. R., Henn, B. M. & Mountain, J. L. Rapid, global demographic expansions after the origins of agriculture. Proc. Natl Acad. Sci USA 108, 6044–6049 (2011).
Stiner, M. C. Thirty years on the "Broad Spectrum Revolution" and paleolithic demography. Proc. Natl Acad. Sci USA 98, 6993–6996 (2001).
Achilli, A. et al. The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS ONE 4, (2009).
Suzuki, H. et al. Evolutionary and dispersal history of Eurasian house mice Mus musculus clarified by more extensive geographic sampling of mitochondrial DNA. Heredity 111, 375–390 (2013).
Yin, M. B. et al. Multiple near-identical genotypes of Schistosoma japonicum can occur in snails and have implications for population-genetic analyses. Int. J. Parasitol. 38 (2008).
Cronn, R. et al. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 36, (2008).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The genome analysis toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 20, 1297–1303 (2010).
Zheng, H.-X. et al. Major population expansion of East Asians began before Neolithic Time: evidence of mtDNA genomes. PLoS ONE 6 (2011).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Guindon, S. & Gascuel, O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUTI and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Kang, L. L. et al. mtDNA lineage expansions in Sherpa population suggest adaptive evolution in Tibetan highlands. Mol. Biol. Evol. 30, 2579–2587 (2013).
Zheng, H.-X., Yan, S., Qin, Z.-D. & Jin, L. MtDNA analysis of global populations support that major population expansions began before Neolithic Time. Sci. Rep. 2 (2012).
Acknowledgements
We thank Prof. Hiroshi Yamasaki from National Insistue of Infectious Diseases of Japan for giving us the Japanese strain samples, Prof. Hong Ma, Hui Li from Fudan University and Prof. Yungang He from CAS-MPG Partner Institute for the discussion and critical comments. This research was supported by National Natural Science Foundation of China (No. 91431104, 31271338 and 31401062) and National Science and Technology Major Project (No. 2012ZX10004-220).
Author information
Authors and Affiliations
Contributions
M.Y., L.J. and W.H. designed the study. J.S., Z.F. and X.Z. collected samples and carried out the genomes sequencing. M.Y. and H.Z. contributed to data analyses and M.Y., H.Z., D.M., L.J. and W.H. wrote the manuscript. All authors read and approved the final version.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yin, M., Zheng, HX., Su, J. et al. Co-dispersal of the blood fluke Schistosoma japonicum and Homo sapiens in the Neolithic Age. Sci Rep 5, 18058 (2016). https://doi.org/10.1038/srep18058
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18058
This article is cited by
-
Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale
Scientific Reports (2020)
-
Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements
Parasites & Vectors (2018)
-
Three new Diplozoidae mitogenomes expose unusual compositional biases within the Monogenea class: implications for phylogenetic studies
BMC Evolutionary Biology (2018)
-
Temporal genetic diversity of Schistosoma japonicum in two endemic sites in China revealed by microsatellite markers
Parasites & Vectors (2016)
-
Cloning and in vitro characterization of a Schistosoma japonicum aquaglyceroporin that functions in osmoregulation
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
