
Abstract
Breast cancer susceptibility gene 1 (BRCA1) located at chromosome 17q12-21 is a classic tumor suppressor gene and has been considered as a significant role in hereditary breast cancers. Moreover, numerous studies demonstrated the methylation status of CpG islands in the promoter regions of BRCA1 gene was aberrant in patients with sporadic breast tumors compared with healthy females or patients with benign diseases. However, these conclusions were not always consistent. Hence, a meta-analysis was performed to get a more precise estimate for these associations. Crude odds ratio with 95% confidence interval were used to assess the association of BRCA1 promoter methylation and the risk or clinicopathologic characteristics of breast cancers under fixed or random effect model. A total of 40 studies were eligible for this present study. We observed the frequency of BRCA1 promoter methylation was statistically significant higher in breast cancers than non-cancer controls. Furthermore, BRCA1 methylation was statistically associated with lymph node metastasis, histological grade 3, ER(-), PR(-), triple-negative phenotype and decreased or lack levels of BRCA1 protein expression. In conclusion, this study indicated that BRCA1 promoter methylation appeared to be a useful predictive or prognostic biomarker for breast cancers in clinical assessment.
Similar content being viewed by others

Introduction
Breast cancer is one of the common malignant tumors for females worldwide. As we know that breast carcinomas belong to a heterogeneous group of tumors, not only for multiple biological behaviors, but also for various therapeutic responses and clinical outcomes. Though the exact pathogenic mechanism is not fully understood as yet, researchers have been demonstrating that multiple factors are involved in the initiation and progression of breast tumors, such as germline mutation and epigenetic alteration. DNA promoter methylation, as one of main ways of epigenetic alteration, has been investigated for breast cancer detection, prognosis and treatment1. There is mounting evidence that methylation status of CpG islands in cancer-related genes promoters, especially tumor suppressor genes, is distinct in breast cancer patients compared with healthy women or patients with benign breast disease2,3,4. For instance, the BRCA1 promoter was prone to methylate in peripheral blood DNA of sporadic breast cancer patients compared with unaffected controls5.
Breast cancer susceptibility gene 1 (BRCA1) located at chromosome 17q12-21 is a classic tumor suppressor gene6 and plays a crucial role in the processes of DNA repair, homologous recombination, checkpoint control of cell cycle and transcription7. Germline mutations of BRCA1 account for 30–40% hereditary breast carcinomas8. However, somatic mutations of BRCA1 are rare in sporadic cases of breast cancer9,10,11. In addition, there are some evidences that lack or low expression of BRCA1 protein is involved in the development of sporadic breast tumors12,13. Furthermore, it’s also noteworthy that several studies have shown a significant association between reduced expression of BRCA1 protein and aberrant methylation status of CpG islands in the BRCA1 promoter6,12,13,14, which indicates that promoter methylation may lead to transcriptional inactivation of BRCA1 gene and contribute to breast carcinogenesis.
Over past few years, the prevalence of the hypermethylated BRCA1 promoter in sporadic breast cancers has been reported to fall in the range from 5.2% to 65.2%13,15. Iwamoto et al. showed that BRCA1 promoter methylation in peripheral blood cells was correlated with elevated risk of BRCA1-methylated breast cancers16. Interestingly, the methylation frequencies of BRCA1 promoter were different in breast cancer tissues, paired adjacent normal tissues and peripheral blood cells derived from breast cancers and unaffected women17. Furthermore, during the past decade, a considerable amount of studies have been going to elucidate the association between BRCA1 promoter methylation and clinicopathological characteristics of breast cancer18,19. However, the research results are not always consistent. Therefore, we performed a meta-analysis to evaluate whether BRCA1 gene promoter methylation is a risk factor for sporadic breast cancers and elucidated the association of BRCA1 promoter methylation with clinicopathological characteristics in patients with breast cancer.
Materials and Methods
Literature search strategy
We performed a comprehensive literature search from PubMed and EMBASE database (last search updated in August 2015) without language restrictions. The following search terms were used, (“BRCA1” or “Breast cancer susceptibility gene 1”) and (“methylation” or “DNA methylation” or “promoter methylation”) and (“breast cancer” or “breast carcinoma” or “breast tumor” “breast carcinogenesis”). In addition, we carried out a manual search for other relevant articles via the reference lists of eligible studies.
Selection criteria
Eligible studies had to meet the following predefined criteria, (1) case-control studies evaluating the association between the prevalence of BRCA1 promoter methylation and sporadic breast cancer risk; or clinical cohort studies evaluating the associations of BRCA1 promoter methylation with clinicopathological features of sporadic breast cancer; (2) sufficient published data for calculating an odds ratio (OR) and 95% confidence interval (CI); (3) studies were confined to human female groups. It’s noteworthy that if the same study population was included in several different studies, we would only bring the most recent or comprehensive study into the meta-analysis.
Data extraction
A standard protocol was applied to extract data. For every eligible study, the following data were extracted: the first author’s name, publication year, original country, methods for detecting methylation, the frequency of BRCA1 promoter methylation in case and control groups, control characteristics, sample materials and so on.
Statistical methods
The strength of the association between the BRCA1 promoter methylation and sporadic breast cancer risk or clinicopathological features was assessed by OR with corresponding 95%CI. A chi-square-based Q test was applied to test heterogeneity among studies. The p value of the Q test was >0.1, which suggested a lack of statistically significant heterogeneity and we used the fixed-effect model (Mantel-Haenszel method)20 to calculate pooled ORs. Otherwise, heterogeneity was present and the random-effect model (DerSimonian-Laird method)21 was more appropriate. Additionally, the degree of heterogeneity was also quantitatively assessed by I2-test, which the value of I2 ranged from 0 to 100% and was generally considered mild heterogeneity for I2 < 25%, moderate heterogeneity for 25%–50%, large heterogeneity for 50%–75% and extreme heterogeneity for I2 > 75%22. Moreover, stratified-analyses were conducted based on ethnicity, methods for detecting methylation and sample materials to explore the potential source of heterogeneity. Sensitivity analyses, by which each study was omitted in each turn to confirm the influence of individual data set to the pooled OR, were implied to evaluate the robustness of the results. Furthermore, we estimated potential publication bias with funnel plot and Egger’s linear regression test. The funnel plot was visual symmetrical and the P-value of Egger’s test was greater than 0.05, which indicated that there were no statistically significant publication bias. All statistical tests in the meta-analysis were two-tailed and P-value  was considered statistically significant unless otherwise noted. Statistical analyses were performed with Review Manager 5.2 software recommended by Cochrane Collaboration and STATA software version 12.0.
was considered statistically significant unless otherwise noted. Statistical analyses were performed with Review Manager 5.2 software recommended by Cochrane Collaboration and STATA software version 12.0.
Results
Study Characteristics
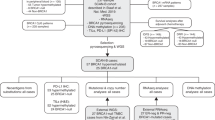
Based on the above selection criteria, 20 case-control studies , involving 2747 cases and 2256 controls2,3,4,5,6,10,15,16,17,23,24,25,26,27,28,29,30,31,32,33, were included to analyze the association between BRCA1 promoter methylation and sporadic breast cancer risk. Among these studies, 12 studies4,6,10,15,17,23,24,25,26,27,28,32 confirmed the status of BRCA1 methylation in tissues derived from breast carcinoma, benign disease or normal breast epithelium. And the objects of 9 studies2,3,5,16,17,29,30,31,33 were the prevalence of BRCA1 methylation in peripheral blood of breast cancer patients compared with cancer-free or healthy females. It’s worth noting that a study17 detected BRCA1 methylation in tissues of tumor and normal breast epithelium and peripheral blood from breast cancer patients and healthy women. Due to the different type of sample materials, we considered this study as two case-control studies. Furthermore, 30 clinical studies3,4,6,12,13,14,16,18,27,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49 with a total of 5058 breast cancer patients met our selection criteria for analyzing the association between BRCA1 promoter methylation and clinicopathological characteristics which included early age (<50 years) at diagnosis, premenopausal status, lymph node metastasis, histological grade 3, estrogen receptor (ER), progesterone receptor (PR), human epidermal receptor 2 (Her2), triple-negative phenotype and the expression of BRCA1 protein. In short, our meta-analysis included 40 eligible articles, among which 20 articles were analyzed for the frequency of BRCA1 promoter methylation in breast cancers compared with non-cancer controls and 30 articles were analyzed for the association between BRCA1 promoter methylation and clinicopathological features. What was noteworthy was that 10 articles3,4,6,16,27,29,30,31,32,33 not only studied the prevalence of BRCA1 promoter methylation, but also elaborated clinicopathological characteristics in breast cancer patients with BRCA1 promoter methylation versus BRCA1-unmethylated tumors. The flow diagram of study selection procedure was shown in Fig. 1. Every study characteristics were summarized in Tables 1 and 2.

Flow diagram of study selection in this meta-analysis.
Meta-analysis results
Association between BRCA1 promoter methylation and the risk of breast cancer
In general, our study indicated that the frequency of BRCA1 promoter methylation was statistically significant elevated in breast cancers compared with non-cancer controls (OR = 3.15, 95%CI 1.97–5.03, P < 0.001, Fig. 2). Because of large heterogeneity (PH < 0.001, I2 = 74%), we explored the potential source of heterogeneity via stratified analysis based on sample materials, methods for detecting methylation and ethnicity. In the subgroup analysis about sample materials, the pooled OR for BRCA1 methylation in breast cancer tissues compared with normal or benign tissues was 4.75 (95%CI 2.37–9.54, P < 0.001) and was higher than the pooled OR (OR = 1.87, 95%CI 1.19–2.96, P = 0.007) in peripheral blood of breast cancers compared with non-cancer controls. Furthermore, the frequency of BRCA1 methylation by MSP method (OR = 6.79, 95%CI 3.05–15.11, P < 0.001) was significantly higher than other methods (OR = 1.53, 95%CI 1.09–2.14, P = 0.01). Meanwhile, the prevalence of BRCA1 methylation in Asians (OR = 4.03, 95%CI 1.07–15.18, P = 0.04) was higher than that in Caucasians (OR = 3.16, 95%CI 1.78–5.62, P < 0.001) and in Australians (OR = 3.27, 95%CI 1.37–7.84, P = 0.008) in breast cancers compared with non-cancer controls. It’s worth mentioning that the degree of heterogeneity was apparently reduced in stratified analysis. The detailed results were summarized in Table 3.

Forest plot for evaluating the association between BRCA1 promoter methylation and breast cancer risk.
Random- effect model was used for the analysis.
Association of BRCA1 promoter methylation with clinicopathological features of breast cancer
The results of our meta-analysis showed that the BRCA1 promoter methylation was statistically significant correlated with lymph node metastasis (OR = 1.25, 95%CI 1.06–1.48, P = 0.009, Fig. 3) and histological grade 3 (OR = 2.29, 95%CI 1.65–3.18, P < 0.001, Fig. 4), but had no correlation with early age (<50 years) at diagnosis (OR = 1.21, 95%CI 0.98–1.50, P = 0.07, Fig. 5) or premenopausal status (OR = 1.21, 95%CI 0.98–1.50, P = 0.08, Fig. 6). As for hormone receptors, strong associations of BRCA1 methylation were found with ER negative (OR = 2.36, 95%CI 1.67–3.33, P < 0.001, Fig. 7) and also with PR negative (OR = 2.14, 95%CI 1.49–3.07, P < 0.001, Fig. 8). In contrast, no association was found between BRCA1 methylation and Her2 status (OR = 1.58, 95%CI 0.98–2.56, P = 0.06, Fig. 9). Interestingly, our study confirmed that the BRCA1 promoter methylation was positively correlated with triple-negative phenotype (OR = 2.79, 95%CI 1.74–4.48, P < 0.001, Fig. 10). Furthermore, it’s worth mentioning that there was statistically significant correlation between BRCA1 methylation and decreased or lack expression levels of BRCA1 protein (OR = 4.44, 95%CI 2.56–7.70, P < 0.001, Fig. 11). The detailed results were summarized in Table 4.

Forest plot for evaluating the association between BRCA1 promoter methylation and lymph node metastasis.
Fix-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and histological grade 3.
Random-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and early age (<50 years) at diagnosis.
Fix-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and premenopausal status.
Fix-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and ER negative.
Random-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and PR negative.
Random-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and Her2 negative.
Random-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and triple-negative phenotype.
Random-effect model was used for the analysis.

Forest plot for evaluating the association between BRCA1 promoter methylation and decreased or lack levels of BRCA1 protein expression.
Random-effect model was used for the analysis.
Sensitivity analyses and publication bias
We performed sensitivity analyses to assess the robustness of the meta–analysis results by omitting each study in turn and no single study could essentially change the results. And the sensitivity analyses demonstrated that the results of our meta-analysis were statistically stable. Figure 12 showed the plot of sensitivity analysis for evaluating the association between BRCA1 promoter methylation and breast cancer risk. The shapes of funnel plots and Egger’s linear regression test were used to evaluate the publication bias of the eligible literatures. In general, the funnel plots were not entirely symmetrical and the P-value of Egger’s test was not always greater than 0.05, indicating there was publication bias in our study. The funnel plot for evaluating the association of BRCA1 promoter methylation with breast cancer risk was shown in Fig. 13 and the detailed results for P-value of Egger’s test were summarily in Table 3 and Table 4.

The plot of sensitivity analysis for evaluating the association between BRCA1 promoter methylation and breast cancer risk.

The funnel plot for evaluating the association of BRCA1 promoter methylation with breast cancer risk.
Discussion
As is well-known that breast carcinoma is a heterogeneous group of tumors. It’s essential to find a reliable biomarker for the early diagnosis and prognosis prediction of breast cancer. In the meta-analysis, the strength of relationships of BRCA1 promoter methylation with breast cancer risk and its clinicopathological features were systematically investigated. The results of our study confirmed that BRCA1 promoter methylation was significantly correlated with the increased risk of breast cancer and associated with lymph node metastasis, histological grade 3, ER(-), PR(-), triple-negative phenotype and BRCA1 protein expression, which indicated that BRCA1 promoter methylation may be utilized as an effective biomarker in the management of breast tumors.
Our meta-analysis demonstrated that the prevalence of BRCA1 promoter methylation in the breast cancer group was statistically significant elevated in comparison with the control group. This suggested a possibility that aberrant methylation of BRCA1 promoter was correlated with an increased risk of breast cancer. And this almost was in line with the report by Wong et al. which confirmed that BRCA1 methylation in peripheral blood DNA was correlated with 3.5-fold evaluated risk of having early-onset breast tumors31. In the stratification analysis based on sample materials, the summary OR was 4.75 in tissues and 1.87 in peripheral bloods, indicating that the association of BRCA1 methylation with breast cancer risk in tissues was stronger than in peripheral bloods. Because there was only one study26 in which the tissue of the control group was derived from patients with benign breast diseases, we omitted the study to evaluate the frequency of BRCA1 promoter methylation in breast cancer tissues compared with normal breast tissues and the pooled OR was 7.23 (95%CI 4.35–12.01, P < 0.00001), which showed that the frequency of BRCA1 methylation in breast carcinoma tissues was 7.23-fold higher than that in normal breast tissues. The result demonstrated that the difference for the frequency of BRCA1 methylation between breast tumors and non-cancerous breast tissues group was smaller than that between cancers and normal breast tissues group. Therefore, it suggested that normal breast tissues had a lower prevalence of BRCA1 methylation than benign and malignant breast tissues, which also implied that the methylation of BRCA1 gene promoter may play a certain role in the initiation of breast carcinoma. Similarly, a recent research confirmed that the BRCA1 promoter methylation of histological normal breast epithelial cells may result in BRCA1-methylated breast tumors47. Additionally, our study demonstrated that the methylation of BRCA1 promoter in peripheral blood DNA was correlated with a 1.87-fold increased risk of breast cancer, which was accordance with the result of a previous study16. The way of extracting DNA from peripheral blood is simple and barely invasive for detecting the methylation of BRCA1 promoter. Therefore, the finding could open a new avenue for screening the risk of breast cancer.
In the subgroup analysis based on ethnicity, there were 15 studies in Caucasians, 4 in Asians and 2 in Australians for the association of BRCA1 methylation with breast cancer risk. Meanwhile, significant difference in the level of BRCA1 methylation was found in Caucasians (OR = 3.16), Asians (OR = 4.03) and Australians (OR = 3.27) in the cancer group compared with the control group, which suggested that different ethnicity and environment had certain impact on the prevalence of BRCA1 methylation. Additionally, the results of stratified analysis based on methods showed that the different methods had different efficiency for detecting methylation. It’s essential to find an appropriate method based on feasibility of clinical practice in order to apply BRCA1 methylation as a biomarker in clinical management.
In our meta-analysis, 18 articles discussed the association of BRCA1 methylation with age at diagnosis in breast cancers. However, it’s meaningless to extract data from these articles to calculate the pooled OR and 95%CI on account of the difference in the definition of early age among these studies. Nevertheless, due to 10 articles considering early age as less than 50 years, we then investigated the correlation between BRCA1 methylation and early age (<50 years) at diagnosis. However, there was no statistically significant association. In addition, no association between BRCA1 methylation and premenopausal status was observed in our study. This was inconsistent with a previous report43 which showed that BRCA1-methylated breast cancers tended to occur at an early age (<50 years) and were more frequently observed in premenopausal or perimenopausal women than postmenopausal women. Furthermore, our results showed that BRCA1 promoter methylation was strongly related to breast cancer patients with high histological grade and lymph node metastasis, revealing that aberrant methylation of BRCA1 promoter may be implicated in the invasion and metastasis of breast cancer. In this sense, a report investigated the methylation profiles of 12 genes in the matched axillary lymph nodes compared with primary tumor tissues and the adjacent normal tissues from the same breast cancer patients and demonstrated that the proportion of BRCA1 methylation was higher in the matched axillary lymph nodes metastasis than normal tissue50. Thus, a hypothesis may be proposed that the methylation status of BRCA1 promoter may be served as a biomarker for screening metastasis in breast tumors.
As expected from previous studies14,29,43, we demonstrated that there was a remarkable correlation between BRCA1 promoter hypermethylation and breast tumors with lack of ER and PR expression. However, no inverse relationship was found between BRCA1 methylation and Her2 status. Interestingly, it’s noteworthy that the triple negative phenotype was more common in BRCA1-methylated breast cancers than unmethylated tumors, which also was in agreement with numerous reports18,32,44,45. Altogether, these indicate that the patients with BRCA1-methylated breast tumors are likely to have little benefit from traditional hormone or targeted therapies. Additionally, a considerable amount of researches investigated that hypermethylation of BRCA1 promoter resulted in the down-regulation of BRCA1 expression12,13,14. Likewise, a statistically significant association of BRCA1 methylation with lack or decreased expression of BRCA1 protein was confirmed in our study. It’s interesting to note that there were positive and negative methylation-expression relationships in diverse gene regions, which differently affected genes expression and prognosis in breast cancer subtypes51. Therefore, it is reasonably predicted that methylation of BRCA1 different promoter region and distinct region of BRCA1 gene play different role in the BRCA1 expression and prognosis in breast tumors and this needs further study.
Taken together, the clinicopathological features in sporadic BRCA1-methylated breast cancers compared with BRCA1-unmethylated tumors in our meta-analysis showed that sporadic breast carcinomas with BRCA1 promoter methylation had molecular and clinicopathologic phenotype similar to those of hereditary BRCA1-mutated breast cancers, which was in line with several reports27,37. Furthermore, several lines of evidences confirmed that the expression pattern of sporadic BRCA1-methylated breast cancers was the same as that of inherited BRCA1 mutations52. Herein, numerous preclinical researches investigated whether the antitumor activity of DNA-damaging agents in BRCA1-mutated breast cancers had a similar activity in BRCA1-methylated tumors and the results demonstrated that the BRCA1 hypermethylation conferred the same extent of sensitivity to poly adeno-sine diphosphate-ribose polymerase-1 (PARP1) inhibitors and platinum-derived drugs as did the BRCA1 mutation53,54,55. Moreover, Xu et al. reported that BRCA1-methylated triple-negative breast tumor patients were sensitive to adjuvant chemotherapy and had a significantly better 10-year disease-free survival (DFS) and disease-specific survival (DSS) than patients with BRCA1-unmethylated triple-negative tumors46. Importantly, a recent meta-analysis including 9 studies with 3205 breast cancer patients indicated that there was significant association of BRCA1 methylation with poor overall survival and DFS of breast tumors56. Hence, BRCA1 promoter methylation may be a potential biomarker for targeted therapy and prognostic assessment.
Despite the advantage of a considerable number of included literatures, our meta-analysis had some limitations that should be thought over. First and most importantly, there was large heterogeneity in the outcome of the association between BRCA1 promoter methylation and breast cancer risk. Nevertheless, stratification analyses based on sample materials, methods for detecting methylation or ethnicities showed to reduce the degree of heterogeneity among studies, which demonstrated that the three factors may be contributed to the heterogeneity. Additionally, there was large heterogeneity among studies for the correlation of BRCA1 methylation with ER status, PR status and triple negative phenotype. The expression of ER and PR protein were almost assayed by immunohistochemical staining, but different antibody source and dilution ratio or even cut-off value for result evaluation should be taken into account for the source of heterogeneity. On the other hand, our included studies did not illustrate specific promoter regions of BRCA1 gene for methylation detection and whether this may cause heterogeneity need to be further studied. Second, it’s noteworthy that there were no related articles for the prevalence of BRCA1 promoter methylation in breast cancers in the African population among our eligible literatures. Therefore, further research is needed to evaluate whether this section of our results may be consistent with studies for the African ethnicity. Third, the control groups included healthy females and patients with benign breast diseases. No uniform definition of control groups may, to some extent, partly affected our research results. Finally, there was apparent publication bias in our study, which may be generated by defective design method of small sample studies or absent publication of small trials with negative results. In addition, language bias may be present on the basis of the fact that only English articles were included for our meta-analysis.
In conclusion, this meta-analysis indicated that BRCA1 promoter methylation was associated with an increased risk of breast cancer. The prevalence of BRCA1 methylation was, especially in mammary tissue, was high in patients with breast cancers compared with healthy females or patients with benign breast diseases. Furthermore, there were significant associations between BRCA1 promoter methylation and clinicopathological characteristics in breast tumors, such as lymph node metastasis, high histological grade, ER-negative, PR-negative, triple-negative phenotype and reduced or lack expression of BRCA1 protein. It’s necessary to need large-scale researches which use uniform criterion of control groups, detection methods for methylation and sample materials, before BRCA1 promoter methylation can be a useful predictive or diagnostic biomarker for patients with breast cancer and applied to novel targeted therapeutic strategies in the future.
Additional Information
How to cite this article: Zhang, L. and Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 5, 17869; doi: 10.1038/srep17869 (2015).
References
Dworkin, A. M. et al. Epigenetic alterations in the breast, implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol 19, 165–71 (2009).
Jing, F. et al. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumor Biol 31, 321–31 (2010).
Sturgeon, S. R. et al. Detection of promoter methylation of tumor suppressor genes in serum DNA of breast cancer cases and benign breast disease controls. Epigenetics 7, 1258–67 (2012).
Jung, E. J. et al. Comparison of methylation profiling in cancerous and their corresponding normal tissues from Korean patients with breast cancer. Ann Lab Med 33, 431–40 (2013).
Bosviel, R. et al. BRCA1 promoter methylation in peripheral blood DNA was identified in sporadic breast cancer and controls. Cancer Epidemiol 36, 177–82 (2012).
Hasan, T. N. et al. Association of BRCA1 promoter methylation with rs11655505 (c.2265C>T) variants and decreased gene expression in sporadic breast cancer. Clin Transl Oncol 15, 555–62 (2013).
Venkitaraman, A. R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108, 171–82 (2002).
Lux, M. P. et al. Hereditary breast and ovarian cancer, review and future perspectives. J Mol Med 84, 16–28 (2006).
Khoo, U. S. et al. Somatic mutations in the BRCA1 gene in Chinese sporadic breast and ovarian cancer. Oncogene 18, 4643–6 (1999).
Esteller, M. et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92, 564–9 (2000).
Turner, N. et al. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4, 814–9 (2004).
Alkam, Y. et al. Protein expression and methylation of DNA repair genes hMLH1, hMSH2, MGMT and BRCA1 and their correlation with clinicopathological parameters and prognosis in basal-like breast cancer. Histopathology 63, 713–25 (2013).
Ignatov, T. et al. BRCA1 promoter methylation is a marker of better response to anthracycline-based therapy in sporadic TNBC. Breast Cancer Res Treat 141, 205–12 (2013).
Bal, A. et al. BRCA1-methylated sporadic breast cancers are BRCA-like in showing a basal phenotype and absence of ER expression. Virchows Arch 461, 305–12 (2012).
Buyru, N. et al. Methylation profiles in breast cancer. Cancer Invest 27, 307–12 (2009).
Iwamoto, T. et al. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat 129, 69–77 (2011).
Cho, Y. H. et al. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res 30, 2489–2496 (2010).
Hsu, N. C. et al. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PLoS One 8, e56256 (2013).
Xu, X. et al. BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat 115, 397–404 (2009).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22, 719–48 (1959).
DerSimonian, R. & Laird, N. Meta-analysis in clinical trials. Control Clin Trials 7, 177–188 (1986).
Higgins, J. P. et al. Measuring inconsistency in meta-analyses. BMJ 327, 557–60 (2003).
Dobrovic, A. & Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res 57, 3347–50 (1997).
Chen, C. M. et al. Methylation target array for rapid analysis of CpG island hypermethylation in multiple tissue genomes. Am J Pathol 163, 37–45 (2003).
Jerónimo, C. et al. Detection of gene promoter hypermethylation in fine needle washings from breast lesions. Clin Cancer Res 9, 3413–7 (2003).
Parrella, P. et al. Nonrandom distribution of aberrant promoter methylation of cancer-related genes in sporadic breast tumors. Clin Cancer Res 10, 5349–54 (2004).
Wei, M. et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res 65, 10692–9 (2005).
Bhavani, V. et al. Role of BRCA1, HSD17B1 and HSD17B2 methylation in breast cancer tissue. Cancer Biomark 5, 207–13 (2009).
Sharma, G. et al. Clinical significance of promoter hypermethylation of DNA repair genes in tumor and serum DNA in invasive ductal breast carcinoma patients. Life Sci 87, 83–91 (2010).
Al-Moghrabi, N. et al. Methylation-related mutations in the BRCA1 promoter in peripheral blood cells from cancer-free women. Int J Oncol 39, 129–35 (2011).
Wong, E. M. et al. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res (Phila) 4, 23–33 (2011).
Ben Gacem, R. et al. Contribution of epigenetic alteration of BRCA1 and BRCA2 genes in breast carcinomas in Tunisian patients. Cancer Epidemiol 36, 190–7 (2012).
Gupta, S. et al. Methylation of the BRCA1 promoter in peripheral blood DNA is associated with triple-negative and medullary breast cancer. Breast Cancer Res Treat 148, 615–22 (2014).
Catteau, A. et al. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer, correlation with disease characteristics. Oncogene 18, 1957–65 (1999).
Niwa, Y. et al. BRCA1 expression status in relation to DNA methylation of the BRCA1 promoter region in sporadic breast cancers. Jpn J Cancer Res 91, 519–26 (2000).
Miyamoto, K. et al. Promoter hypermethylation and post-transcriptional mechanisms for reduced BRCA1 immunoreactivity in sporadic human breast cancers. Jpn J Clin Oncol 32, 79–84 (2002).
Birgisdottir, V. et al. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res 8, R38 (2006).
Jing, F. et al. Multigene methylation in serum of sporadic Chinese female breast cancer patients as a prognostic biomarker. Oncology 75, 60–6 (2008).
Chen, Y. et al. BRCA1 promoter methylation associated with poor survival in Chinese patients with sporadic breast cancer. Cancer Sci 100, 1663–7 (2009).
Galizia, E. et al. BRCA1 expression in triple negative sporadic breast cancers. Anal Quant Cytol Histol 32, 24–9 (2010).
Xu, X. et al. Gene promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat 121, 685–92 (2010).
Saelee, P. et al. Clinicopathological Significance of BRCA1 Promoter Hypermethylation in Thai Breast Cancer Patients. Asian Pac J Cancer Prev 15, 10585–9 (2014).
Singh, A. K. et al. Epigenetic silencing of BRCA1 gene associated with demographic and pathologic factors in sporadic breast cancer, a study of an Indian population. Eur J Cancer Prev 20, 478–83 (2011).
Stefansson, O. A. et al. CpG island hypermethylation of BRCA1 and loss of pRb as co-occurring events in basal/triple-negative breast cancer. Epigenetics 6, 638–49 (2011).
Jacot, W. et al. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer 13, 523 (2013).
Xu, Y. et al. Promoter methylation of BRCA1 in triple-negative breast cancer predicts sensitivity to adjuvant chemotherapy. Ann Oncol 24, 1498–505 (2013).
Otani, Y. et al. BRCA1 promoter methylation of normal breast epithelial cells as a possible precursor for BRCA1-methylated breast cancer. Cancer Sci 105, 1369–76 (2014).
Yamashita, N. et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin Breast Cancer S1526–8209, 00125–1 (2015).
Zhu, X. et al. Hypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancer. Breast Cancer Res Treat 150, 479–486 (2015).
Barekati, Z. et al. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer 12, 244 (2012).
Győrffy, B. et al. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int J Cancer 138, 87–97 (2015).
Hedenfalk, I. et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med 344, 539–48 (2001).
Veeck, J. et al. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J Clin Oncol 28, e563–4 (2010).
Stefansson, O. A. et al. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics 7, 1225–9 (2012).
Cai, F. et al. Pyrosequencing analysis of BRCA1 methylation level in breast cancer cells. Tumour Biol 35, 3839–44 (2014).
Wu, L. et al. Promoter methylation of BRCA1 in the prognosis of breast cancer: a meta-analysis. Breast Cancer Res Treat 142, 619–27 (2013).
Acknowledgements
The Project-sponsored by SRF for ROCS, SEM. This work was supported by grants from National Natural Science Foundation of China (nos. 30873044 and 81272372).
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, L., Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci Rep 5, 17869 (2016). https://doi.org/10.1038/srep17869
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17869
This article is cited by
-
Contribution of constitutional BRCA1 promoter methylation to early-onset and familial breast cancer patients from Pakistan
Breast Cancer Research and Treatment (2023)
-
Identifying the BRCA1 c.-107A > T variant in Dutch patients with a tumor BRCA1 promoter hypermethylation
Familial Cancer (2023)
-
A phase 1 study of veliparib (ABT-888) plus weekly carboplatin and paclitaxel in advanced solid malignancies, with an expansion cohort in triple negative breast cancer (TNBC) (ETCTN 8620)
Breast Cancer Research and Treatment (2023)
-
Methylation as a critical epigenetic process during tumor progressions among Iranian population: an overview
Genes and Environment (2021)
-
Loss of BRCA1 expression and morphological features associated with BRCA1 promoter methylation status in triple-negative breast cancer
Journal of Human Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
