
Abstract
Germanium is an extremely important material used for numerous functional applications in many fields of nanotechnology. In this paper, we study the crystallization of amorphous Ge using atomistic simulations of critical nano-metric nuclei at high temperatures. We find that crystallization occurs by the recurrent transfer of atoms via a diffusive process from the amorphous phase into suitably-oriented crystalline layers. We accompany our simulations with a comprehensive thermodynamic and kinetic analysis of the growth process, which explains the energy balance and the interfacial growth velocities governing grain growth. For the 〈111〉 crystallographic orientation, we find a degenerate atomic rearrangement process, with two zero-energy modes corresponding to a perfect crystalline structure and the formation of a Σ3 twin boundary. Continued growth in this direction results in the development a twin network, in contrast with all other growth orientations, where the crystal grows defect-free. This particular mechanism of crystallization from amorphous phases is also observed during solid-phase epitaxial growth of 〈111〉 semiconductor crystals, where growth is restrained to one dimension. We calculate the equivalent X-ray diffraction pattern of the obtained nanotwin networks, providing grounds for experimental validation.
Similar content being viewed by others


Introduction
Growth of semiconductor crystals from glassy or vapor phases is an extremely important process for many applications in nano technology1,2. In general, crystallization from a disordered structure is ultimately a diffusive process3,4 and –as such– strongly temperature dependent. Growth, however, is highly susceptible to the formation of crystal defects, which can be copious, mediated by imperfections, both related to the environmental variables of the physico-chemical treatment5 and to structural heterogeneities associated with the substrate, such as impurities, lattice mismatch, etc.6. Defect formation is typically also temperature dependent and thus a compromise must be found to balance reasonable growth rates while keeping acceptably-low defect concentrations. Since the early times of solid-phase crystallization, great emphasis has been placed on suppressing the emergence of these imperfections7,8, as many properties of crystallized systems strongly depend on achieving pristine structures and a defect-free finish9,10.
Among the panoply of possible defects found during crystal growth, growth twins stand out as one of the more prolific ones due to low formation energies and a variety of possible genesis pathways6,11,12. In this work we are concerned with twinning in diamond-cubic structures, such as Si and Ge, whose growth, solidification and recrystallization have been studied extensively6,7,13,14. It is believed that twinning may appear as a consequence of internal transformations to rotate the crystal structure towards energetically favorable interfacial orientations12,15. However, twinning is also observed during epitaxial recrystallization in Si, Ge and their alloys, particularly along the 〈111〉 growth direction16. The chief difficulty in studying twin nucleation in these systems is that it is generally impossible to see them form in situ. Several authors have hypothesized twins form at a very early stage, when the crystal nucleus is extremely small. However, twinned overgrowth may also be observed forming on a nucleus of relatively-large size11,15. In both of these cases, atomistic simulation suggests itself as the ideal avenue to unravel the nature of twin nucleation and growth.
Here we report molecular dynamics simulations of Ge recrystallization from amorphous substructures at high homologous temperatures (fraction of the melting temperature Tm). Our simulations are framed within the study of amorphization-crystallization (a → c) processes in laser spot heating of GST-based* phase-changing materials (PCMs)17,18. In GST materials, the main role of Ge is to accelerate the recrystallization process and thus here we study pure Ge as the point of reference for Ge-based PCMs19. We start from a critical nucleus of crystalline Ge (c-Ge) embedded in an amorphous (a-Ge) medium at an initial temperature of T0 = 1100 K, which is representative of the conditions found within the laser spot. We find that grain growth is characterized by the formation of intricate twin networks facilitated by near zero surface and stacking fault energies20. We accompany our simulations with a full thermodynamic analysis to explain the mechanisms behind the observed behavior.
Simulation details
Molecular dynamics simulations
We use a Stillinger-Weber potential parameterized by Posselt and Gabriel for Ge20, which reproduces the experimental values for the cohesive energy and lattice constant for the diamond-cubic structure and yields reasonable values for the energetics of other crystalline phases and the structure of the liquid. Our simulations are run in the isobaric-isenthalpic ensemble NpH–where N is the number of particles, p is the pressure and H is the enthalpy– using periodic boundary conditions in three dimensions. The NpH ensemble was chosen to allow for local temperature increases due to latent heat release during the crystallization process. To simulate the effect of laser spot heating, the system is equilibrated to an initial temperature of T0 = 1100 K, which is approximately the temperature at the center of the spot18. The starting configuration is generated from a perfect diamond structure with lattice constant a0 = 0.5654 nm, corresponding to the value at 1100 K, oriented along the [100], [011] and  directions. The simulation comprises 62a1 × 44a2 × 44a3 supercells (≈35 nm per side, where a1 = a0,
directions. The simulation comprises 62a1 × 44a2 × 44a3 supercells (≈35 nm per side, where a1 = a0,  ) containing 1,920,512 atoms. A central spherical region of radius of 2 nm (the nucleus) is then kept frozen, while the outer region is melted by fixing the temperature at 3000 K using a Langevin thermostat during 100 ps and then quenched. At the same time the NpH ensemble maintains zero pressure globally. Finally, the entire system (the a-Ge block containing the c-Ge nucleus) is further equilibrated at 1100 K during an additional 10 ps. The thermostat is then turned off during the subsequent crystallization simulations. We have shown that this results in a glass transition temperature of approximately 810 K, in good agreement with laboratory experiments for Ge21. The procedure just described was used by the authors22 to generate planar a/c interfaces to calculate free energies and interface mobilities.
) containing 1,920,512 atoms. A central spherical region of radius of 2 nm (the nucleus) is then kept frozen, while the outer region is melted by fixing the temperature at 3000 K using a Langevin thermostat during 100 ps and then quenched. At the same time the NpH ensemble maintains zero pressure globally. Finally, the entire system (the a-Ge block containing the c-Ge nucleus) is further equilibrated at 1100 K during an additional 10 ps. The thermostat is then turned off during the subsequent crystallization simulations. We have shown that this results in a glass transition temperature of approximately 810 K, in good agreement with laboratory experiments for Ge21. The procedure just described was used by the authors22 to generate planar a/c interfaces to calculate free energies and interface mobilities.
Calculation of the critical nucleus size
In classical nucleation theory, the critical nucleus size r* is governed by the balance between the volumetric and interfacial driving forces expressed, respectively, as the derivative of the net free energy release ΔG0,a→c and a surface energy penalty ΔGs with respect to the radius of the nucleus. In principle, this balance must also account for the expansion of Ge upon crystallization, which is approximately 10% less dense than its amorphous counterpart (cf. Fig. 3b in ref. 22) at p = 0 and T0 = 1100 K. However, the procedure detailed in the previous section to seed an amorphous matrix with crystalline grains removes any differential strains by construction. This allows us to write:

Temperature dependence of Δg0,a→c expressed on a per atom basis (referred to the left vertical axis).
Also shown are the internal energies u of both amorphous and crystalline Ge as a function of temperature (right vertical axis). The heat capacity Cp is calculated from the slope of u(T), which for c-Ge results in a value of ≈2.69 × 10−4 eV · K−1 per atom (≈0.36 J · g−1 · K−1), in excellent agreement with experimental measurements47.

Surface free energy as a function of temperature for the three surface normals representing the vertices of the standard triangle.

(Free) energy landscape governing the crystallization process.

where Δg0,a→c is the volumetric free energy density at zero pressure and γ is the surface (free) energy density, which is orientation dependent: γ ≡ γ(θ), with θ representing the surface normal with respect to the crystal orientation. We have calculated the atomic free energy densities g0,a and g0,c of the amorphous and crystalline phases using thermodynamic integration, see ref. 22 for details. The variation of Δg0,a→c = g0,a − g0,c with temperature in units of energy per atom is provided in Fig. 1. When this difference is zero, there is phase coexistence, which by definition occurs at the melting point, here Tm = 1350 K. The driving force per unit volume for the a → c transformation at zero pressure is readily obtained as: Δg0,a→c = ρcΔg0,a→c. From the figure, at T0 = 1100 K, Δg0,a→c = −0.073 eV per atom, while the atomic density of the crystalline phase at the same temperature is ρc = 4.36 × 1028 m−3 (after ref. 22). From this, Δg0,a→c ≈ −5.07 × 108 J · m−3.
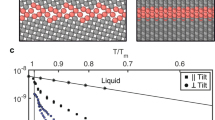
As mentioned earlier, ΔGs is orientation dependent. However, crystalline Ge displays cubic symmetry, which allows us to reduce the orientation space to that contained in the standard stereographic triangle whose vertices in the first octant are the intersects of the unit sphere with the [001], [110] and [111] directions22. Thus, we restrict our study of the orientation dependence of γ(θ) to those three orientations#. The interfacial free energies are shown in Fig. 2 as a function of temperature, where a surface orientation anisotropy can be clearly distinguished at low temperatures. At T0 however, this anisotropy is smeared out by the high thermal diffusivity of the amorphous phase above the glass transition temperature, cf. Fig. 2 in ref. 22 and we find an orientation-independent value of γ ≈ 0.08 J · m−2.
The critical grain size is found by minimizing eq. (1): dΔG/dr = 0, which results in

Replacing γ and Δg0,a→c for their respective values, we obtain that r* ≈ 0.32 nm. This value is approximately 55% of the magnitude of the lattice constant a0 and suggests stable crystalline grains with only a handful of atoms in them, probably implying that very localized fluctuations suffice to produce crystal growth seeds.
Interface mobility
The growth rate of the crystalline phase at the expense of the amorphous phase is governed by the schematic energy landscape shown in Fig. 3. The excess atomic flux a → c relative to c → a transitions is governed by Δg0,a→c (shown in the figure) and results in net interface velocity v(T) and grain growth. Mathematically, this can be expressed to first order as4,23:

where v0 is a prefactor and EB is an activation energy for the transformation (shown in the figure). EB represents the energy for the detachment/reattachment process, which is diffusive in nature. We have devised a special procedure to calculate EB, for which a value of 0.42 eV was obtained for the [100] orientation22 at 0 K. A low value of EB may result in faster growth speeds at low temperatures, but it also results in faster detachment (a ← c) rates at higher temperatures, where the difference of free energies decreases, resulting in lower effective growth speeds.
The interface velocities can also be obtained by direct atomistic simulation as described by Reina et al.22. Figure 4 shows results for four distinct orientations at T0 with the 〈111〉 being the slowest one, which ultimately controls grain growth.

Interface velocity as a function of surface orientation at the initial (1100 K) and final (1250 K) simulation temperatures.
Simulation results
Next we present the simulations of supercritical grain growth at T0. While the critical radius calculated earlier suggests a very small stability threshold, we have found that –in practice– a minimum radius of 2 nm was needed to have positive grain growth on the timescales captured in the MD simulations. The discrepancy can be attributed to a number of factors, chief of which is the magnitude and frequency of thermal fluctuations at these high temperatures and small volumes, which lead to low signal-to-noise ratios in terms of the stable critical size. Other factors such as nonsphericity and finite size effects, may also play a non-negligible role. Consequently, in the following we show results of 2-nm radius supercritical c-Ge nuclei in an amorphous medium.
Three-dimensional growth of critical grains
In a 3D (spherical) nucleus, in principle all growth orientations are sampled, which means that in materials with sizable interface energy anisotropies and/or interface velocities, some growth directions will be preferred over others. A complete animation of the grain growth process starting from (super)critical nuclei is provided in the Supporting Information. A snapshot of the simulation at the point of maximum growth –which occurs 9.1 ns after the system is equilibrated at T0– is shown in Fig. 5. Atoms in the image are colored using structure analysis as implemented in the OVITO visualization package24, which assigns dark blue to atoms with diamond cubic structure and orange to atoms with hexagonal diamond crystal structure.

Structure of the crystalline grain after 9.1 ns of growth in the NpH ensemble.
(a) Atomistic structure: Only atoms possessing an ordered crystallographic structure are shown. Dark blue and orange spheres represent atoms with diamond cubic and twin plate structure, respectively. Light blue spheres represent atoms belonging to the amorphous/crystalline interface and dislocation cores. The circular region in the center of the image indicates the extent of the critical grain at the beginning of the simulation. (b) Through-thickness view of the nanotwin structure corresponding to (a).
Closer examination of the atoms with hexagonal diamond structure reveals that they belong to 〈111〉 twin (Σ3) boundaries, typical of the diamond cubic lattice structure. Figure 6 shows a region around one such boundary in local detail, where the mirror symmetry characteristic of twin plates can be clearly identified. Moreover, using boundary analysis available in OVITO, we have examined the atomistic structures in Figs 5(a) and 6 and find the emergence of a network of twinned regions, as showcased in Fig. 5(b). The figure shows a through-thickness view of the entire grain at the exact same time as Fig. 5(a). As depicted, in the lower half of the grain twins are elongated along 〈211〉 directions, while in the upper half a more or less three-dimensional arrangement is formed.

 view of the atomistic configuration of a twinned region.
view of the atomistic configuration of a twinned region.
As discussed earlier, grain growth is controlled by the magnitude of the driving force and the interface mobility. Both of these quantities are temperature dependent. The exothermic nature of the a → c reaction (i.e. internal energy density difference Δua→c < 0) results in a local energy deposition that increases the global system temperature. Thus, the temperature of the system correlates directly with the volume of material transformed. Such correlation is clearly visible in Fig. 7, where both quantities are exactly proportional to each other with a proportionality constant of ≈8.01 × 10−27 m3 K−1. The temperature is seen to increase from T0 to a final value of approximately 1250 K. This effectively arrests the growth process, as dictated by the sharp decrease in mobility at such temperature (cf. Fig. 4). We remark that this arrest is partially an artifact of the simulations which limits unrestricted heat flow due to periodic boundary effects.

Evolution of the temperature with time during growth of a critical nucleus under Nph simulations conditions.
The crystallized volume is also shown as a function of time. Selected snapshots of the grain structure corresponding to three distinct instants (2, 5 and 8 ns) are shown.
Analysis of growth of a/c bicrystals
The appearance of twins during the growth stage of critical Ge grains may obey the energetics of two different scenarios. On the one hand, there is ample evidence in the literature that in Ge twinning emerges if the growth orientation deviates appreciably from the preferred growth direction, understood as that which results in the lowest interfacial energy. Twins would then appear to alter the internal crystal orientation and bring it closer to the preferred one9,25. However, other works have pointed out that if a new facial orientation was the only advantage gained by twinning, twinned crystals should not be much larger than twice the size of a single crystal, something at odds with observations of twinned crystals being more than ten times as large as untwinned ones15. As well, this hypothesis is strongly weakened by the lack of a noticeable surface energy anisotropy at 1100 K according to our calculations (cf. Fig. 2).
The alternative scenario is that twins are a manifestation of a growth mode that relies on the indistinct formation of ordered atomic layers with the correct stacking sequence and stacking faults. This is the same growth mode observed under the so-called solid-phase epitaxial recrystallization (SPER) process of {111}-oriented crystals, as well as by liquid epitaxial growth of crystals with the same orientation via chemo-physical vapor deposition. There is ample evidence of twin formation in the literature for both of these processes in Ge, particularly at high temperatures26,27,28. This mechanism is controlled by the value of the stacking fault energy γSF†, which ranges between 0.07 and 0.09 J · m−2 according to several measurements29,30,31. By contrast, the interatomic potential employed in our simulations predicts zero stacking fault energy20. Evidently then, the model for Ge employed here offers no impediment to the favorable formation of epitaxial twins. However, while at low temperatures this might clearly result in an overestimation of the volume fraction of stacking faults and/or twins when conditions are conducive to their formation32,33, it is reasonable to assume that values of γSF on the order of the experimentally-measured ones result in zero effective stacking fault energy at a temperature of 1100 K via thermal softening.
To ascertain which mechanism is responsible for the observed formation of nanotwin networks, next we carry out MD simulations of a/c bi-crystals at T0 oriented along three selected directions: [111] (low mobility, cf. Figs 2 and 4) and [100] and [110] (high mobility). These are qualitatively similar to other simulations of the SPER process using atomistic methods34,35,36. The three surface orientations simulated here are schematically shown in Fig. 8 relative to a  view of the Ge diamond cubic lattice.
view of the Ge diamond cubic lattice.

Schematic view along the  direction of the Ge diamond cubic lattice, with the
direction of the Ge diamond cubic lattice, with the  plane highlighted.
plane highlighted.
Image obtained with Wolfram CDF Player48.
[111] amorphous/crystalline bi-crystals
The starting microstructures (a/c bi-crystals) are generated in the manner described in ref. 22. The computational cell has dimensions of 20.8 × 19.4 × 39.2 nm containing 698,880 atoms. The system is again equilibrated at 1100 K and let to evolve in the NpH ensemble. Two animations illustrating the process are provided in the Supporting Information. Growth of the crystalline phase proceeds via the formation of an intricate twin network, an image of which is shown in Fig. 9. Twin boundaries are shown as green-colored surfaces and are seen to form a quasi-hexagonal network as dictated by the topological structure of a set of interconnected {111} surfaces. The structures are reminiscent of coral-like porous networks in synthesized ceramic materials37.

Twinning network formed from the growth of a crystalline Ge half-crystal along the [111] direction.
[100] and [110] amorphous/crystalline bi-crystals
The size of the computational cells employed to study growth along the [100] and [110] directions was 20.4 × 20.4 × 39.6 nm with 725,760 atoms and 20.4 × 20.8 × 40.0 nm with 748,800 atoms, respectively. As shown in the corresponding animations (Supporting Information), crystallization along these directions results in growth of a homogeneous Ge crystal, forming essentially no defects. Here, atom rearrangements from the amorphous into the crystalline phase occurs by forming atomic planes with the correct stacking sequence.
XRD analysis
An important part of the analysis of the simulations is their experimental validation. Ge crystals can be examined by a variety of sources, from transmission electron microscopy (TEM), to Raman spectroscopy (RS) and X-ray diffraction analysis (XRD). However, twin boundaries are not sources of strain and are thus difficult to detect via conventional TEM analysis. In contrast, they act as scattering agents to X-rays and do leave an imprint on diffraction patterns. Therefore, we have calculated the equivalent XRD signature for pure crystalline and amorphous samples, as well as for the multitwinned structure shown in Fig. 9, using the Debyer code38 considering a X-ray source with wave length of 1.542 Å at 0 K. The resulting pattern is shown in Fig. 10, where the intensity peaks represent the different scattering directions. The figure reveals clear differences in the footprints of the three structures considered, namely, no structure for the amorphous system, well marked peaks for the ideal crystal and softened peaks for the twinned crystal. The XRD pattern showed in the figure is in excellent agreement with experimental results for pure crystalline Ge but only in modest agreement for amorphous Ge39. This may indicate that the generated amorphous structures may not be fully optimized in terms of their atomic configuration, likely a result of using unphysically-high heating and cooling rates to entrap a liquid structure into a disordered solid. With regard to multitwinned structures, Fig. 10 provides a pathway for their detection in future experiments.

Diffraction patterns for amorphous, perfect crystal and twinned Germanium.
The atomic structures were minimized before being analyzed with Debyer38 considering a X-ray source with wave length of 1.542 Å.
Discussion and Conclusions
Although it is clear from the literature that twinned Ge crystals may emerge during crystallization at low temperatures to favor low energy interface orientations (and thus decrease the critical nucleus size), our simulations conclusively show that the origin of the twinning network observed during crystallization of Ge grains from amorphous structures at T0 lies in the energetic degeneracy observed for the stacking sequence of {111} planes. Although this effect is favored by construction in our simulations (due to a zero stacking fault energy predicted by our atomic model), the overall effect of γSF in materials such as Ge at these high temperatures is likely to be negligible in any case. The result is the spontaneous formations of multiply twinned structures along each of the three equivalent 〈111〉 directions.
The other notable observation is that grain growth at 1100 K is controlled by low interface mobilities. This together with a small critical radius for stable crystalline nuclei, suggests the development of nano crystalline or very fine-grained structures, as is indeed the case experimentally. The reason for this is that 1100 K is near the tipping point where the mobility sharply decreases from its maximum value. This is compounded by latent heat deposition released during the exothermic a → c process, which increases the temperature beyond that tipping point. The phenomenon where crystallization is fueled by the intrinsic latent heat release is well known and referred to as explosive crystallization40,41,42. For this, however, an increase in temperature should result in growth acceleration by a surge in interface velocity, which is not the case in the temperature regime considered here. In all, high nucleation rates due to small critical radii, slow mobilities due to high temperatures, plus high-twinning propensities result in the notoriously fine-grained nanostructures reported for laser-induced Ge crystallization18,41,42,43. We have recently proposed a thermodynamically-consistent phase field model to predict these microstructures22. However, intrinsic twinning was not a feature of those simulations and we believe that the present atomistic simulations provide a new piece of physics that must be incorporated into such higher-level models.
Twin boundaries may also act as scattering agents for elastic and electromagnetic waves and may impact the value of fundamental constants such as the thermal conductivity or electric susceptibility. Indeed, it has been observed that the appearance of twins during epitaxial growth of Si wafers resulted in faulty devices, while for other defects, such as e.g. extrinsic stacking faults, it did not44,45,46 (albeit perfect Ge crystals have been grown in the [111] direction as well33). This may be of importance in GST materials where high contrast between amorphous and crystalline phases in terms of these properties is desired.
Additional Information
How to cite this article: Sandoval, L. et al. Formation of Nanotwin Networks during High-Temperature Crystallization of Amorphous Germanium. Sci. Rep. 5, 17251; doi: 10.1038/srep17251 (2015).
References
Greene, J. A review of recent research on the growth and physical properties of single crystal metastable elemental and alloy semiconductors. Journal of Vacuum Science & Technology B 1, 229–237 (1983).
Shay, J. L. & Wernick, J. H. Ternary Chalcopyrite Semiconductors: Growth, Electronic Properties and Applications: International Series of Monographs in The Science of The Solid State vol. 7 (Elsevier, 2013).
Jonsson, H. Theoretical studies of atomic-scale processes relevant to crystal growth. Annu. Rev. Phys. Chem. 51, 623 (2000).
Bourgoin, J. C. & Asomoza, R. Solid phase growth of silicon and germanium. J. Cryst. Growth 69, 489 (1984).
Hurle, D. & Rudolph, P. A brief history of defect formation, segregation, faceting and twinning in melt-grown semiconductors. Journal of crystal growth 264, 550–564 (2004).
Drosd, R. & Washburn, J. Some observations on the amorphous to crystalline transformation in silicon. J. Appl. Phys. 53, 397 (1982).
Brown, R. A. Theory of transport processes in single crystal growth from the melt. AICHE J. 34, 881 (1988).
Dash, W. C. Growth of silicon crystals free from dislocations. J. Appl. Phys. 30, 459 (1959).
Kohn, J. A. Twinning in diamond-type structures: A proposed boundary-structure model. Am. Mineral. 43, 263 (1958).
Hwang, C.-W., Ryu, M.-K., Kim, K.-B., Lee, S.-C. & Kim, C.-S. Solid phase crystallization of amorphous si1- xgex films deposited on sio2 by molecular beam epitaxy. J. Appl. Phys. 77, 3042 (1995).
Donnelly, T. W. Kinetic considerations in the genesis of growth twinning. Am. Mineral. 52, 1 (1967).
Bolling, G. F., Rutter, J. W. & Tiller, W. A. Growth twins in germanium. Can. J. Phys. 34, 234 (1956).
Billig, E. Growth of monocrystals of germanium from an undercooled melt. In P. Roy. Soc. Lond. A Mat. vol. 229, 346–363 (The Royal Society, 1955).
Cullis, A. G. & Booker, G. R. The epitaxial growth of silicon and germanium films on (111) silicon surfaces using uhv sublimation and evaporation techniques. J. Cryst. Growth 9, 132 (1971).
Carstens, H. Kinetic considerations in the genesis of growth twinning: A discussion. Am. Mineral. 53, 342 (1968).
Darby, B. L. et al. Substrate orientation dependence on the solid phase epitaxial growth rate of ge. J. Appl. Phys. 113, 033505 (2013).
Nikolova, L. et al. Nanocrystallization of amorphous germanium films observed with nanosecond temporal resolution. Appl. Phys. Lett. 97, 203102 (2010).
Nikolova, L. et al. Complex crystallization dynamics in amorphous germanium observed with dynamic transmission electron microscopy. Phys. Rev. B 87, 064105 (2013).
Wuttig, M. & Salinga, M. Phase-change materials: Fast transformers. Nat. Mater. 11, 270 (2012).
Posselt, M. & Gabriel, A. Atomistic simulation of amorphous germanium and its solid phase epitaxial recrystallization. Phys. Rev. B 80, 045202 (2009).
Angell, C. A. The amorphous state equivalent of crystallization: new glass types by first order transition from liquids, crystals and biopolymers. Solid State Sci. 2, 791 (2000).
Reina, C., Sandoval, L. & Marian, J. Mesoscale computational study of the nanocrystallization of amorphous ge via a self-consistent atomistic phase-field model. Acta Mater. 77, 335 (2014).
Liu, J., Xu, X., Brush, L. & Anantram, M. A multi-scale analysis of the crystallization of amorphous germanium telluride using ab initio simulations and classical crystallization theory. Journal of Applied Physics 115, 023513 (2014).
Stukowski, A. Visualization and analysis of atomistic simulation data with ovito - the open visualization tool. Modelling Simul. Mater. Sci. Eng. 18, 015012 (2010).
Billig, E. Growth twins in crystals of low co-ordination number. J. Inst. Metals. 83, 53 (1954).
Claeys, C. & Simoen, E. Extended Defects in Germanium: Fundamental and Technological Aspects (Springer Science & Business Media, 2008).
Darby, B. L. Amorphization and solid phase epitaxial growth of germanium. Ph.D. thesis, University of Florida (2012).
Capper, P. & Mauk, M. (eds.) Liquid Phase Epitaxy of Electronic, Optical and Optoelectronic Materials (John Wiley & Sons, 2007).
Art, A., Aerts, E., Delavignette, P. & Amelinckx, S. The Stacking-Fault Energy in Germanium. Appl. Phys. Lett. 2, 40 (1963).
Gomez, A., Cockayne, D. J. H., Hirsch, P. B. & Vitek, V. Dissociation of near-screw dislocations in germanium and silicon. Philos. Mag. 31, 105 (1975).
Denteneer, P. J. H. & van Haeringen, W. Stacking-fault energies in semiconductors from first-principles calculations. J. Phys. C: Solid State 20, L883 (1987).
Biswas, S., Singh, A., Morris, M. A. & Holmes, J. D. Inherent control of growth, morphology and defect formation in germanium nanowires. Nano Lett. 12, 5654 (2012).
Nguyen, V. H., Dobbie, A., Myronov, M. & Leadley, D. R. High quality relaxed germanium layers grown on (110) and (111) silicon substrates with reduced stacking fault formation. J. Appl. Phys. 114, 154306 (2013).
Roland, C. & Gilmer, G. H. Growth of germanium films on si(001) substrates. Phys. Rev. B 47, 16286 (1993).
Akis, R. & Ferry, F. Kinetic monte-carlo simulations of germanium epitaxial growth on silicon. J. Comput. Electron. 5, 451 (2006).
Martin-Bragado, I. Importance of twin defect formation created by solid-phase epitaxial growth: An atomistic study. Scripta Mater. 66, 186 (2012).
Caruso, R. A., Giersig, M., Willig, F. & Antonietti, M. Langmuir 14, 6333 (1998).
Debyer: software for calculation of diffraction patterns for atomistic models. https://code.google.com/p/debyer/wiki/debyer.
Chockla, A. M. & Korgel, B. A. Seeded germanium nanowire synthesis in solution. J. Mater. Chem. 19, 996 (2009).
Leamy, H. J. et al. Explosive crystallization of amorphous germanium. Appl. Phys. Lett. 38, 137 (1981).
Grigoropoulos, C., Rogers, M., Ko, S. H., Golovin, A. A. & Matkowsky, B. J. Explosive crystallization in the presence of melting. Phys. Rev. B 73, 184125 (2006).
Nikolova, L. et al. In situ investigation of explosive crystallization in a-ge: Experimental determination of the interface response function using dynamic transmission electron microscopy. J. Appl. Phys. 116, 093512 (2014).
Kuo, C.-C. Characterization of polycrystalline ge thin films fabricated by short-pulse xef excimer laser crystallization. J. Russ. Laser Res. 29, 167 (2008).
Lee, M. L., Antoniadis, D. A. & Fitzgerald, E. A. Challenges in epitaxial growth of sige buffers on si (111), (110) and (112). Thin Solid Films 508, 136 (2006).
Vlek, L. & Sik, J. Defect Engineering During Czochralski Crystal Growth and Silicon Wafer Manufacturing, Modern Aspects of Bulk Crystal and (Intech, 2012).
Zhuang, H., Zhang, L., Fuchs, R., Staedler, T. & Jiang, X. When epitaxy meets plasma: a path to ordered nanosheets arrays. Sci. Rep. 3 (2013).
Okhotin, A. S., Pushkarskii, A. S. & Gorbachev, V. V. Thermophysical Properties of Semiconductors (“Atom” Publ. House, Moscow, 1972). In Russian.
Wolfram CDF Player (Wolfram Research, Inc., Champaign, IL, 2014), version 10.0 edn.
Acknowledgements
We thank the DTEM group at Lawrence Livermore National Laboratory for useful suggestions and guidance. This work was partially performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory and Los Alamos National Laboratory. LLNL is operated by Lawrence Livermore National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE, under contract DE-AC52-07NA27344. LANL is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE, under contract DE-AC52-O6NA25396.
Author information
Authors and Affiliations
Contributions
L.S. and J.M. designed the numerical simulations and free energy calculations. L.S. carried out the simulations and post-processed the data. J.M. analysed the results and wrote the first version of the manuscript. C.R. helped write the paper, reviewed the results and provided theoretical interpretation of the data. All authors discussed and approved the conclusions and reviewed the submitted manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sandoval, L., Reina, C. & Marian, J. Formation of Nanotwin Networks during High-Temperature Crystallization of Amorphous Germanium. Sci Rep 5, 17251 (2015). https://doi.org/10.1038/srep17251
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17251
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
