
Abstract
Members of the Mitis group of streptococci possess teichoic acids (TAs) as integral components of their cell wall that are unique among Gram-positive bacteria. Both, lipoteichoic (LTA) and wall teichoic acid, are formed by the same biosynthetic pathway, are of high complexity and contain phosphorylcholine (P-Cho) residues. These residues serve as anchors for choline-binding proteins (CBPs), some of which have been identified as virulence factors of the human pathogen Streptococcus pneumoniae. We investigated the LTA structure of its close relative Streptococcus oralis. Our analysis revealed that S. oralis Uo5 LTA has an overall architecture similar to pneumococcal LTA (pnLTA) and can be considered as a subtype of type IV LTA. Its structural complexity is even higher than that of pnLTA and its composition differs in number and type of carbohydrate moieties, inter-residue connectivities and especially the P-Cho substitution pattern. Here, we report the occurrence of a saccharide moiety substituted with two P-Cho residues, which is unique as yet in bacterial derived surface carbohydrates. Finally, we could link the observed important structural variations between S. oralis and S. pneumoniae LTA to the divergent enzymatic repertoire for their TA biosynthesis.
Similar content being viewed by others


Introduction
Peptidoglycan, wall teichoic acids (WTA) and lipoteichoic acids (LTA) are the major polysaccharides of the Gram-positive cell wall. The teichoic acids (TAs) of Streptococcus pneumoniae are unique in comparison to TAs of many other Gram-positive bacteria in several structural aspects. The composition of pneumococcal LTA (pnLTA) is the most complex of all LTAs investigated so far1. Pneumococcal WTA (pnWTA) and pnLTA exhibit identical structures within their repeating units (RUs)2 and are both decorated with phosphorylcholine (P-Cho) moieties3,4. This substituent is present only in cell walls of a few Gram-positive bacteria like Streptococcus oralis, Streptococcus mitis, Streptococcus pseudopneumoniae and Streptococcus infantis5,6,7,8,9, which are close relatives of S. pneumoniae. These species belong to the Mitis group of streptococci10, which mainly consists of commensals that reside in the oral cavity and the nasopharynx of humans. Only S. pneumoniae is frequently associated with diseases such as otitis media, pneumonia and meningitis, whereas e.g. S. mitis and S. oralis rarely cause disease such as endocarditis11,12. The choline-containing TAs of S. pneumoniae anchor choline-binding proteins (CBPs), an important family of cell surface proteins, at the cell wall and are involved in the interaction with host cells. TAs have further been described to be involved in processes like the regulation of cell wall hydrolases, the regulation of cell wall elongation and cell division, cation homoeostasis or the resistance to lysozymes and antimicrobial peptides13,14. Some CBPs—which are associated with the choline moiety of TAs by non-covalent interactions—have been identified as virulence factors specific for S. pneumoniae15,16. Therefore, the elucidation of structural differences between TAs of the pathogen versus those of the commensal species are of great importance.
We describe the comprehensive structural analysis of S. oralis Uo5 LTA, for which we combined different methodologies such as chemical degradations, high-resolution mass spectrometry (MS) as well as one- and two-dimensional, homo- and heteronuclear nuclear magnetic resonance (NMR) spectroscopy.
Results
As starting material for our structural analysis, we used LTA of S. oralis Uo5Δcps, which has been isolated following our previously published workflow17. The use of an unencapsulated variant is beneficial in terms of LTA extraction and yield. The final purification was performed with hydrophobic interaction chromatography (HIC) and fractions containing phosphate18 (Fig. S1A) were combined and lyophilized. In a first step, we analysed the components of S. oralis Uo5Δcps LTA and generated defined part structures by hydrofluoric acid (HF) treatment to investigate the structure of its lipid anchor and its de-phosphorylated RU. Afterwards, we analysed the interconnection of the RUs as well as the presence and nature of phosphate-containing residues using de-O-acylated LTA obtained after hydrazine treatment. Additional ester-bound substituents were finally identified using the intact LTA.
Compositional analysis
D-galactose (D-Galp), D-glucose (D-Glcp), N-acetyl-D-galactosamine (D-GalpNAc), 2-acetamido-4-amino-2,4,6-trideoxygalactose (AATGalp), ribitol (Rib-ol), glycerol (Gro), phosphate (P) and alanine (Ala) have been identified as constituents of S. oralis Uo5Δcps LTA. The fatty acid analysis revealed the presence of 16:0, 16:1, 18:0 and 18:1 acids in an approximate ratio of 5.4 : 1.0 : 4.1 : 1.1, together with traces of 14:0 acid.
Preparation and structural analysis of the lipid anchor and LTA part structures
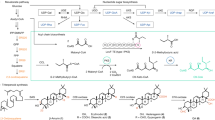
To elucidate the nature of the lipid anchor we treated the isolated LTA with 48% HF for two days at 4 °C. This usually cleaves all phosphodiester bonds and leads to the formation of monomerized polysaccharide (monoPS) units as well as a diacyl-glycerol (DAG) with all carbohydrate residues up to the first phosphate in the TA chain. In S. pneumoniae this procedure results in the formation of 119 (Fig. 1), whereas only 2 (Fig. 1) represents the biological lipid anchor. The other residues belong to the first RU of the pnLTA17,20. Therefore, 1 has to be considered as a lipid anchor-containing trisaccharide-DAG (tsDAG). Additionally, the monoPS units generated this way cannot be considered as the de-phosphorylated carbohydrate backbone of the biological RU. For pnLTA HF treatment leads mainly to pseudo-pentasaccharide 319,20 (Fig. 1), whereas the biological RU of pnLTA corresponds to 417,20 (Fig. 1). The HF treated LTA of S. oralis Uo5Δcps was applied to HIC (Fig. S1B) as described for the purification of intact LTA (Fig. S1A). At early retention time (18.5 to 33.5 min; fractions 7–11 in Fig. S1B) the monomerized LTA repeats (monoPS units; 6a, 6b, 7a and 7b in Fig. 1) and other fatty acid-free part structures were eluted. To collect the lipid anchor-containing tsDAG of S. oralis Uo5Δcps (5; Fig. 1), fractions between 78.5 and 102.5 min (fractions 27–34 in Fig. S1B) were combined. The mass spectrum of this latter pool is depicted in Fig. S2. The presence of lyso-tsDAG (respective monoacyl variants of 5) was interpreted as a side product caused by the long HF treatment, which is required to generate the monoPS units in sufficient yield. After 8 h HF treatment (without HIC purification), the MS analysis almost exclusively displayed signals of intact 5 (calculated masses depending on the fatty acids composition: 1076.690 Da, 1104.722 Da and 1130.737 Da; Fig. S3). However, the monoPS unit formation is insufficient under these conditions. The exact structure of the carbohydrate part of S. oralis Uo5Δcps tsDAG (5; Fig. 1) was analyzed by NMR (Table S1) and was similar to the pneumococcal tsDAG (1), but with a β-D-Galp-moiety instead of the β-D-Glcp-moiety. The combined and lyophilized fractions of the early eluate of the HIC purification (fractions 7–11; Fig. S1B) were subsequently submitted to gel permeation chromatography (Bio-Gel P-10). A section of the chromatogram is shown in Fig. S4. All pools have been investigated separately by MS, with only P-10 pools 1 and 2 containing LTA derived polysaccharides. The completely dephosphorylated monoPS units 7a and 7b were identified in P-10 pool 2 (Fig. S4), the corresponding mass spectrum is shown in Fig. S5 and the assigned peaks are listed in Table S2. Very interestingly, in P-10 pool 1 (Fig. S4) monoPS units 6a and 6b (Fig. 1) with one P-Cho residue still present were identified. The corresponding mass spectrum is shown in Fig. 2 and the assigned peaks are listed in Table S2. The respective 31P NMR spectrum of P-10 pool 1 (Fig. 3A) provides proof for a homogeneous preparation comprising one specific P-Cho residue that has not been cleaved off. A section of the 1H,13C-HSQC NMR spectrum is depicted in Fig. 3B, the 1H and 13C NMR chemical shift data for 6a and 6b are summarized in Table S3. Some minor signals (labeled with * in Fig. 3B) are originating from 7a and 7b, mainly eluting in P-10 pool 2 (Fig. S4). Integration of suitable 1H NMR signals revealed a ratio of 6a : 6b of approximately 0.3 : 0.7. This result was in good agreement with the MS analysis, in which these two forms have been observed as well (Fig. 2), with 6b as the predominant species. A similar ratio has been observed for 7a and 7b (Fig. S5) as well. Furthermore, an additional acetyl or alanine group has been observed, whereas the acetyl substitution seemed to be present predominantly in the molecules containing two galactosyl residues (6b, 7b). However, since HF treatment can cleave off these substituents and acidic catalyzed rearrangements are possible, the presence and position of these residues has to be analysed using native LTA and will be discussed later.

LTA part structures of S. pneumoniae and S. oralis Uo5.
Compilation of the structures of the respective lipid anchor-containing trisaccharide-DAG (tsDAG; 1 and 5, both including lipid anchor 2; R = fatty acid) and monomerized LTA repeats (monoPS units; 3, 6a,b and 7a,b) isolated after HF treatment, as well as observed repeating units in hydrazine treated or native LTA (4, 8 and 9a,b).

Section of the charge deconvoluted ESI-FT-ICR-MS spectra (acquired in positive ion mode) of 6a,b obtained after 2 d HF treatment of LTA of S. oralis Uo5∆cps and subsequent purification by HIC and GPC (P-10 pool 1 in Fig. S4).
Besides the major molecule 6b with a MWfound of 1030.408 Da (MWcalc: 1030.403 Da), a second compound with one hexose less (6a; MWfound: 868.356 Da; MWcalc: 868.353 Da) was observed in minor amounts as well. For both molecules, variants with a bound alanine or acetyl residue could be identified with very small signal intensity.

NMR analysis of 6a,b obtained after 2 d HF treatment of LTA of S. oralis Uo5∆cps and subsequent purification by HIC and GPC (P-10 pool 1 in Fig. S4; mass spectrum shown in Fig. 2).
(A) Section (δP 5-(−5)) of the 31P NMR. (B) The respective 1H,13C-HSQC NMR spectrum (δH 5.40–3.00; δC 115–45) including assignment of signals. The presence of two molecular species—already observed in the mass spectrometrical analysis—is clearly visible and can be assigned to the absence or presence of the α-Gal moiety (E). Signals labeled with * originate from 7a,b, which are present in small amount. All NMR chemical shift data of 6a,b are listed in Table S3.
Hydrazine treatment and MS analysis
We have recently shown that hydrazine treatment of pneumococcal LTA17 can be employed to reduce the structural heterogeneity caused by differing substitution pattern of fatty acids, alanine and other ester bound residues. That has two major advantages: 1) less complex MS spectra; 2) NMR spectra show a significant higher resolution due to the absence of aggregates formed due to the lipophilic nature of LTA molecules. The mass spectrum of hydrazine treated (de-O-acylated) LTA of S. oralis Uo5Δcps (10) is shown in Fig. 4, the corresponding structure is depicted in Fig. 5A. The monoisotopic mass of 4959.69 Da in Fig. 4 corresponds to 10 (Fig. 5A) with n = 2 (4959.69 Da = Gro + Glc + 2 × RU 8 + 2 × RU 9a). In line with the observed ratios of 6a : 6b and 7a : 7b, larger LTA molecules were detected that comprised additional RUs of structure 9a, which can be recognized based on the specific mass difference of 1257.4 Da. Consequently, we assigned the signals at 6217.123 Da (n = 3), 7474.559 Da (n = 4) and 8732.011 Da (n = 5) to the respective LTA molecules with longer chains (n is equivalent to the number of RU with structure 9a). The fact, that within 5 only an O-3-unsubstituted β-Gal has been observed (Fig. S2, Table S1), clearly reveals that the first biological RU must correspond to 8 (Fig. 1). In Fig. S6 the indicated section of Fig. 4 is enlarged, which shows a minor population of LTA molecules with five RUs comprising only one RU that lacks the α-Gal substituent (10 with n = 3; 1 RU corresponds to 8 and 4 RU to 9a). This structural variant has also been identified in LTA molecules with four or six RUs, respectively.

Section of the charge deconvoluted the ESI-FT-ICR-MS spectra (acquired in negative ion mode) of hydrazine-treated LTA of S. oralis Uo5∆cps (10).
The monoisotopic mass of 4959.69 Da corresponds to 10 with n = 2 (structure shown in Fig. 5A), the assigned higher masses to respective molecules with longer chains (6217.123 Da (n = 3); 7474.559 Da (n = 4); 8732.011 Da (n = 5)).

NMR analysis of 10 (mass spectrum shown in Fig. 4).
(A) Section (δH 5.40–3.30; δC 115–40) of the 1H,13C-HSQC NMR spectrum including structure and assignment of signals. (B) Section (δP 2-(−2)) of the 31P NMR including assignment of signals. All NMR chemical shift data of 10 are listed in Table 1.
NMR analysis of the hydrazine treated LTA (10)
A section of the 1H,13C HSQC NMR of 10 including the assignment of signals is depicted in Fig. 5A, the respective 31P NMR spectrum is shown in Fig. 5B. The detailed NMR chemical shift data of 10 are listed in Table 1. The anomeric region of the 1H,13C HSQC NMR shown in Fig. 5A reflects the complexity of the S. oralis Uo5 LTA. The anomeric signal of the O-3-substituted β-Gal (C1) can be found at δH 4.55/δC 105.3, whereas the two signals of O-3-unsubstituted β-Gal moieties (C#1) are slightly shifted in the proton spectra to δH 4.49/4.48. The respective 13C shifts remain comparable (δC 105.4/105.2). These two signals for C#1 are present due to a slightly different chemical surrounding (presence in the RU vs. in the proximity of the lipid anchor). The different β-Gal-moieties imply diversity in the AATGal-moieties as well (B/B#). Additionally, the terminal β-GalNAc (Dterm) can be distinguished from the β-GalNAc within the RUs (D). The signals for the two P-Cho-substituents of the terminal β-GalNAc (Dterm) can be differentiated in the 31P NMR (Fig. 5B) resulting from the ones of the comparable residues bound to the β-GalNAc placed in the RU (D) very well, since both of them are significantly shifted. Two-dimensional 1H,31P-HMQC and 1H,1H-COSY NMR experiments revealed that the S. oralis Uo5 LTA contains 1,2-linked ribitol moieties, in contrast to the S. pneumoniae LTA, which contains 1,5-linked ribitol17,19,21. Moreover, the 13C NMR chemical shift data for the ribitol moiety in 10 (Table 1) as well as in 6a,b (Table S3) match very well with described 13C NMR values for such an 1,2-linked ribitol moiety as discovered in WTA of Listeria monocytogenes serotype 622.
Analysis of the native LTA
Finally, we investigated the native S. oralis Uo5Δcps LTA by NMR and mass spectrometry. A representative 1H NMR spectrum of this LTA is shown in Fig. S7. Here, the alanine-residues—which have been cleaved off during hydrazine treatment, but were present in the monoPS units after HF treatment—can be identified by their methyl group at δH 1.60 (δC 15.9). The exact position of this alanine-substitution could not be determined due to its minor occurrence. Most interestingly, another substituent that had been cleaved off during hydrazine treatment could be identified. The signal at δH 5.58 (δC 66.8) was assigned to the H-4 of β-Galp, which was further substituted with an acetyl substituent at the hydroxy group at C-4, causing the shift of the signal for H-4 in the 1H NMR. MS analysis of the native LTA (Fig. 6) indicated that all RUs containing the α-Galp substituent also bear this acetyl group, since the mass of the RU in the native LTA is 1299.4 Da (corresponding to 9b; Fig. 1). The mass difference of 42.0 Da compared to RU 9a present in 10 (Fig. 4) matches an additional acetyl group. The molecules with a combined fatty acid composition of 32:0 have been chosen in Fig. 6 for calculations of the RU size, which exemplifies the composition of all LTA molecules (Table S4). Furthermore, we recognized a variation in the number of alanine residue per LTA of zero to three by this MS analysis (representative magnified section in Fig. S8). We identified 32 molecular species within the LTA fraction that are summarized in Table S4.

Section of the charge deconvoluted ESI-FT-ICR-MS spectra (acquired in negative ion mode) of S. oralis Uo5∆cps LTA.
Repeating units (RUs) were detected with an average mass of 1299.4 Da. The RU size was determined on LTA molecules comprising a fatty acid composition with 32 carbon atoms and full saturation. The isolated LTA fraction comprised a complex mixture of molecular species as depicted in Fig. S8 and assigned molecules are summarized in Table S4.
Finally, combination of the results from the NMR spectroscopy and MS of native as well as chemically degraded LTA (part structures) allowed us to resolve the structure of S. oralis Uo5 LTA, which is depicted in comparison to the pneumococcal LTA23 in Fig. 7. The glycolipid anchor of the S. oralis Uo5 LTA is the same as in the pneumococcal LTA: α-D-Glcp(1 → 3)-DAG (2; Fig. 1). The subsequent repeating unit (RU 1) consists of the pseudo-tetrasaccharide 8 (Fig. 1). The following RUs (RU 2 to n + 1) have a structurally identical chain but are further substituted at the β-D-Galp with an α-D-Galp-(1 → )-moiety in position O-3 and an acetyl residue in position O-4 (9b; Fig. 1). Our analysis revealed that in addition to RU 1 another RU in the molecule lacks the α-D-Galp and acetyl substitution (Figs 2, 3B and 5A). Based on our analysis, we conclude that this is the terminal RU, even if the final proof for this has yet to come. Our attempts to address this question by MS/MS-experiments or the investigation of other LTA part structures (e.g. after Smith degradation) failed so far.

Structure for S. oralis Uo5 LTA compared to S. pneumoniae LTA.
In the S. oralis Uo5 LTA, the repeating units (RUs) are composed of a pseudo-tetrasaccharide chain ((→ 4)-[3,6-O-di-P-Cho]-β-D-GalpNAc-(1 → 2)-Rib-ol-(1-P → 6)-(β-D-Galp-(1 → 3)-β-AATGalp-(1 →) (8 in Fig. 1)), RU 2 to n + 1 bear an additional α-1-linked D-Galp moiety at O-3 and an acetyl residue at O-4 of the β-D-Galp (9b in Fig. 1). Hydroxyl groups of the ribitol-1-P can be partially substituted with alanine. The chain length is 4 to 7 RUs. In S. pneumoniae LTA23, all RUs comprise the pseudo-pentasaccharide ((→ 4)-6-O-P-Cho-α-D-GalpNAc-(1 → 3)-6-O-P-Cho-β-D-GalpNAc-(1 → 1)-Rib-ol-5-P-(O → 6)-β-D-Glcp-(1 → 3)-AATGalp-(1 →)) (4 in Fig. 1), the terminal RU can occur with or without 6-O-P-Cho-substitution (X = H or P-Cho). Known strains with the content of only one P-Cho per RU lack the P-Cho at β-D-GalpNAc (R′ = H). Hydroxyl groups of Rib-ol-5-P can be partially substituted with D-Ala. The chain length of pnLTA is 4 to 8 RUs in general. Notably, in pnLTA the first repeating unit is β-1-linked via the AATGalp to the lipid anchor, all other RUs are α-1-linked to the previous one. In S. oralis Uo5 LTA all AATGalp residues are β-configurated.
Implication of the structural analysis for understanding the type IV LTA biosynthesis
Unlike other Gram-positive bacteria, the TAs in S. pneumoniae, S. oralis and S. mitis share a common biosynthetic pathway, which has been bioinformatically analysed recently24. These investigations indicated, that S. oralis Uo5 LTA has the same linker as S. pneumoniae LTA (2; Fig. 1)24, what we confirmed by structural analysis (Table S1). The biosynthetic steps of TA repeating unit formation in S. oralis Uo5 are summarized in Fig. 8 in comparison with the genetic machinery present in S. pneumoniae. Hereby, the close link between the deviating enzymatic repertoire for TA biosynthesis in S. pneumoniae R6 and S. oralis Uo5 and therefore varying structural elements in their TAs is demonstrated. The first sugar moiety of the biological TA repeating unit in S. oralis Uo5 is an AATGal as in S. pneumoniae. All enzymes required for the synthesis of its activated precursor (Sor_1861, Sor_0469) as well as for the transfer to the undecaprenyl-phosphate linker in the membrane (Sor_0468) are present in the S. oralis Uo5 genome with high identity (93–96%) to the respective enzymes in S. pneumoniae strain R6 (Spr_0092, Spr_1654, Spr_1655)24. The second carbohydrate moiety in pnTAs is a β-Glcp, which is transferred by Spr_0091, whereas our structural analysis revealed a β-Galp being present in the S. oralis Uo5 LTA at this position. This β-Galp is incorporated by the glycosyl transferase Sor_1862, which has 93% identity to SP70585_0164, representing a glycosyl transferase which transfers a β-Galp to the AATGal moiety in TAs of a pneumococcal serotype 5 strain24,25. Subsequently, a phosphate-bridged ribitol is incorporated by LicD3. S. oralis LicD3 (Sor_0760) has 93% identity with the pneumococcal LicD3 (Spr_1225)24. In contrast to pnTAs, in which the structural element α-GalpNAc-(1 → 3)-β-GalpNAc-(1 → ) is present at the ribitol, only a β-GalpNAc-(1 → ) moiety has been identified in the S. oralis Uo5 TA repeating unit. The glycosyl transferase Sor_0761 has 42% similarity with Spr_1223, but no homolog or similar enzyme to Spr_1224 has been found in the S. oralis Uo5 genome24. Therefore, it is most likely that Spr_1223 incorporates the β-GalpNAc and Spr_1224 the α-GalpNAc into pnTAs, respectively. Notably, the ribitol-phosphate in pnTAs is 1,5-linked, whereas a 1,2-linkage is present in S. oralis Uo5 LTA. This is in accordance with the low similarity of Sor_0761 and Spr_1223. The pneumococcal choline uptake system consisting of LicA, B and C is present in S. oralis Uo5 with high identity (88–94%) as well. In S. pneumoniae, P-Cho is attached by the LicD1 (Spr_1151) and LicD2 (Spr_1152) proteins to the O-6-positions of the GalpNAc moieties. In S. oralis no homologs to LicD1 and LicD2 have been identified. Instead, LicD4 (Sor_0762) is present, which contains a C-terminal region of 35% and 32% sequence identity compared to pneumococcal LicD1 and LicD2, respectively24. Certainly, LicD4 incorporates at least the P-Cho substituent at the O-6-position of β-GalpNAc in S. oralis Uo5. If the P-Cho substituent at the O-3-position is incorporated by LicD4 as well has to be investigated further. Another potential candidate for this is a protein (Sor_0763) encoded downstream of licD4 with very limited homology to a LicD domain. Neither the genes encoding the proteins responsible for the addition of the α-Galp and the acetyl residue to the β-Galp, nor the time point of these modifications in the biosynthesis pathway have been identified so far.

Biosynthesis pathway for the teichoic acid repeating unit in S. pneumoniae (black; Spr numbers represents S. pneumoniae R6 proteins) highlighting similar or homologous genes in S. oralis (red; Sor numbers represents S. oralis Uo5 proteins).
The biosynthesis pathway of TAs in S. pneumoniae R6 and identity to S. oralis Uo5 proteins is according to reference24. (§) The gene encoding this protein was identified in the genome of an S. pneumoniae serotype 5 strain, which has been shown to incorporate Gal instead of Glc into its WTA25. (#) Neither the genes encoding the proteins responsible for the addition of the α-Gal and acetyl residue to the β-Gal, nor the time point of these modifications in the biosynthesis pathway have been identified so far.
Discussion
The present study provides the first structural elucidation of an S. oralis LTA. For our analysis, we generated an unencapsulated variant of strain Uo5 (S. oralis Uo5Δcps). The structure of the isolated LTA is of higher complexity than in any other reported teichoic acid. Due to a close genetic relationship to S. pneumoniae, a ribitol-phosphate containing type IV LTA1 was anticipated. Combined structural analysis by NMR spectroscopy and mass spectrometry of natural as well as chemically generated LTA part structures allowed us to resolve its structure (Fig. 7). We could show that the S. oralis Uo5 LTA possesses an overall architecture similar to the pnLTA, but differs with regard to the number of GalpNAc moieties, inter-residue connectivities, positions of the P-Cho substituents and has additional, off-stoichiometric branch substituents. With help of the detailed structural analysis, we could improve the functional assignments for genes involved in the S. oralis Uo5 TA biosynthesis in comparison to those identified in S. pneumoniae (Fig. 8). This led in turn to a more specific assignment of enzyme functionalities in S. pneumoniae as well, since it appears now obvious that Spr_1223 incorporates the β-GalpNAc and Spr_1224 the α-GalpNAc into pnTAs.
Our results indicate that in S. oralis Uo5 additional modifications take place, that—causally determined by the different TA structure—do not happen in S. pneumoniae. As shown in Fig. S6, we observed a small portion of LTA molecules comprising only one RU of structure 8 (all other RU correspond to 9b), whereas the majority of LTA molecules contained two of them. Presumably, these modifications (removal of one α-Galp moiety and one acetyl residue) of the TA chain occur after transfer through the cell membrane and onto the glycolipid anchor. In S. pneumoniae, the terminal RU is modified under certain conditions, in this case with respect to its P-Cho content17. Besides the TAs of S. pneumoniae and S. oralis, the modification of glycans with P-Cho residues is known for some capsular polysaccharides of S. pneumoniae as well as for lipopolysaccharides of a few organisms, especially H. influenzae. These P-Cho moieties have been found to be attached to the O-2, O-3, O-4 or O-6 positions of different hexoses as well as to O-7 of a heptose8. However, to the best of our knowledge, this is the first report of a bacterial derived glyco(lipid) structure containing a sugar residue with two P-Cho substituents. Therefore, the identification and investigation of the responsible P-Cho transferring enzymes will be of interest. Further studies have to show, whether the positions of the P-Cho residues have an influence on the attachment and the number of CBPs in S. oralis compared to S. pneumoniae and S. mitis. Several physiological relevant CBPs are highly conserved between S. pneumoniae and S. mitis26, being in line with the structurally identical TA repeating unit discovered for S. mitis strain SK1377. This is further corroborated by the bioinformatic analysis of the genome of S. mitis strain B6, which predicted a very similar structure compared to the pneumococcal TA, merely with the exchange of the β-D-Glcp by a galactose24. In contrast, only a small number of CBPs has been identified in S. oralis Uo5, including those playing a principal role in cell physiology: LytB, LytC, CbpF and two paralogues of CbpD27,28. As discovered in S. pneumoniae17,29, the LTA of S. oralis Uo5 was found to be partially substituted with alanine at its ribitol moieties. D-alanine substituents in the repeating units of LTA polymers, both in S. pneumoniae and in S. aureus, have been thought to play essential roles in their biological activity, especially with respect to their pro-inflammatory potency and activation of the Toll-like receptor 2 (TLR2)30,31,32. Meanwhile it has been demonstrated that lipoproteins (LPs) are the predominant TLR2 stimuli in LTA preparations of S. aureus and S. pneumoniae, respectively and not the LTA itself17,33,34. Another study suggested that, like in many other low-G + C Gram-positive bacteria, teichoic acids of S. pneumoniae contain D-alanine residues in order to protect the bacterium against the actions of cationic antimicrobial peptides35. Some of the players involved in the TA repeating unit biosynthesis of S. oralis Uo5 remained elusive so far and seem to be unique for S. oralis. Particularly, the enzymes for the addition and potential removing of the α-D-Galp-(1 → )-moiety in position O-3 and the acetyl residue in position O-4 of the β-D-Galp have to be identified. All together, the detailed knowledge of these complex structural features of S. oralis LTA will help to decipher the molecular requirements for the interaction of TAs and CBPs in human pathogens like S. oralis and S. pneumoniae.
Methods
Bacterial strain and growth conditions
Bacteria were grown at 37 °C without aeration in C medium36 supplemented with 0.1% yeast extract (C + Y medium) or on D-agar plates37 supplemented with 3% defibrinated sheep blood. The growth of bacteria in liquid culture was monitored by measuring the optical density at 600 nm.
Genetic transformation and DNA techniques
The transformation of S. oralis Uo5 for strain construction was carried out as described previously for S. pneumoniae strains38. Transformants were selected by plating on D-agar supplemented with 3% defibrinated sheep blood and 200 μg/ml kanamycin.
All DNA manipulations were performed using standard procedures39. DNA was amplified with iProof high-fidelity DNA polymerase (Bio-Rad Laboratories).
Construction of S. oralis Uo5Δcps
To construct the Uo5Δcps mutant, in which the capsule locus (genes from sor_1646 to sor_1661) was completely deleted, a kanamycin cassette flanked by DNA corresponding to the upstream and downstream regions of the cps locus was designed. A DNA fragment corresponding to the upstream region of cps was amplified by PCR from Uo5 strain using the primers U05_cps_pcr1_f and U05_cps_pcr1_r, whereas downstream fragment of cps was amplified using the primers U05_cps_pcr3_f and U05_cps_pcr3_r. A 1010-bp fragment containing kanamycin resistance gene aphIII, including the promotor and terminator regions, was amplified from plasmid pMP1 DNA using the primers U05_cps_pcr2_f and U05_cps_pcr2_r. The three PCR fragments were purified and used as a template in a fusion PCR using primers U05_cps_pcr1_f and U05_cps_pcr3_r. The resulting PCR fragment was purified und transformed into Uo5 competent cells. The correct integration was verified by PCR and sequencing. In S. oralis, insertion of the kanamycin cassette produced a 14,537-bp deletion. The oligonucleotides used for construction of these DNA fragments were obtained from Eurofins MWG Operon and are listed in Table S5.
Cell wall preparation
S. oralis Uo5Δcps was grown in 5-liter batches in C + Y medium. Bacterial cells were harvested at an OD600 of appr. 1.0 by centrifugation (7,500 × g, 10 min) at room temperature. The cell pellet was washed with citric buffer (50 mM, pH 4.7) and resuspended in citric buffer containing 4% sodium dodecyl sulfate. The cell suspension was incubated for 20 min at 100 °C, stored at −80 °C and lyophilized subsequently.
LTA extraction and purification
LTA extraction and purification by hydrophobic interaction chromatography (HIC) was performed as described for pnLTA17. Cells were resuspended in distilled water prior to extraction, thus restoring the concentration of citric buffer (50 mM, pH 4.7) and SDS (4%). Cells were disrupted in a cell homogenizer (Vibrogen Cell Mill VI 6, Edmund Bühler GmbH) with 0.1 mm glass beads (Carl Roth GmbH). Afterwards, glass beads were removed by centrifugation (2,000 × g, 10 min, 20 °C) and washed twice with citric buffer. To the combined supernatants SDS (20% solution) was added to have a final concentration of 4%. This solution was incubated for 30 min at 100 °C and centrifuged (30,000 × g, 15 min, 4 °C) subsequently. The pellet was washed four times with citric buffer, all supernatants combined and lyophilized. The resulting solid was washed SDS-free with ethanol (10,650 × g, 15 min, 20 °C). The resulting sediment was resuspended in citric buffer and extracted with an equal volume of butan-1-ol (Merck) for 30 min at RT under vigorous stirring. Phases were separated at 2,100 × g and 4 °C for 15 min. The aqueous phase was removed and the extraction procedure was repeated twice. The combined LTA containing water phases were lyophilized prior to dialysis for 5 d at 4 °C against 50 mM ammonium acetate buffer (pH 4.7; 3.5 kDa cut-off; buffer change usually every 24 h). A final lyophilization leads to crude LTA which is further purified by hydrophobic-interaction chromatography (HIC) performed on a HiPrep octyl-sepharose column (GE Healthcare; 16 × 100 mm, bed volume 20 ml). The crude LTA material was dissolved in less starting buffer (15% propan-1-ol (Merck) in 0.1 M ammonium acetate (pH 4.7)) as possible, centrifuged (13,000 × g, 5 min, 20 °C) and the obtained supernatant was subjected to HIC using a linear gradient from 15% to 60% propan-1-ol in 0.1 M ammonium acetate (pH 4.7). LTA-containing fractions were identified by a photometric phosphate test18 and appropriate phosphate-containing fractions were combined (Fig. S1A), lyophilized and repeatedly washed with water on freeze-drying to remove residual buffer. The extraction of 10 l bacterial culture yielded 21.7 mg purified LTA.
Hydrazine-treatment of LTA
LTA preparations were dissolved 5 μg/μl in anhydrous hydrazine (N2H4; ICN Biomedicals) and then incubated for 1 h at 37 °C and 100–150 rpm (orbital shaker). The reaction was quenched by adding the same volume of acetone and dried under a stream of nitrogen. The latter step was repeated twice and the crude O-deacylated LTAs were purified by gel permeation chromatography (GPC) on a column using Bio-Gel P-10 (45–90 μm, BioRad; column size: 1.5 × 120 cm; buffer: 150 mM ammonium acetate (pH 4.7)) and Toyopearl TSK-40S (Tosoh Bioscience; column size: 2.5 × 50 cm; buffer: 50 mM pyridine/acetic acid/water (8/20/2000 v/v/v)) in a sequential order17.
Preparation of the lipid anchor and monomerized polysaccharides
To generate the lipid anchor as well as TA derived polysaccharides, LTA was treated with 48% hydrofluoric acid (HF) at 4 °C for 2 d (20 μl HF/mg LTA). After drying under a stream of nitrogen at room temperature, the resulting residue was resuspended in water and lyophilized. With this material a purification by HIC as used for the purification of native LTA was performed (Fig. S1B). The polysaccharides eluted in the void volume, the lipid anchor in a comparable retention time period as the natural LTA. After lyophilization, a gel permeation chromatography (GPC) on a column using Bio-Gel P-10 (45–90 μm, BioRad; column size: 1.5 × 120 cm; buffer: 150 mM ammonium acetate (pH 4.7)) was performed with the polysaccharide containing material (Fig. S4); polysaccharides were monitored by a Knauer differential refractometer.
Analytical chemistry
Compositional analysis of the LTA (50–200 μg) was done by gas-liquid chromatography–mass spectrometry (GLC–MS) after methanolysis (0.5 M HCl/CH3OH, 30 min, 85 °C) and per-O-acetylation (pyridine/Ac2O (1:1, v/v), 85 °C, 15 min) with or without prior HF treatment (48% HF, 4 °C, 3 d). The configuration of the aldopyranosides was determined by GLC–MS of acetylated (S)-2-butyl glycosides in comparison to authentic standards40. Fatty acids were recovered from the chloroform phase after alkaline hydrolysis of LTA (1 M NaOH in 50% aqueous CH3OH, 2 h, 85 °C) and detected as fatty acid methyl esters (diazomethane in CHCl3/CH3OH (95:5, v/v)), 5 min, room temperature). For quantification, n-heptadecanoic acid (17:0; Sigma) was used as internal standard. Reported data for fatty acid ratios are the mean of five independent hydrolysis of the same LTA batch. All GLC–MS analyses were performed on an Agilent Technologies 6890N gas chromatograph coupled to a 5975 inert XL Mass Selective Detector. A 30-m HP-5MS column (Hewlett-Packard) was used and the applied temperature gradient started at 70 °C (1.5 min), was increased linearly with 60 °C/min to 150 °C, kept there for 3 min and then increased linearly with 5 °C/min to 320 °C.
Mass spectrometry
For Electrospray Ionization Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry (ESI-FT-ICR-MS) analysis a 7 Tesla APEX Qe instrument (Bruker Daltonics, Bremen, Germany) has been used. For measurement in the negative-ion mode samples were dissolved in a water/propan-2-ol/7 M triethylamine/acetic acid mixture (50:50:0.06:0.02, v/v/v/v), for the positive-ion mode a water/propan-2-ol/30 mM ammonium acetate/acetic acid mixture (15:15:1:0.04, v/v/v/v) was used. Spectra were acquired in broadband acquisition mode either with micro-ESI using a flow rate of 2 μl/min with a capillary voltage set to −3.8 kV or with nano-ESI using the Triversa Nanomate (Advion, USA) as ion source with a spray voltage set to −1.1 kV. The mass scale was externally calibrated with glycolipids of known structure, all spectra were smoothed and charge deconvoluted. The given mass numbers refer to the monoisotopic mass of the neutral molecules.
NMR spectroscopy
Deuterated solvents were purchased from Deutero GmbH (Kastellaun, Germany). NMR spectroscopic measurements were performed in D2O at indicated temperatures on a Bruker AvanceIII 700 MHz (equipped with an inverse 5 mm quadruple-resonance Z-grad cryoprobe). Acetone was used as an external standard for calibration of 1H (δH = 2.225) and 13C (δC = 30.89) NMR spectra41, 85% phosphoric acid was used as an external standard for calibration of 31P NMR spectra (δP = 0.0) at the respective temperatures. Analysis of 5 was performed in CD3OD, spectra were calibrated using the residual solvent peak (δH = 3.31, δC = 49.0)41. All data were acquired and processed using Bruker TOPSPIN V 3.0 or 3.1. 1H NMR assignments were confirmed by 2D 1H,1H-COSY and -TOCSY experiments, 13C NMR assignments were indicated by 2D 1H,13C-HSQC, based on the 1H NMR assignments. Interresidue connectivity and further evidence for 13C assignment were obtained from 2D 1H,13C-HMBC and 1H,13C-HSQC-TOCSY. Connectivity of phosphate groups were assigned by 2D 1H,31P-HMQC and 1H,31P-HMQC-TOCSY.
Additional Information
How to cite this article: Gisch, N. et al. Lipoteichoic acid of Streptococcus oralis Uo5: a novel biochemical structure comprising an unusual phosphorylcholine substitution pattern compared to Streptococcus pneumoniae. Sci. Rep. 5, 16718; doi: 10.1038/srep16718 (2015).
Change history
22 April 2016
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has not been fixed in the paper.
References
Percy, M. G. & Gründling, A. Lipoteichoic acid synthesis and function in Gram-positive bacteria. Annu Rev Microbiol 68, 81–100 (2014).
Fischer, W., Behr, T., Hartmann, R., Peter-Katalinić, J. & Egge, H. Teichoic acid and lipoteichoic acid of Streptococcus pneumoniae possess identical chain structures. A reinvestigation of teichoid acid (C polysaccharide). Eur J Biochem 215, 851–857 (1993).
Tomasz, A. Choline in the cell wall of a bacterium: novel type of polymer-linked choline in Pneumococcus. Science 157, 694–697 (1967).
Fischer, W. Pneumococcal lipoteichoic and teichoic acid. Microb Drug Resist 3, 309–325 (1997).
Gillespie, S. H. et al. Species of alpha-hemolytic streptococci possessing a C-polysaccharide phosphorylcholine-containing antigen. Infect Immun 61, 3076–3077 (1993).
Horne, D. S. & Tomasz, A. Possible role of a choline-containing teichoic acid in the maintenance of normal cell shape and physiology in Streptococcus oralis. J Bacteriol 175, 1717–1722 (1993).
Bergström, N., Jansson, P.-E., Kilian, M. & Skov Sørensen, U. B. Structures of two cell wall-associated polysaccharides of a Streptococcus mitis biovar 1 strain. A unique teichoic acid-like polysaccharide and the group O antigen which is a C-polysaccharide in common with pneumococci. Eur J Biochem 267, 7147–7157 (2000).
Young, N. M., Foote, S. J. & Wakarchuk, W. W. Review of phosphocholine substituents on bacterial pathogen glycans: synthesis, structures and interactions with host proteins. Mol Immunol 56, 563–573 (2013).
Kilian, M. et al. Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS One 3, e2683 (2008).
Whatmore, A. M. et al. Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis and Streptococcus mitis: characterization of “atypical” pneumococci and organisms allied to S. mitis harboring S. pneumoniae virulence factor-encoding genes. Infect Immun 68, 1374–1382 (2000).
Maeda, Y. et al. Population structure and characterization of viridans group streptococci (VGS) including Streptococcus pneumoniae isolated from adult patients with cystic fibrosis (CF). J Cyst Fibros 10, 133–139 (2011).
Mitchell, J. Streptococcus mitis: walking the line between commensalism and pathogenesis. Mol Oral Microbiol 26, 89–98 (2011).
Weidenmaier, C. & Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol 6, 276–287 (2008).
Neuhaus, F. C. & Baddiley, J. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in Gram-positive bacteria. Microbiol Mol Biol Rev 67, 686–723 (2003).
Bergmann, S. & Hammerschmidt, S. Versatility of pneumococcal surface proteins. Microbiology 152, 295–303 (2006).
Denapaite, D. et al. The genome of Streptococcus mitis B6 - what is a commensal? PLoS One 5, e9426 (2010).
Gisch, N. et al. Structural reevaluation of Streptococcus pneumoniae lipoteichoic acid and new insights into its immunostimulatory potency. J Biol Chem 288, 15654–15667 (2013).
Lowry, O. H., Roberts, N. R., Leiner, K. Y., Wu, M.-L. & Farr, A. L. The quantitative histochemistry of brain. I. Chemical methods. J Biol Chem 207, 1–17 (1954).
Behr, T., Fischer, W., Peter-Katalinić, J. & Egge, H. The structure of pneumococcal lipoteichoic acid. Improved preparation, chemical and mass spectrometric studies. Eur J Biochem 207, 1063–1075 (1992).
Seo, H. S., Cartee, R. T., Pritchard, D. G. & Nahm, M. H. A new model of pneumococcal lipoteichoic acid structure resolves biochemical, biosynthetic and serologic inconsistencies of the current model. J Bacteriol 190, 2379–2387 (2008).
Fischer, W. In Streptococcus pneumoniae: Molecular biology & mechanisms of disease (ed A. Tomasz ) 155–177 (Mary Ann Liebert, Inc., 2000).
Uchikawa, K., Sekikawa, I. & Azuma, I. Structural studies on teichoic acids in cell walls of several serotypes of Listeria monocytogenes. J Biochem 99, 315–327 (1986).
Gisch, N., Peters, K., Zähringer, U. & Vollmer, W. In Streptococcus pneumoniae: Molecular mechanisms of host-pathogen interactions (eds J. S. Brown, S. Hammerschmidt & C. J. Orihuela ) Ch. 8, 145–167 (Elsevier, 2015).
Denapaite, D., Brückner, R., Hakenbeck, R. & Vollmer, W. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist 18, 344–358 (2012).
Vialle, S., Sepulcri, P., Dubayle, J. & Talaga, P. The teichoic acid (C-polysaccharide) synthesized by Streptococcus pneumoniae serotype 5 has a specific structure. Carbohydr Res 340, 91–96 (2005).
Hakenbeck, R., Madhour, A., Denapaite, D. & Brückner, R. Versatility of choline metabolism and choline-binding proteins in Streptococcus pneumoniae and commensal streptococci. FEMS Microbiol Rev 33, 572–586 (2009).
Tettelin, H. et al. In Streptococcus pneumoniae: Molecular mechanisms of host-pathogen interactions (eds J. S. Brown, S. Hammerschmidt & C. J. Orihuela ) Ch. 5, 81–107 (Elsevier, 2015).
Madhour, A., Maurer, P. & Hakenbeck, R. Cell surface proteins in S. pneumoniae, S. mitis and S. oralis. Iran J Microbiol 3, 58–67 (2011).
Draing, C. et al. Comparison of lipoteichoic acid from different serotypes of Streptococcus pneumoniae. J Biol Chem 281, 33849–33859 (2006).
Morath, S., Geyer, A. & Hartung, T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med 193, 393–397 (2001).
Schröder, N. W. et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP) and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem 278, 15587–15594 (2003).
Morath, S., von Aulock, S. & Hartung, T. Structure/function relationships of lipoteichoic acids. J Endotoxin Res 11, 348–356 (2005).
Hashimoto, M. et al. Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol 177, 3162–3169 (2006).
Zähringer, U., Lindner, B., Inamura, S., Heine, H. & Alexander, C. TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213, 205–224 (2008).
Kovács, M. et al. A functional dlt operon, encoding proteins required for incorporation of d-alanine in teichoic acids in Gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae. J Bacteriol 188, 5797–5805 (2006).
Lacks, S. & Hotchkiss, R. D. A study of the genetic material determining an enzyme in Pneumococcus. Biochim Biophys Acta 39, 508–518 (1960).
Alloing, G., Granadel, C., Morrison, D. A. & Claverys, J.-P. Competence pheromone, oligopeptide permease and induction of competence in Streptococcus pneumoniae. Mol Microbiol 21, 471–478 (1996).
Mascher, T., Heintz, M., Zahner, D., Merai, M. & Hakenbeck, R. The CiaRH system of Streptococcus pneumoniae prevents lysis during stress induced by treatment with cell wall inhibitors and by mutations in pbp2x involved in β-lactam resistance. J Bacteriol 188, 1959–1968 (2006).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual. 2nd edn (Cold Spring Harbor Laboratory Press, 1989).
Leontein, K., Lindberg, B. & Lönngren, J. Assignment of absolute configuration of sugars by g.l.c. of their acetylated glycosides formed from chiral alcohols. Carbohydr Res 62, 359–362 (1978).
Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J Org Chem 62, 7512–7515 (1997).
Acknowledgements
We gratefully acknowledge B. Buske and H. Moll (GLC–MS), H. Käßner (NMR) and B. Kunz (MS) for excellent technical assistance. We also thank U. Klein for isolation of streptococcal cell walls, R. Brückner for providing plasmid pMP1 and U. Zähringer for valuable discussions and critical reading of the manuscript. This work was supported by a grant of the Deutsche Forschungsgemeinschaft to RH (Ha1011/13–1) as well as in-house funding of the Research Center Borstel.
Author information
Authors and Affiliations
Contributions
N.G., D.S. and D.D. conceived and designed the experiments; N.G., S.T., N.H. and D.D. performed the experiments; N.G., D.S., R.H. and D.D. analysed the data; N.G., D.S., R.H. and D.D. wrote the paper. All authors discussed the results and approved of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gisch, N., Schwudke, D., Thomsen, S. et al. Lipoteichoic acid of Streptococcus oralis Uo5: a novel biochemical structure comprising an unusual phosphorylcholine substitution pattern compared to Streptococcus pneumoniae. Sci Rep 5, 16718 (2015). https://doi.org/10.1038/srep16718
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16718
This article is cited by
-
Lipoteichoic acid deficiency permits normal growth but impairs virulence of Streptococcus pneumoniae
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
