
Abstract
The heritability of schizophrenia (SCZ) has been estimated to be as high as 80%, suggesting that genetic factors may play an important role in the etiology of SCZ. Cav1.2 encoded by CACNA1C and Cav1.3 encoded by CACNA1D are dominant calcium channel-forming subunits of L-type Voltage-dependent Ca2+ channels, expressed in many types of neurons. The CACNA1C has been consistently found to be a risk gene for SCZ, but it is unknown for CACNA1D. To investigate the association of CACNA1D with SCZ, we designed a two-stage case-control study, including a testing set with 1117 cases and 1815 controls and a validation set with 1430 cases and 4295 controls in Han Chinese. A total of selected 97 tag single nucleotide polymorphisms (SNPs) in CACNA1D were genotyped and single-SNP association, imputation analysis and gender-specific association analyses were performed in the two independent datasets. None was found to associate with SCZ. Further genotype and haplotype association analyses indicated a similar pattern in the two-stage study. Our findings suggested CACNA1D might not be a risk gene for SCZ in Han Chinese population, which add to the current state of knowledge regarding the susceptibility of CACNA1D to SCZ.
Similar content being viewed by others

Introduction
Schizophrenia (SCZ) is a devastating mental disorder which affects approximately 1% of the general population worldwide. Evidence from family, adoption and twins studies supported high heritability in the development of SCZ (80%), which results from multiple loci with small effects1. New treatments are desperately needed to enhance the currently unacceptable rates of morbidity, non-response and relapse. An important limiting factor is our lack of understanding of the molecular mechanism of system-level pathophysiology. Since the heritability of SCZ is very high, characterizing mechanisms of genetic risk is a promising route.
L-type voltage-gated Ca2+ channels (VGCCs), which comprises isoforms Cav1.1, Cav1.2, Cav1.3 and Cav1.4, contribute to some patterns of synaptic plasticity, including long-term potentiation and depression2,3. Whereas the expressions of Cav1.1 and Cav1.4 are restricted to skeletal muscle and retinal neurons respectively, Cav1.2 and Cav1.3 have a widely overlapping expression profile in the mammalian neurones4. They have previously been found in some brain areas involved in mood and anxiety (e.g. hippocampus, amygdala, prefrontal cortex cortex)5. Furthermore, they are both able to couple electrical activity to transcriptional regulation6,7. Cav1.3 channels activate at more negative voltages enabling them to modulate neuronal firing behavior and to serve pacemaker function in neurons8,9, which plays an important role in neuronal signaling and suggests that pharmacological modulation would also affect brain function to some extent. Therefore, these channels may be considered as potential new therapeutic targets for the treatment of some central nervous system (CNS) disorders.
Recent applications of genome-wide association studies (GWAS) and next generation sequencing have shown an overlap between the genetic variant that is susceptible to different psychiatric disorders10,11. Smoller et al. (2013) performed a meta-analysis of the GWAS data from 33,332 cases and 27,888 controls of European ancestory, which were distributed among the 5 major psychiatric disorders in Psychiatric Genomics Consortium (major depressive disorder, bipolar disorder, schizophrenia, autism spectrum disorders and attention-deficit/hyperactivity disorder). In that study, they reported SNPs in 2 L-type voltage-gated calcium-channel subunits, CACNA1C and CACNB2, showed genome-wide significance in Europeans, which provided evidence that genetic variation in calcium channel signaling could increase the risk of these 5 neuropsychiatric disorders12. Accumulating evidence have been added for the association of CACNA1C with SCZ13,14,15,16,17,18 and CACNA1C is considered to be one of the most robust findings for genetic susceptibility of mental diseases13,19,20,21, implicating that variants within CACNA1C belong to a wide class of susceptibility factors shared across the psychiatric spectrum12,22,23.Based on the important modulatory role of L-type VGCCs (Cav1.2 and Cav1.3) in neurons, it would be very interesting to know relationship between SCZ and CACNA1D gene, encoding the alpha subunit of the L-type VGCC Cav1.3. To investigate the role of CACNA1D in SCZ susceptibility and its possible related biology function in SCZ, we designed a case-control study to identify whether selected common SNPs (97 tagSNPs) of CACNA1D were involved in SCZ by using two independent data sets of Han Chinese individuals.
Results
Allelic and genotypic association analysis
97 SNPs in the CACNA1D gene were genotyped in the testing dataset (1117 SCZ cases and 1815 controls). The allele and genotype distributions of all SNPs in both cases and controls, including the result of Hardy–Weinberg equilibrium (HWE) test, are shown in Table 1 and Table S1. They were highly polymorphic in both samples and the allelic and the genotype distributions of them were all in HWE (P > 0.05).
We firstly conducted a single SNP association analysis in the testing dataset. When all the samples were considered, we observed potential associations for 6 SNPs (rs709323, rs3774458, rs1460118, rs3774530, rs3774601, rs3774605; p = 0.073731, p = 0.079330, p = 0.061349, p = 0.080180, p = 0.056828, p = 0.090101, respectively) (Table 1), although they were not statistically significant (0.1 > p ≥ 0.05). Genotype association analysis for 6 SNPs suggested a similar pattern with a trend of association in 3 SNPs (rs709323, rs3774458, rs3774601; p = 0.082293, p = 0.075241, p = 0.053895, respectively). The G allele of rs3774601 was more frequent in patients than that in controls (OR = 1.13, 95% CI: 1.00-1.28). The other 91SNPs did not differ significantly in both genotype and allele distributions. According to small effect sizes conferred by common alleles requiring the use of large samples, the overall state of a given SNP is best summarized by association-analysis of different populations. Therefore, we performed a single SNP association analysis for the above 6 SNPs in the validation dataset (1430 SCZ cases and 4295 controls). However, no potential or significant associations with SCZ were found in them (Table 1). To examine whether gender would play a role in the potential association as suggested in the testing dataset, we analyzed our data by separating males and females in the testing and validation datasets. Furthermore, we found no potential or significant associations with SCZ in females or males (Table S2).
Imputation
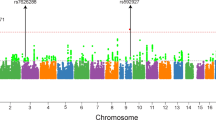
A total of 745 SNPs were successfully imputed. After applying the quality control procedure, 168 imputed SNPs were included in the association test. The full results of association test based on the typed and imputed SNPs were summarized in Table S3. The most significant SNP is an imputed SNP rs4687587 (p = 0.00085), however, considering the multiple comparison burden, this finding failed to pass Bonferroni correction threshold (0.05/130). The regional association plot based on HapMap CHB dataset was shown in Fig. 1.

Regional association plot for both imputed and genotyped SNPs in HapMap CHB.
This plot shows association results of imputed and genotyped SNPs within CACNA1D gene in Han Chinese cohort. The diamonds represent the imputed SNPs and the circles represent the genotyped SNPs (marked by arrow), which are rs709323, rs3774458, rs1460118, rs3774530, rs3774601 and rs3774605 from left to right in order. The horizontal axis is the genomic context of the region studied (along with the genes). The left vertical axis represents negative logarithm of p-value and the right vertical axis is the recombination frequency of the region. Red diamond shows an imputed SNP, rs4687587, which has minimum p-value.
Haplotypic association analysis
To perform haplotype-based association analyses, we examined LD structure within the genotype data of 6 SNPs in validation dataset and identified one haplotype-block. Block 1 was 3 kb long and consisted of 2 SNPs (rs709323 and rs3774458) (Fig. 2). We next carried out haplotypic association analysis of the haplotype block, as was shown in Fig. 2. Tests of the two-marker haplotype association with the use of rs709323 and rs3774458 provided no evidence of association with SCZ (global p = 0.225 in GENECOUNTING and 0.324 in HAPLOSTATS) (Table 2). We also noticed that the haplotype frequencies estimated by two different programs, GENECOUNTING and Haplo Stats, were the same. Therefore, the potential bias reported by Curtis and Xu24 that minor differences in haplotype frequency estimates can produce very large differences in heterogeneity test statistics may not affect our analysis.

Estimation of LD between each pair of 6 SNPs in the validation dataset.
LD structure (D’) between marker pairs was indicated by the shaded matrices.
Discussion
The predominant hypothesis in recent years has been that the genetic architecture of SCZ involves several common variants of small effects and possibly also rare variants with much larger effects25. It is reported that Cav1.2 and Cav1.3 have a broad and overlapping expression profile in the mammalian neuronal system4 and are both able to couple electrical activity to transcriptional regulation6,7. Moreover, as predominant isoforms of L-type VGCCs, they were found to be present in brain areas implicated in mood and anxiety (e.g. hippocampus, amygdala, prefrontal cortex)5,26. Previous studies have focused on the genetic variants within CACNA1C gene19,20,27. Recently, emerging evidence has supported that the role of variations within CACNA1C gene may also contribute to the risk of mental disorders14,18,28. In the present study, we evaluated the potential associations between genetic variations in the CACNA1D gene and the risk of SCZ.
We firstly dissected the association of CACNA1D polymorphisms with SCZ in two-stage case-control study of the Han Chinese population. We detected 97 tagSNPs of CACNA1D gene in the testing samples. Trends of associations were found in allele and genotype frequencies between patients and controls at 6 SNPs (rs709323, rs3774458, rs1460118, rs3774530, rs3774601 and rs3774605). Given that false association may arise from case–control study, because of the influence of small sample size, the second stage study in 5725 subjects was conducted as an effective approach to follow up the findings from the first stage study. However, no association was found in the samples, even though the statistical power of our study was enough to detect the different frequencies between patients and controls. Additionally, we identified an imputed SNP (rs4687587, p = 0.00085) with significant p value. However, considering the multiple comparison burden, the SNP failed to pass the Bonferroni correction. Imputation as a supplemental tool is widely used in common SNP based genetic association analysis, especially large scale analysis like GAWS. Although this method is proved to be powerful in some studies, the imputation accuracy is always a problem need to take care of and can be affected by several factors including the reference data utilized, the original data quality and density of the genotyped marker. To reduce the potential effects on association tests due to the inaccuracy of imputation, we applied several quality control criteria including the control of average maximum posterior probability. Our negative finding of imputation based analysis replicated the negative finding based on the genotyped markers. Furthermore, there was still no gender-specific association between these SNPs and SCZ. The ability to draw conclusions regarding associations based on the analysis of individual SNPs is limited29. Therefore, we performed haplotype analysis, which uses additional information on linkage between typed markers. The results of haplotype frequency estimation showed none of significant association with SCZ (p > 0.1, global permutation). To avoid the inaccuracy of haplotype frequency estimation which could lead to unreliable results, we used two different programs (GENECOUNTING and Haplo Stats) to conduct the haplotype association analyses and the same results were obtained.
L-type VGCCs are found to involve in neuronal development and in the establishment of connectivity maintenance during development30. The increasing findings in genetic studies indicates that the CACNA1C gene is one of shared susceptibility factors for major psychiatric disorders and may have played an important role in the pathogenesis of these diseases at some level. However, the mechanisms illustrating how genetic variants within CACNA1C gene capture risk for developing psychiatric disorders are still unknown14,18,28. Ca2+ influx through L-type VGCCs is considered as a privileged signal, which transmits information of synaptic activation to the transcriptional machinery of the cell’s nucleus31. Some different signaling pathways, including activation of CREB (cAMP-response-element-binding protein), are involved in activity-dependent nuclear signaling via L-type VGCCs31. As an important signal integrator, CREB is responsible for critical central nervous system functions, such as learning, memory and depression-like effects and both Cav1.2 and Cav1.3 can activate CREB in cultured neurons32. It has been reported that specific activation of Cav1.3 could induce depression-like behaviors33 and lead to activation of brain regions involved in anxiety and fear circuits34. Recently, CACNA1D genetic mutations have already been found to be involved in some neurological disorders including autism35,36,37,38 while a previous study indicated a possible role of Cav1.3 in the etiology of Parkinson’s disease39. When interpreting our results, we recognized that we designed the study based on the “Common Disease-Common Variant” hypothesis and we had not sequenced the CACNA1D gene yet to completely evaluate the effect of rare variants on SCZ susceptibility. It could be possible that some rare variants might contribute to the risk of SCZ in a certain unpredicted way or in LD with other undiscovered markers involved with unknown machinery conferring the risk for SCZ. Thus, additional follow-up studies are required including high density mapping and targeted deep sequencing to undercover fundamental characteristics of pathogenic CACNA1D mutations and any potential association with SCZ.
A major limitation of the current study was that we did not perform further analyses for the possible risks of these SNPs that were involved in the clinic phenotype because of the lack of additional subgroups and clinic parameters of the high number of patients in the study. Moreover, although our moderate sample size was small compared with GWAS samples, it was larger than most of individual association studies. Total relatively, the statistical power to detect the moderate effect size for complex diseases such as schizophrenia was not strong and all findings would need to be confirmed by further studies with enlarged sample size. Most recently, a large-scale GWAS17 has provided supportive evidence for our negative results and also implicated that rare variants in CACNA1D gene should be further studied and paid more attention by researchers. Therefore, as a two-stage designed case–control association study, the objective was reached and could be considered reasonable and reliable. Additionally, although we selected subjects with no migration history within the previous three generations to control population stratification caused by genetic factors, we did not focus much on other possible factors leading to population stratification; thus, hidden stratification interference could not be completely ruled out. All our interesting findings, therefore, should be considered preliminary and these results should be interpreted with caution.
In summary, our studies did not support CACNA1D as a susceptible gene for SCZ in Han Chinese population. Our results contribute to a better understanding of the complex neurobiological mechanisms underlying SCZ and add to the current state of knowledge regarding the susceptibility of CACNA1D to SCZ. However, the present findings require replication to clarify the pathological mechanisms of L-type VGCCs for their functional roles in SCZ and to eventually make use in clinical practice. Future studies should not only include larger samples of patients and controls but also focus on the investigation and comparison of different patient samples that are more homogeneous in certain clinical phenotypes, which would be an important next step to demonstrate whether specific effects of CACNA1D genotype exist in subgroups of SCZ patients or not.
Methods
Participants
Two separate datasets were included in this study and a two-stage approach was utilized for the discovery single marker analyses. Subjects containing 1117 SCZ cases (536 males, mean age = 36.0 ± 8.84; 581 females, mean age = 37.2 ± 8.54) and 1815 healthy controls (942 males, mean age = 35.7 ± 7.74; 873 females, mean age = 36.3 ± 7.46 were considered the testing set, while subjects containing 1430 SCZ cases (764 males, mean age = 34.4 ± 7.07; 666 females, mean age = 35.8 ± 6.81) as well as 4295 healthy controls (2280 males, mean age = 34.5 ± 7.02; 2015 females, mean age = 35.3 ± 6.95) were categorized as the validation set. All patients were recruited from the inpatient and outpatient clinical services of a psychiatric unit at Xi’an Mental Health Center and all unrelated healthy controls from local volunteers.
All diagnoses were assigned by a standard procedure. After providing written informed consent, each subject was assessed with the Structured Clinical Interview for DSM-IV Axis I disorder (SCID), which was administered by two or more experienced psychiatrists. Standard diagnostic assessments were supplemented with clinical information obtained by a review of medical records and interviews with family informants. Detailed information on the onset and course of clinical disorders, the presence of personality disorders and mental retardation and a brief description of the subject’s psychosocial and occupational functioning during the course of illness were presented to a consensus diagnostic group including at least three trained psychiatrists with DSM-IV (Diagnostic and statistical manual of mental disorders, 4th revision) diagnostic experience, as well as other trained SCID raters. All available information (personal history, hospital record and family-history report) was used to arrive at a consensus DSM-IV diagnosis. Research subjects with substance-induced psychotic disorders, learning disabilities, head injuries and other symptomatic psychoses were excluded from the present study. All subjects were recruited from the cities of Xi’an in Shaanxi Province. Based on self-report and medical records regarding their own and their paternal grandparents’ place of birth, we excluded anyone not born in Xi’an or whose families for three generations were not born in Xi’an. This study was performed in accordance with the ethical guidelines of the Declaration of Helsinki (version 2002) and was approved by the Xi’an Jiaotong University Ethics Committee. All of the participants have completed written informed consent forms.
SNP selection and genotyping
We searched for all the SNPs with minor allele frequencies (MAF) ≥ 0.05 between 5 kb upstream and 5 kb downstream of CACNA1D gene in the HapMap CHB database by Haploview (Version 4.2) and found 153 SNPs in total. MAF ≥ 0.05 with pair-wise tagging and r2 ≥ 0.840 were used as the criteria when selecting tagSNPs. Finally, we selected 97 tagSNPs covering the region of CACNA1D gene in the study.
Peripheral blood was drawn from a vein into a sterile tube containing ethylenediamine tetraacetic acid (EDTA). Plasma samples were stored at −80 °C. Genomic DNA was isolated from peripheral blood leukocytes according to the manufacturer’s protocol (Genomic DNA kit, Axygen Scientific Inc., California, USA). DNA was stored at −80 °C for SNP analysis. SNPs genotyping was performed using the Sequenom MassARRAY platform with the iPLEX GOLD chemistry (Sequenom, San Diego, CA, USA) following the manufacturer’s protocols. Polymerase chain reaction (PCR) primers and locus-specific extension primers were designed using MassARRAY Assay Design software package (v. 3.1). DNA template of 50 ng was used in each multiplexed PCR well. PCR products were treated with shrimp alkaline phosphatase (USB, Cleveland, OH, USA) before the iPLEX GOLD primer extension reaction. The single base extension products were desalted with SpectroCLEAN resin (Sequenom) and then an aliquot of 10 nL of the desalted product was spotted onto a 384-format SpectroCHIP with the MassARRAY Nanodispenser. Mass determination was done with the MALDI-TOF mass spectrometer. The MassARRAY Typer 4.0 software was employed for data acquisition. Because the final genotype call rate of each SNP was greater than 99.1% and the overall genotyping call rate was 99.6%, the reliability of further statistical analyses was ensured.
Statistical analysis
Hardy–Weinberg equilibrium (HWE) was separately tested among the patient and control groups to examine the genotype distributions of each SNP mentioned above by using GENEPOP v4.0. Allelic and genotypic association tests were performed using CLUMP v2.4 with 10000 simulations, which employs an empirical Monte Carlo test of significance through simulation and does not require correction for multiple alleles41. To increase the density of the SNP markers in the validation dataset, we implemented imputation using genetic software IMPUT242 with HapMap dataset from combined sample set as reference. The follow up association analysis was performed with software snptest43. We implemented frequentist association tests for each imputed SNPs and adding gender as a covariate. To ensure the accuracy of imputation, we only used the imputed SNPs with average maximum posterior probability larger than 0.5. For the power considerations, in the association test, we only included those SNPs with observed statistical information larger than 0.1 and with MAF larger than 0.01. The pair-wise linkage disequilibrium (LD) analysis was applied to detect the inter-marker relationship using D′and r2 values in the Haploview v4.2 software program. The haplotype frequencies were estimated using GENECOUNTING v2.2 and haplotypic association analysis was performed for the common haplotypes (frequency >0.01). In addition, a permutation algorithm was applied in the testing framework to find the maximum of the haplotype-specific score statistics and the associated p value for this maximum. The Haplo Stats package v1.4.4, which implements the methods of Schaid et al.44, was used for these analyses. To investigate the possible potential effect of gender for a trend of association of SNPs, the samples were stratified by gender. Power calculations are a fundamental component of the design of genetic association studies. We used Genetic Power Calculation (http://pngu.mgh.harvard.edu/~purcell/gpc/) to perform the power calculations. Our sample size had >80% power in the two-stage samples to detect a significant association at the false-positive rate of 5%, disease prevalence of 1%, MAF >5% and a presumed odds ratio (OR) of 1.4 (results of general power analysis in Table S4 & S5).
Additional Information
How to cite this article: Guan, F. et al. Evaluation of genetic susceptibility of common variants in CACNA1D with schizophrenia in Han Chinese. Sci. Rep. 5, 12935; doi: 10.1038/srep12935 (2015).
References
O’Donovan, M. C. et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nature Genetics 40, 1053–1055 (2008).
Wickens, J. R. & Abraham, W. C. The involvement of L-type calcium channels in heterosynaptic long-term depression in the hippocampus. Neuroscience Letters 130, 128–132 (1991).
Kapur, A., Yeckel, M. F., Gray, R. & Johnston, D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. Journal of Neurophysiology 79, 2181–2190 (1998).
Sinnegger-Brauns, M. J. et al. Expression and 1, 4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Molecular Pharmacology 75, 407–414 (2009).
Ludwig, A., Flockerzi, V. & Hofmann, F. Regional expression and cellular localization of the α1 and β subunit of high voltage-activated calcium channels in rat brain. The Journal of Neuroscience 17, 1339–1349 (1997).
Moosmang, S. et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. The Journal of Neuroscience 25, 9883–9892 (2005).
Zhang, H. et al. Cav1.2 and Cav1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. European Journal of Neuroscience 23, 2297–2310 (2006).
Olson, P. A. et al. G-protein-coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. The Journal of Neuroscience 25, 1050–1062 (2005).
Chan, C. S. et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086 (2007).
Gershon, E. S., Alliey-Rodriguez, N. & Liu, C. After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. The American Journal of Psychiatry 168, 253–256 (2011).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nature Reviews Genetics 10, 241–251 (2009).
Smoller, J. W. et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
Moskvina, V. et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Molecular Psychiatry 14, 252–260 (2008).
Green, E. K. et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Molecular Psychiatry 15, 1016–1022 (2009).
Nyegaard, M. et al. CACNA1C (rs1006737) is associated with schizophrenia. Molecular Psychiatry 15, 119–121 (2010).
Hamshere, M. L. et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8 and extensive replication of associations reported by the Schizophrenia PGC. Molecular Psychiatry 18, 708–712 (2012).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Guan, F. et al. MIR137 gene and target gene CACNA1C of miR-137 contribute to schizophrenia susceptibility in Han Chinese. Schizophrenia Research 152, 97–104 (2014).
Ripke, S. et al. Genome-wide association study identifies five new schizophrenia loci. Nature Genetics 43, 969–976 (2011).
Sklar, P. et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature Genetics 43, 977–983 (2011).
Williams, H. J. et al. Most genome-wide significant susceptibility loci for schizophrenia and bipolar disorder reported to date cross-traditional diagnostic boundaries. Human Molecular Genetics 20, 387–391 (2011).
Craddock, N., O’Donovan, M. C. & Owen, M. J. Psychosis genetics: modeling the relationship between schizophrenia, bipolar disorder and mixed (or “schizoaffective”) psychoses. Schizophrenia Bull 35, 482–490 (2009).
Bhat, S. et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Progress in Neurobiology 99, 1–14 (2012).
Curtis, D. & Xu, K. Minor differences in haplotype frequency estimates can produce very large differences in heterogeneity test statistics. BMC Genetics 8, 38 (2007).
Wang, W. Y., Barratt, B. J., Clayton, D. G. & Todd, J. A. Genome-wide association studies: theoretical and practical concerns. Nature Reviews Genetics 6, 109–118 (2005).
Hell, J. W. et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. The Journal of Cell Biology 123, 949–962 (1993).
Liu, Y. et al. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Molecular Psychiatry 16, 2–4 (2010).
Bigos, K. L. et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Archives of General Psychiatry 67, 939–945 (2010).
Korostishevsky, M. et al. Is the G72/G30 locus associated with schizophrenia? single nucleotide polymorphisms, haplotypes and gene expression analysis. Biological Psychiatry 56, 169–176 (2004).
Spitzer, N. C. Electrical activity in early neuronal development. Nature 444, 707–712 (2006).
Deisseroth, K., Mermelstein, P. G., Xia, H. & Tsien, R. W. Signaling from synapse to nucleus: the logic behind the mechanisms. Current Opinion in Neurobiology 13, 354–365 (2003).
Rajadhyaksha, A. M. & Kosofsky, B. E. Psychostimulants, L-type calcium channels, kinases and phosphatases. The Neuroscientist 11, 494–502 (2005).
Sinnegger-Brauns, M. J. et al. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type Ca2+ channels. The Journal of Clinical Investigation 113, 1430–1439 (2004).
Hetzenauer, A., Sinnegger-Brauns, M. J., Striessnig, J. & Singewald, N. Brain activation pattern induced by stimulation of L-type Ca2+ -channels: contribution of CaV1.3 and CaV1.2 isoforms. Neuroscience 139, 1005–1015 (2006).
O’Roak, B. J. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
Scholl, U. I. et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nature Genetics 45, 1050–1054 (2013).
Pinggera, A. et al. CACNA1D De Novo Mutations in Autism Spectrum Disorders Activate Cav1.3 L-Type Calcium Channels. Biological Psychiatry 77, 816–822 (2015).
Ilijic, E., Guzman, J. N. & Surmeier, D. J. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiology of Disease 43, 364–371 (2011).
de Bakker, P. I. et al. Efficiency and power in genetic association studies. Nature Genetics 37, 1217–1223 (2005).
Sham, P. C. & Curtis, D. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Annals of Human Genetics 59, 97–105 (1995).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nature Genetics 39, 906–913 (2007).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nature Reviews Genetics 11, 499–511 (2010).
Schaid, D. J., Rowland, C. M., Tines, D. E., Jacobson, R. M. & Poland, G. A. Score tests for association between traits and haplotypes when linkage phase is ambiguous. The American Journal of Human Genetics 70, 425–434 (2002).
Acknowledgements
This research was totally supported by and National Natural Science Foundation of China (No. 81273351 and 81401563), China Postdoctoral Science Foundation Funded Project (No. T70927 and M532029), Ph.D. Programs Foundation of Ministry of Education of China (No. 2013021120078) and Fundamental Research Funds for the Central Universities (No. 08142024 and 08143003). The funding sources had no role in the design of this study, the collection, analysis and interpretation of data, the writing of the report, or the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
F.G. has done sample collection, genotyping, data analysis and written the manuscript which was critically revised by X.L. and L.L. has helped in DNA sample preparation and genotyping; C.Q. and G.C. conducted subject screening; T.Y. and T.L. have clinically characterized the patients whose biological samples have been included in the study; T.Z. has helped in data analysis; F.G. and X.L. have conceptualized and led this project including arranging for the required funds. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Guan, F., Li, L., Qiao, C. et al. Evaluation of genetic susceptibility of common variants in CACNA1D with schizophrenia in Han Chinese. Sci Rep 5, 12935 (2015). https://doi.org/10.1038/srep12935
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep12935
This article is cited by
-
Zebrafish Larvae Carrying a Splice Variant Mutation in cacna1d: A New Model for Schizophrenia-Like Behaviours?
Molecular Neurobiology (2021)
-
Evaluation of genetic susceptibility between systemic lupus erythematosus and GRB2 gene
Scientific Reports (2019)
-
Relationship of common variants in MPP7, TIMP2 and CASP8 genes with the risk of chronic achilles tendinopathy
Scientific Reports (2019)
-
IRAK2 and TLR10 confer risk of Hashimoto’s disease: a genetic association study based on the Han Chinese population
Journal of Human Genetics (2019)
-
Relationship of common variants in VEGFA gene with osteonecrosis of the femoral head: A Han Chinese population based association study
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
