
Abstract
Up to now, no consensus exists about the electronic nature of phosphorus (P) as donor for SiO2-embedded silicon nanocrystals (SiNCs). Here, we report on hybrid density functional theory (h-DFT) calculations of P in the SiNC/SiO2 system matching our experimental findings. Relevant P configurations within SiNCs, at SiNC surfaces, within the sub-oxide interface shell and in the SiO2 matrix were evaluated. Atom probe tomography (APT) and its statistical evaluation provide detailed spatial P distributions. For the first time, we obtain ionisation states of P atoms in the SiNC/SiO2 system at room temperature using X-ray absorption near edge structure (XANES) spectroscopy, eliminating structural artefacts due to sputtering as occurring in XPS. K energies of P in SiO2 and SiNC/SiO2 superlattices (SLs) were calibrated with non-degenerate P-doped Si wafers.  results confirm measured core level energies, connecting and explaining XANES spectra with h-DFT electronic structures. While P can diffuse into SiNCs and predominantly resides on interstitial sites, its ionization probability is extremely low, rendering P unsuitable for introducing electrons into SiNCs embedded in SiO2. Increased sample conductivity and photoluminescence (PL) quenching previously assigned to ionized P donors originate from deep defect levels due to P.
results confirm measured core level energies, connecting and explaining XANES spectra with h-DFT electronic structures. While P can diffuse into SiNCs and predominantly resides on interstitial sites, its ionization probability is extremely low, rendering P unsuitable for introducing electrons into SiNCs embedded in SiO2. Increased sample conductivity and photoluminescence (PL) quenching previously assigned to ionized P donors originate from deep defect levels due to P.
Similar content being viewed by others


Introduction
About 60 years ago, impurity doping of bulk Si was established to introduce majority charge carriers, creating p/n junctions as fundamental building blocks of Si-based electronic devices. The discovery of size-controlled solid-state growth of SiNCs from Si-rich SiO2 (SiOx)1 led to discussions about conventional dopants in SiNC/SiO2 systems. P is of particular interest due to high solubility and diffusivity in Si2. Detailed insight into the behaviour of P within the SiNC/SiO2 material system is crucial. It clarifies whether conventional SiNC doping is able to further advance miniaturization of Si-based electronic structures and electronic SiNC manipulation.
Many works have claimed doping of SiNCs with P donors3,4,5,6,7,8, but very few provided unambiguous evidence and detailed data on doping probabilities9,10 as gauge for working (active) dopants. Experimental evidence of successful P doping in SiNC/SiO2 samples like quantum dot solar cells11 or standard capacitance-voltage curves requiring a bulk semiconductor space charge region12 likely occur due to interconnected SiNC/amorphous Si networks13 where conventional doping does work to some extent. SiNCs separated by ultrathin SiO2 barriers are dominated by defect-assisted conduction14, though electric conductivities can be tremendously increased by massive P incorporation in the 0.5 to 8 atom-% range (0.25 to  cm−3)3,4,5,6,7,8. Such high P concentrations enter the composition range of ternary compounds (SiOxPy) with different properties as compared to Si, SiOx and SiO2. We note that Pearson and Bardeen15 observed the semiconductor to metal transition of bulk Si for donor (P) and acceptor (boron; B) concentrations around 0.25 atom-% (
cm−3)3,4,5,6,7,8. Such high P concentrations enter the composition range of ternary compounds (SiOxPy) with different properties as compared to Si, SiOx and SiO2. We note that Pearson and Bardeen15 observed the semiconductor to metal transition of bulk Si for donor (P) and acceptor (boron; B) concentrations around 0.25 atom-% ( cm−3). With P concentrations in the 0.5 to 8 atom-% range, clustering with dopant inactivation, defect formation and massive out-diffusion occur already in bulk type Si layers for structure sizes of
cm−3). With P concentrations in the 0.5 to 8 atom-% range, clustering with dopant inactivation, defect formation and massive out-diffusion occur already in bulk type Si layers for structure sizes of  nm in ultra-large scale integration (ULSI)16,17. Local P density fluctuations in SiNCs prevent to provide exactly one active dopant per SiNC18. The vast majority of SiNCs are undoped and very few SiNCs have multiple dopants. Latter leads to significant random deterioration of their electronic properties by exchange coupling19. Massive P densities in SiNC systems lead to P localized in SiO2, in SiOx surrounding SiNCs and P gettered by dangling bonds (DBs) at NC interfaces, all being critical for the electronic structure. So far, unpaired electrons bound to P were investigated by electron paramagnetic resonance (EPR) at very low temperatures9,10. Thermal broadening of EPR resonances prevented measurements at room temperature (T = 300 K). XANES is not restricted to low temperatures and yields information on the electronic state of all P at T = 300 K. Excited P K shell electrons in XANES have tremendously increased mean free paths as compared to X-ray photoelectron spectroscopy (XPS) due to their high kinetic energy
nm in ultra-large scale integration (ULSI)16,17. Local P density fluctuations in SiNCs prevent to provide exactly one active dopant per SiNC18. The vast majority of SiNCs are undoped and very few SiNCs have multiple dopants. Latter leads to significant random deterioration of their electronic properties by exchange coupling19. Massive P densities in SiNC systems lead to P localized in SiO2, in SiOx surrounding SiNCs and P gettered by dangling bonds (DBs) at NC interfaces, all being critical for the electronic structure. So far, unpaired electrons bound to P were investigated by electron paramagnetic resonance (EPR) at very low temperatures9,10. Thermal broadening of EPR resonances prevented measurements at room temperature (T = 300 K). XANES is not restricted to low temperatures and yields information on the electronic state of all P at T = 300 K. Excited P K shell electrons in XANES have tremendously increased mean free paths as compared to X-ray photoelectron spectroscopy (XPS) due to their high kinetic energy  20. We boosted sampling depths further by using XANES in fluorescence yield mode, allowing for non-destructive probing depths three orders of magnitude above XPS values. Due to the low
20. We boosted sampling depths further by using XANES in fluorescence yield mode, allowing for non-destructive probing depths three orders of magnitude above XPS values. Due to the low  of P L-III shell electrons, XPS is extremely surface sensitive. Probing samples below their original surface by XPS requires sputtering off top material, introducing artefacts as function of chemical species like sputter yield and atom re-coordination and re-ordering.
of P L-III shell electrons, XPS is extremely surface sensitive. Probing samples below their original surface by XPS requires sputtering off top material, introducing artefacts as function of chemical species like sputter yield and atom re-coordination and re-ordering.
We report on h-DFT calculations of P at central lattice and interstitial sites in completely OH-terminated SiNCs, of saturated P at the surface of such NCs, in SiO0.9 as sub-oxide shell around SiNCs and in SiO2, delivering insights into the specific electronic structure due to P. The spatial distribution of P atoms in SiNC/SiO2 systems is derived from APT data and their statistical processing to yield the P distribution profile from the SiO2 matrix to the interior of the SiNCs. We discuss P data from h-DFT and XANES together with P spatial statistics from APT and obtain a detailed picture of the electronic behaviour of prospective P donors depending on their positions and bond geometries in SiNC/SiO2 systems. The 1s core level energies from h-DFT are used to assign XANES signals to respective P configurations in h-DFT approximants.
Results
Hybrid DFT calculations
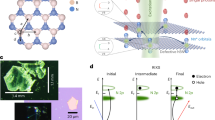
Figure 1 shows optimized approximants of a SiO2 reference ( -quartz), of SiO2 with P on a central Si site (SiO2:P), of a SiO0.9 reference and of P-doped SiO0.9 (SiO0.9:P). Fig. 1 further shows optimized approximants of a fully OH-terminated SiNC of 15 Å size as NC reference (OH-SiNC) and this NC with saturated (penta-valent) P substituting a corner Si atom (OH-SiNC
-quartz), of SiO2 with P on a central Si site (SiO2:P), of a SiO0.9 reference and of P-doped SiO0.9 (SiO0.9:P). Fig. 1 further shows optimized approximants of a fully OH-terminated SiNC of 15 Å size as NC reference (OH-SiNC) and this NC with saturated (penta-valent) P substituting a corner Si atom (OH-SiNC  P(OH)3), an OH group on such corner Si atom (OH-SiNC-P(OH)4) and a H atom at the OH group substituted by P(OH)4 (OH-SiNC-O-P(OH)4). Fig. 1 also shows optimized approximants of fully OH-terminated 15 Å SiNCs with P on a central Si lattice site (OH-SiNC-P[Si]) and on a central interstitial cite (OH-SiNC-P[is]). Interstitial P coordinates relative to its 1-nn Si atoms were used from experiment21. Convergence of structural optimization of the approximant was accepted for residual forces on interstitial P and its 1-nn Si atom (and all other atoms) of 309 μeV/Å (11.3 μHa/Å) which is ca. 1.3% of the convergence threshold of maximum residual forces, see to Methods section at end of article. The atomic displacement associated with this minute residual force was 0.0032 Å (0.32 pm) which is ca. 94% of the convergence threshold of residual displacements of 0.003403 pm – a rather large value for such residual force. This variance is an indication of a somewhat flat energy landscape. Thereby, it is rather difficult to calculate an exact diffusion path of interstitial P. This may explain why dopant atoms on Si lattice sites were considered in ab-initio thermodynamic diffusion simulations22,23,24,25, but dopant atoms on interstitial positions were not included. Further details on DFT calculations can be found in the Methods section at the end of the article.
P(OH)3), an OH group on such corner Si atom (OH-SiNC-P(OH)4) and a H atom at the OH group substituted by P(OH)4 (OH-SiNC-O-P(OH)4). Fig. 1 also shows optimized approximants of fully OH-terminated 15 Å SiNCs with P on a central Si lattice site (OH-SiNC-P[Si]) and on a central interstitial cite (OH-SiNC-P[is]). Interstitial P coordinates relative to its 1-nn Si atoms were used from experiment21. Convergence of structural optimization of the approximant was accepted for residual forces on interstitial P and its 1-nn Si atom (and all other atoms) of 309 μeV/Å (11.3 μHa/Å) which is ca. 1.3% of the convergence threshold of maximum residual forces, see to Methods section at end of article. The atomic displacement associated with this minute residual force was 0.0032 Å (0.32 pm) which is ca. 94% of the convergence threshold of residual displacements of 0.003403 pm – a rather large value for such residual force. This variance is an indication of a somewhat flat energy landscape. Thereby, it is rather difficult to calculate an exact diffusion path of interstitial P. This may explain why dopant atoms on Si lattice sites were considered in ab-initio thermodynamic diffusion simulations22,23,24,25, but dopant atoms on interstitial positions were not included. Further details on DFT calculations can be found in the Methods section at the end of the article.

Optimized approximants calculated by h-DFT.
Top row shows SiO2 reference – Si29O40(OH)36 (a), SiO2:P – Si28PO40(OH)36 (b), SiO0.9 reference – Si74O55(OH)35H29 (c), SiO0.9:P – Si73PO55(OH)35H29 (d) and fully OH-terminated 15 Å NC reference OH-SiNC – Si84(OH)64 (e). The bottom row shows OH-SiNC with corner >Si(OH)2 substituted by >P(OH)3 referred to as OH-SiNC>P(OH)3 – Si83P(OH)3(OH)62 (f), with OH group at corner Si atom substituted by P(OH)4 referred to as OH-SiNC-P(OH)4 – Si84P(OH)4(OH)63 (g), with OH group at corner Si atom substituted by OP(OH)4 referred to as OH-SiNC-O-P(OH)4 – Si84OP(OH)4(OH)63 (h), OH-SiNC with central Si atom substituted for tetravalent P referred to as OH-SiNC-P[Si] – Si83P(OH)64 (i) and with P on an interstitial site in the NC center referred to as OH-SiNC-P[is] – Si84(OH)64-is-P (k). Atom colors: Si is gray, P is black, O is red and H is white. 1-nn O atoms of P in SiO2 and SiO0.9 approximants shown in cyan. Approximants shown along [001] lattice vector group except SiO2 and SiO2:P.
Electronic Structure of P in SiO2.
The SiO2 HOMO-LUMO gap is 7.83 eV which is 89% of the experimental value of ca. 8.8 eV26. We consider P on tetragonal Si sites in SiO2 and SiO. P is surrounded by SiO2 at least to its  next neighbour (5-nn) atom. Oxidation enthalpies27 are 916 kJ/mol (9.49 eV/Si atom) for the chemical reaction Si + O2 → SiO2 and 1493 kJ/mol (7.74 eV/P atom) for the reaction
next neighbour (5-nn) atom. Oxidation enthalpies27 are 916 kJ/mol (9.49 eV/Si atom) for the chemical reaction Si + O2 → SiO2 and 1493 kJ/mol (7.74 eV/P atom) for the reaction  , indicating that pentavalent P configurations (P(–O–)5) should not be favoured over tetravalent Si (Si(–O–)4), leaving P with a DB in analogy to P donors in bulk Si. The DB of P is strongly associated with
, indicating that pentavalent P configurations (P(–O–)5) should not be favoured over tetravalent Si (Si(–O–)4), leaving P with a DB in analogy to P donors in bulk Si. The DB of P is strongly associated with  -HOMO and
-HOMO and  -LUMO, describing one state with its two spin configurations
-LUMO, describing one state with its two spin configurations  and
and  (Fig. 2a). We compare the energies of frontier MOs with HOMO and LUMO energies of the OH-SiNC reference (Fig. 2, green lines). The
(Fig. 2a). We compare the energies of frontier MOs with HOMO and LUMO energies of the OH-SiNC reference (Fig. 2, green lines). The  -LUMO energy of SiO2:P is 0.01 eV above the LUMO of the OH-SiNC approximant while the
-LUMO energy of SiO2:P is 0.01 eV above the LUMO of the OH-SiNC approximant while the  -HOMO of SiO2:P is 0.41 eV below the HOMO of the OH-SiNC approximant. The barrier height for electron (hole) transport is given by the conduction (valence) band offset between Si and SiO2 of 3.2 eV (4.5 eV)26. These values show that P in SiO2 reduces the transport barrier for electrons (holes) by 97% (85%), causing an extreme increase in electron conductivity and a considerably increased hole conductivity. These defect levels are an important electronic aspect of P in SiO2: It causes a massive increase in SiO2 conductivity while not working as a donor. Several works build their evidence of SiNC doping on conductivities increasing with P concentrations of 0.5 to 8 atom-%3,4,6,7,8. From the SiO2:P approximant we get an atomic ratio of P/
-HOMO of SiO2:P is 0.41 eV below the HOMO of the OH-SiNC approximant. The barrier height for electron (hole) transport is given by the conduction (valence) band offset between Si and SiO2 of 3.2 eV (4.5 eV)26. These values show that P in SiO2 reduces the transport barrier for electrons (holes) by 97% (85%), causing an extreme increase in electron conductivity and a considerably increased hole conductivity. These defect levels are an important electronic aspect of P in SiO2: It causes a massive increase in SiO2 conductivity while not working as a donor. Several works build their evidence of SiNC doping on conductivities increasing with P concentrations of 0.5 to 8 atom-%3,4,6,7,8. From the SiO2:P approximant we get an atomic ratio of P/ atom-% P, whereby we consider H terminating outermost O bonds as 1/4 Si.
atom-% P, whereby we consider H terminating outermost O bonds as 1/4 Si.

Electronic DOS of oxides containing P.
Results for SiO2:P (a) and SiO0.9:P (b), shown with DOS of pure SiO2 (top) and pure SiO0.9 (bottom) approximants. Dark (bright) green lines show HOMO (LUMO) of OH-SiNC.
Electronic Structure of P in SiO
Approximants for SiO0.9 and SiO0.9:P are based on  -quartz. Every second O bridge Si–O–Si is substituted by a bond Si–Si. As with SiO2:P, we have a DB on P occupied with one electron in the SiO0.9:P approximant at a central Si lattice site, again resulting in two different spin orientations per MO (
-quartz. Every second O bridge Si–O–Si is substituted by a bond Si–Si. As with SiO2:P, we have a DB on P occupied with one electron in the SiO0.9:P approximant at a central Si lattice site, again resulting in two different spin orientations per MO ( ). Frontier MOs are similar to SiO2:P, describing the DB of P with one electron occupying the
). Frontier MOs are similar to SiO2:P, describing the DB of P with one electron occupying the  -HOMO. The
-HOMO. The  -HOMO –
-HOMO –  -LUMO gap of 1.96 eV is 0.76 eV below Egap=2.72 eV of the OH-SiNC reference. The HOMO in SiO0.9:P is located 1.05 eV above the HOMO of the OH-SiNC reference. Hence, P presents a deep recombination center in SiOx shells (Fig. 2b) which cover SiNCs with a thickness of 1 to 1.5 mono layers (MLs)28. This finding is supported by PL quenching reported for high P concentrations mentioned above3,29.
-LUMO gap of 1.96 eV is 0.76 eV below Egap=2.72 eV of the OH-SiNC reference. The HOMO in SiO0.9:P is located 1.05 eV above the HOMO of the OH-SiNC reference. Hence, P presents a deep recombination center in SiOx shells (Fig. 2b) which cover SiNCs with a thickness of 1 to 1.5 mono layers (MLs)28. This finding is supported by PL quenching reported for high P concentrations mentioned above3,29.
The LUMO of SiO0.9:P facilitates electron transport by diminishing the electron barrier. As for the SiO2:P approximant, electron (hole) barriers are decreased down to 32% (removed completely). For SiO0.9 and SiO0.9:P approximants, a helical arrangement of Si atoms along the  vector (Fig. 1c,d) dominates MOs from E − Evac = 0.2 to -8.5 eV. The inner bonds of these Si backbones can resist electron transfer to O to some extent, diminishing the splitting of their bonding and anti-bonding MOs. Experiments yield
vector (Fig. 1c,d) dominates MOs from E − Evac = 0.2 to -8.5 eV. The inner bonds of these Si backbones can resist electron transfer to O to some extent, diminishing the splitting of their bonding and anti-bonding MOs. Experiments yield  (SiO) ≈ 2.48 eV30, our calculations overestimate this value by 54%. This may be due to the very balanced local stoichiometry of the SiO0.9 reference and SiO0.9:P approximants as well as their high space group symmetry which allows for mentioned Si helices. Local Si segregation suggests that SiO is not uniform13 which can lower the band gap. The P concentration can be calculated as for the SiO2:P approximant, yielding 0.56 atom-% for SiO0.9:P.
(SiO) ≈ 2.48 eV30, our calculations overestimate this value by 54%. This may be due to the very balanced local stoichiometry of the SiO0.9 reference and SiO0.9:P approximants as well as their high space group symmetry which allows for mentioned Si helices. Local Si segregation suggests that SiO is not uniform13 which can lower the band gap. The P concentration can be calculated as for the SiO2:P approximant, yielding 0.56 atom-% for SiO0.9:P.
Electronic Structure: Saturated P at SiNC interfaces
Tetravalent P atoms substantially gain binding energy when gettering their DBs at NC interfaces and maximize binding energies of Si atoms providing DBs. It is thus energetically unfavourable for P at the NC interface to have a DB. This finding is supported by a maximum P density at SiNC interfaces derived from APT below.
We show the DOS of the OH-SiNC reference approximant along with the DOS of all three approximants containing bond-saturated P at the interface (Fig. 3). Fully gettered P at NC interfaces does not introduce defect levels within the HOMO-LUMO gap of the SiNC. The DOS of OH groups has an energy gap of 8.0 eV, corresponding to 91% of the experimental band gap of SiO226. The DOS of the SiNC approximants expose a small shift of HOMO and LUMO to higher binding energies, correlating with an increasing number of O atoms31,39.

Electronic DOS of SiNCs with P at interface.
Data of OH-SiNC reference (Si84(OH)64) and its versions with bond-saturated P at NC interface, see Figure 1 for details.
Electronic Structure of P within SiNCs
We consider P on a central Si lattice site OH-SiNC-P[Si] and on a central interstitial site OH-SiNC-P[is]. P on a Si lattice site generates a HOMO 0.51 eV below the LUMO energy (Fig. 4a). While this HOMO presumably becomes a donor state for vanishing quantum confinement, its ionization energy Eion=0.51 eV is too big to ionize SiNCs with a reasonable probability at T = 300 K;  . Even for SiNCs at the upper size limit of quantum confinement,
. Even for SiNCs at the upper size limit of quantum confinement,  will be too small for providing electrons to SiNCs; experimental values10 for
will be too small for providing electrons to SiNCs; experimental values10 for  Å are
Å are  . Interstitial P introduces two gap states, a HOMO 0.57 eV above the HOMO of the 1.5 nm SiNC and a LUMO 0.46 eV below the LUMO of the SiNC (Fig. 4b). Both states due to P cannot donate electrons but provide efficient carrier recombination with a transition energy of 1.72 eV. As this transition is optically active at a wavelength of ca. 720 nm, it must be considered for PL spectra of P-doped SiNC/SiO2 species. Both cases of P in OH-SiNC introduce recombination levels into SiNCs.
. Interstitial P introduces two gap states, a HOMO 0.57 eV above the HOMO of the 1.5 nm SiNC and a LUMO 0.46 eV below the LUMO of the SiNC (Fig. 4b). Both states due to P cannot donate electrons but provide efficient carrier recombination with a transition energy of 1.72 eV. As this transition is optically active at a wavelength of ca. 720 nm, it must be considered for PL spectra of P-doped SiNC/SiO2 species. Both cases of P in OH-SiNC introduce recombination levels into SiNCs.

Electronic DOS of OH-SiNC approximant with P residing inside SiNC.
P located on a central Si substitutional site (a) and on a central interstitial site (b), shown with DOS of reference OH-SiNC approximant.
Atom Probe Tomography
We show the APT scan of a SiNC SL in SiO2 where SiNCs are enclosed by iso-surfaces with atomic concentrations of Si  , i.e.
, i.e.  atom-% Si (Fig. 5a). With the molar ratio of
atom-% Si (Fig. 5a). With the molar ratio of  in SiO2, we derive the molar SiO2 partition
in SiO2, we derive the molar SiO2 partition  of SiNCs via
of SiNCs via  Ignoring the P partition of ca. 1 atom-%, we get
Ignoring the P partition of ca. 1 atom-%, we get  mol-% SiO2 and
mol-% SiO2 and  mol-% Si for volumes enclosed by iso-surfaces. We note that the real
mol-% Si for volumes enclosed by iso-surfaces. We note that the real  value is lower due to APT projection artefacts. Detailed statistical analyses of APT data32 revealed that about 15% of the P atoms are found within SiNCs, whereas about 30% are trapped at the interface and about 55% reside in the surrounding SiO2 matrix. This relatively low P concentration in SiNCs can be explained by self-purification22,23,24,25, by solubilities of P in Si and SiO2 and by the high relative SiO2 volume of 85% in our samples. Zooming into the APT scan shows P atoms within SiNCs (Fig. 5b). A notable P concentration within SiNCs appears to disprove self-purification. However, interstitial P21 should have a much higher probability to exist in SiNCs as compared to P built into SiNC lattice sites. It does not require bond breakage and can exploit the fast diffusivity and high saturation density of P. An inclusion of such P configurations into ab-initio thermodynamic diffusion simulations would complement existing self-purification models which only consider foreign atoms at SiNC lattice sites. Tomogram data from APT used for a cluster analysis32 comprised numerous SiNCs in SiO2:P. The resulting proxigram shows the radial concentration of Si, O and P (Fig. 5c). We found a strong accumulation of P atoms in the SiNC/SiO2 interface shell with SiOx≈1 and also an increased P concentration within SiNCs.
value is lower due to APT projection artefacts. Detailed statistical analyses of APT data32 revealed that about 15% of the P atoms are found within SiNCs, whereas about 30% are trapped at the interface and about 55% reside in the surrounding SiO2 matrix. This relatively low P concentration in SiNCs can be explained by self-purification22,23,24,25, by solubilities of P in Si and SiO2 and by the high relative SiO2 volume of 85% in our samples. Zooming into the APT scan shows P atoms within SiNCs (Fig. 5b). A notable P concentration within SiNCs appears to disprove self-purification. However, interstitial P21 should have a much higher probability to exist in SiNCs as compared to P built into SiNC lattice sites. It does not require bond breakage and can exploit the fast diffusivity and high saturation density of P. An inclusion of such P configurations into ab-initio thermodynamic diffusion simulations would complement existing self-purification models which only consider foreign atoms at SiNC lattice sites. Tomogram data from APT used for a cluster analysis32 comprised numerous SiNCs in SiO2:P. The resulting proxigram shows the radial concentration of Si, O and P (Fig. 5c). We found a strong accumulation of P atoms in the SiNC/SiO2 interface shell with SiOx≈1 and also an increased P concentration within SiNCs.

P-doped SiNC SL in SiO2 scanned by APT.
Composition of SL, volumes with ≥70 atom-% Si are covered by red iso-surfaces, individual P atoms are shown in green (a). P atoms within 3 nm SiNC (b). Proxigram derived from SiNCs in left graph, showing radial concentration distribution of Si, O and P, latter with error bars for standard deviation (c). Zero of distance scale defined by interface located at SiO0.3 (85 mol-% Si and 15 mol-% SiO2, ignoring P content). Concentrations scanned along normal vector of interface into SiNCs, stopping at center of smallest SiNCs (size ca. 2.7 nm) to avoid signal back-folding. Horizontal dashed line shows average P concentration.
XANES spectroscopy
We measure P K spectra to determine the P oxidation stage by its K shell electron binding energy (Fig. 6), using a non-degenerate P-doped Si wafer (donor density =  cm−3 or 0.004 to 0.02 atom-%) for calibration. We assign XANES results to P environments using 1s core levels calculated by h-DFT with all-electron MO-BSs. P 1s core level energies from h-DFT correspond to 97.864% of P K XANES energies, see table 1. We calibrated h-DFT values by a factor of 1.02183 as supported by h-DFT P 1s core level energies of P2O5 (P+5) and P2O3 (P+3) approximants calculated with the same h-DFT route (Fig. 7).
cm−3 or 0.004 to 0.02 atom-%) for calibration. We assign XANES results to P environments using 1s core levels calculated by h-DFT with all-electron MO-BSs. P 1s core level energies from h-DFT correspond to 97.864% of P K XANES energies, see table 1. We calibrated h-DFT values by a factor of 1.02183 as supported by h-DFT P 1s core level energies of P2O5 (P+5) and P2O3 (P+3) approximants calculated with the same h-DFT route (Fig. 7).

XANES spectra of SiNC/SiO2 samples.
Normalized K shell spectra of P in SiNC/SiO2 SLs (2 nm, 3 nm, 4 nm, 5 nm), annealed bulk SiOx sample SiOx:P and P doped SiO2 sample SiO2:P shown together with doped Si wafer Si:P. Dashed gray lines show P oxidation stages.

P2O3 and P2O5 approximants for XANES calibration. Approximants of P4O6 (a) and P4O10 (b) cages constituting P2O3 (P+3) and P2O5 (P+5), respectively27.
Discussion
Origin of PL quenching of SiNCs containing P
Diffusion of P through Si proceeds at high rates during SiNC segregation anneal with  °C. Experimental data shown above and DFT calculations22,23,24,25 indicate that P appears to be within SiNCs on interstitial sites with a probability of nearly 100%. Auger recombination was assumed to cause PL quenching in SiNC/SiO2 material systems with high P concentrations3. Our findings do not support this assumption. With extremely low P ionization probabilities, the difference in free carrier densities of doped and intrinsic SiNCs is virtually nil. The Auger recombination
°C. Experimental data shown above and DFT calculations22,23,24,25 indicate that P appears to be within SiNCs on interstitial sites with a probability of nearly 100%. Auger recombination was assumed to cause PL quenching in SiNC/SiO2 material systems with high P concentrations3. Our findings do not support this assumption. With extremely low P ionization probabilities, the difference in free carrier densities of doped and intrinsic SiNCs is virtually nil. The Auger recombination  rate is33,34
rate is33,34  , where
, where  (
( ) are the density of free electrons (holes) and
) are the density of free electrons (holes) and  is the Auger scattering coefficient (
is the Auger scattering coefficient ( cm6/s for bulk Si34). Under high injection conditions (
cm6/s for bulk Si34). Under high injection conditions ( ), Auger recombination is
), Auger recombination is  which explains its strong increase at high free carrier densities35,36. P located within SiNCs or within SiOx shells around SiNCs are deep defect centers which appear to provide the most efficient and fastest path for non-radiative carrier recombination. This process explains PL quenching already at reasonably high P densities29 still below values reported elsewhere3,4. Co-doping with B was shown in experiment to
which explains its strong increase at high free carrier densities35,36. P located within SiNCs or within SiOx shells around SiNCs are deep defect centers which appear to provide the most efficient and fastest path for non-radiative carrier recombination. This process explains PL quenching already at reasonably high P densities29 still below values reported elsewhere3,4. Co-doping with B was shown in experiment to  PL intensities while transition energies decreased below those of undoped SiNCs3. Localized states of B and P at or within SiNCs provide strong radiative transitions as donor electrons directly relax into acceptor states. PL energies decreasing with co-doping 37 are a clear indication of this mechanism.
PL intensities while transition energies decreased below those of undoped SiNCs3. Localized states of B and P at or within SiNCs provide strong radiative transitions as donor electrons directly relax into acceptor states. PL energies decreasing with co-doping 37 are a clear indication of this mechanism.
P ionization in SiNC/SiO2 samples
P in bulk Si has four bonds to its 1-nn Si atoms, acquiring 0.09 electrons (2.2% bond ionicity). The P charge is  for neutral donors and
for neutral donors and  for ionized donors. P donors in bulk Si have
for ionized donors. P donors in bulk Si have  eV38, yielding a doping (ionization) probability at T = 300K of
eV38, yielding a doping (ionization) probability at T = 300K of  . The average charge of all P atoms in bulk Si is then
. The average charge of all P atoms in bulk Si is then  , corresponding to oxidation stage zero (P0). This value refers to the XANES peak at 2144.8 eV of the P doped Si wafer reference (Si:P), see Fig. 6. All P-doped SiNC/SiO2 samples (2 to 5 nm SiNC/SiO2 SLs, bulk) show peaks at 2143.7 eV. The 1.1 eV shift to lower binding energies shows that P in SiNC/SiO2 is much less positively ionized, corresponding to P−1. This result is corroborated by the Mulliken charges of P obtained from h-DFT and the analytical value of P in bulk Si (table 1). A hint of a signal shoulder might exist for all SiNC samples at the XANES peak for P0 at 2144.8 eV. An indication of a signal occurs for the smallest SiNC size of 2 nm, suggesting a slightly increased doping probability for ultrasmall SiNCs also observed by EPR10, though the ultrasmall SiNC size notably increases the signal background for XANES and presumably EPR. Our results show that P does not provide electrons to SiNCs embedded in SiO2 with reasonable probabilities.
, corresponding to oxidation stage zero (P0). This value refers to the XANES peak at 2144.8 eV of the P doped Si wafer reference (Si:P), see Fig. 6. All P-doped SiNC/SiO2 samples (2 to 5 nm SiNC/SiO2 SLs, bulk) show peaks at 2143.7 eV. The 1.1 eV shift to lower binding energies shows that P in SiNC/SiO2 is much less positively ionized, corresponding to P−1. This result is corroborated by the Mulliken charges of P obtained from h-DFT and the analytical value of P in bulk Si (table 1). A hint of a signal shoulder might exist for all SiNC samples at the XANES peak for P0 at 2144.8 eV. An indication of a signal occurs for the smallest SiNC size of 2 nm, suggesting a slightly increased doping probability for ultrasmall SiNCs also observed by EPR10, though the ultrasmall SiNC size notably increases the signal background for XANES and presumably EPR. Our results show that P does not provide electrons to SiNCs embedded in SiO2 with reasonable probabilities.
Conclusion
We carried out DFT calculations for the SiNC/SiO2 system to monitor the electronic nature of P. On a lattice site within OH-terminated SiNCs, P introduces a deep donor level with Eion = 0.51 eV; ionisation for small SiNCs is virtually nil, but is likely to increase for SiNCs with diminishing quantum confinement. However, formation energies of P on Si lattice sites22,23,24 suggest that P in SiNCs occurs almost exclusively on interstitial sites which is indirectly corroborated by experiments showing an extremely small density of P atoms with unpaired electrons even for 10 nm SiNCs9,10. On a central interstitial site within SiNCs, P cannot donate an electron (Eion >2 eV), but forms two deep defect levels with a recombination transition at 1.72 eV. At SiNC interfaces, fully saturated P have no impact on frontier molecular orbitals, leaving HOMO and LUMO energies virtually unchanged. In SiO shells around SiNCs, P is again unable to donate an electron, but induces a deep defect level which triggers massive recombination. This defect causes PL quenching – as opposed to Auger recombination – and increases SiO shell conductivities which were both interpreted as evidence for successful SiNC doping in the literature3,8. Although P atoms in SiO2 are deep defects which cannot donate electrons, they tremendously improve inter-NC conductivities in particular for electrons by diminishing electron (hole) barriers by 97% (85%) of the conduction (valence) band offset between bulk phases of Si and SiO2. Massively increased conductivities were assumed to prove successful SiNC doping6,8. APT analyses revealed an enrichment of P at SiNC interfaces, which appears to be due to DB saturation and support h-DFT analyses of fully O-saturated P at SiNC interfaces. SiNCs were found to contain significant amounts of P. While this appears to contradict self-purification theory, interstitial P with considerably more favourable thermodynamics and its high diffusivity and saturation density has not been considered in self-purification modeling. Core level (K shell) electron energies of P in SiO2 and SiNC/SiO2 samples were measured by XANES at room temperature. In contrast to bulk Si, P atoms in SiNC/SiO2 samples could not donate electrons into 2 to 5 nm size NCs in SLs or in annealed bulk SiOx films with reasonable probabilities, confirming our h-DFT results. We conclude that conventional doping of SiNCs with P does not provide majority charge carriers to SiNCs embedded in SiO2. Alternative approaches for majority carrier introduction into embedded SiNCs and ultrasmall Si nanovolumes such as embedding material effects39 have to be explored to advance SiNC-based nanoelectroncis and ULSI.
Methods
Sample Preparation
Size-controlled SiNCs in SiO2 were fabricated by deposition of P-doped Si-rich oxide (SiO0.93)/ intrinsic SiO2 SLs by plasma enhanced chemical vapor deposition and subsequent annealing (1150 °C, 1 h). During deposition, P was incorporated by adding 1% PH3 to Ar, resulting P concentrations were ca. 1 atom-% as found by secondary ion mass spectroscopy (SIMS)29,32. All samples were fabricated on low B-doped Si wafers (20 Ωcm) with 30 nm SiO2 layers to prevent P diffusion into Si substrates during anneal. For APT, P doped SLs with 30 bilayers and 5 nm nominal NC size were fabricated. Samples with 50 bilayers and nominal NC sizes from 2 to 5 nm in steps of 1 nm were chosen for XANES. In addition, 300 nm thick P-doped SiO2 and SiO0.93 samples were fabricated as references.
Hybrid Density Functional Theory (h-DFT) Calculations
Approximants were calculated with non-periodic boundary conditions and underwent geometrical optimization with the B3LYP h-DF40,41 and the 6-31G(d) all-electron molecular-orbital basis set (MO-BS)42,43,44 using the GAUSSIAN 03 and GAUSSIAN 09 suites45,46. RMS and peak force convergence limits were 15.4 meV/Å ( Ha/Å) and 23.1 meV/Å (
Ha/Å) and 23.1 meV/Å ( Ha/Å), respectively. Electronic structures were computed with the same route; B3LYP/6-31G(d) //
Ha/Å), respectively. Electronic structures were computed with the same route; B3LYP/6-31G(d) //  B3LYP/6-31G(d). Additional information is available on accuracy tests and tests of functional group termination as approximation of the dielectric31,39,47. During all calculations, no MO symmetry constraints were applied and tight convergence criteria were set for the self-consistent field routine.
B3LYP/6-31G(d). Additional information is available on accuracy tests and tests of functional group termination as approximation of the dielectric31,39,47. During all calculations, no MO symmetry constraints were applied and tight convergence criteria were set for the self-consistent field routine.
Alternative approaches to the B3LYP h-DF with similar accuracy are the Heyd-Scuseria-Ernzerhof h-DF (HSE06)48 and the Becke-Johnson exchange potential49,50, latter used within the Perdew-Burke-Ernzerhof (PBE) DF51 generalized gradient approximation (GGA) scheme.
Characterisation
We examined the position of P within the SiNC/SiO2 system by APT using a Cameca LEAP 4000X HR instrument with a reflectron-type time-of-flight mass spectrometer and a pulsed UV laser (355 nm, 10 ps pulse length, 70 pJ pulse energy, 100 kHz repetition rate). During the analyses (chamber pressure  mbar), specimens were cooled to temperatures of around 76 K. The mass resolution of the system was
mbar), specimens were cooled to temperatures of around 76 K. The mass resolution of the system was  , around 36% of all atoms are detected. Specimen tips have been prepared by the cut-and-lift-out technique using an ALTURA 875 dual-beam Focused Ion Beam instrument32.
, around 36% of all atoms are detected. Specimen tips have been prepared by the cut-and-lift-out technique using an ALTURA 875 dual-beam Focused Ion Beam instrument32.
The P K-edge absorption in XANES was measured at the SUL-X beamline at the Angströmquelle Karlsruhe (ANKA). Monochromatic X-rays were obtained using a Si(111) double crystal monochromator with an energy resolution of about 0.2 eV at 2150 eV with fixed exit. Scans were carried out using a shallow incident angle to maximize the SL or thin layer volume of samples for excitation. Absorption was measured by monitoring the P K fluorescence emission using a seven element Si(Li) fluorescence detector (SGX Sensortech). The signal is normalized to the incident photon flux measured simultaneously by a custom made ionization chamber (ADC, US) filled with N2 at a pressure of 50 mbar. Energies were calibrated to 2152 eV at the white line maximum of the P K-edge XANES spectrum of NaH2PO2 ⋅ 2 H2O. The energy step size across the XANES region was 0.2 eV. XANES peaks of our samples show a full width half maximum of ca. 2 eV. P K XANES spectra have been pre- and post-edge background corrected and normalized to the edge jump with the ATHENA program of the IFEFIT package52.
fluorescence emission using a seven element Si(Li) fluorescence detector (SGX Sensortech). The signal is normalized to the incident photon flux measured simultaneously by a custom made ionization chamber (ADC, US) filled with N2 at a pressure of 50 mbar. Energies were calibrated to 2152 eV at the white line maximum of the P K-edge XANES spectrum of NaH2PO2 ⋅ 2 H2O. The energy step size across the XANES region was 0.2 eV. XANES peaks of our samples show a full width half maximum of ca. 2 eV. P K XANES spectra have been pre- and post-edge background corrected and normalized to the edge jump with the ATHENA program of the IFEFIT package52.
Additional Information
How to cite this article: König, D. et al. Location and Electronic Nature of Phosphorus in the Si Nanocrystal – SiO2 System. Sci. Rep. 5, 9702; doi: 10.1038/srep09702 (2015).
References
Zacharias, M. et al. Size-controlled highly luminescent silicon nanocrystals: A SiO/SiO2 superlattice approach. Appl. Phys. Lett. 80, 661–663 (2002).
Böer, K. W. Survey of Semiconductor Physics, Vol. 2. Van Nostrand Reinhold: New York, 1992).
Fujii, M., Yamaguchi, Y., Takase, Y., Ninomiya, K. & Hayashi, S. Photoluminescence from impurity codoped and compensated Si nanocrystals. Appl. Phys. Lett. 87, 211919 (2005).
Pi, X. D., Gresback, R., Liptak, R. W., Campbell, S. A. & Kortshagen, U. Doping efficiency, dopant location and oxidation of Si nanocrystals. Appl. Phys. Lett. 92, 123102 (2008).
Hao, X. J. et al. Effects of phosphorus doping on structural and optical properties of silicon nanocrystals in a SiO2 matrix. Thin Sol. Films 517, 5646–5652 (2009).
Hao, X. J. et al. Phosphorus-doped silicon quantum dots for all-silicon quantum dot tandem solar cells. Solar Energy Mater. & Solar Cells 93, 1524–1530 (2009).
Perego, M., Bonafos, C. & Fanciulli, M. Phosphorus doping of ultra-small silicon nanocrystals. Nanotechnology 21, 025602 (2010).
Khelifi, R. et al. Efficient n-type doping of Si nanocrystals embedded in SiO2 by ion beam synthesis. Appl. Phys. Lett. 102, 013116 (2013).
Stegner, A. R. et al. Electronic Transport in Phosphorus-Doped Silicon Nanocrystal Networks. Phys. Rev. Lett. 100, 026803 (2008).
Stegner, A. R. et al. Doping efficiency in freestanding silicon nanocrystals from the gas phase: Phosphorus incorporation and defect-induced compensation. Phys. Rev. B 80, 165326 (2009).
Perez-Wurfl, I. et al. Si nanocrystal p-i-n diodes fabricated on quartz substrates for third generation solar cell applications. Appl. Phys. Lett. 95, 153506 (2011).
Lin, D., Ma, L., Conibeer, G. & Perez-Wurfl, I. Study on electrical properties of Si quantum dots based materials. Phys. Stat. Sol. B 248, 472–476 (2011).
Liedke, B., Heinig, K.-H., Mücklich, A. & Schmidt, B. Formation and coarsening of sponge-like Si-SiO2 nanocomposites. Appl. Phys. Lett. 103, 133106 (2013).
Gutsch, S. et al. Charge transport in Si nanocrystal/SiO2 superlattices. J. Appl. Phys. 113, 133703 (2013).
Pearson, G. L. & Bardeen, J. Electrical Properties of Pure Silicon and Silicon Alloys Containing Boron and Phosphorus. Phys. Rev. 75, 865–883 (1948).
Kambham, A. K., Kumar, A., Florakis, A. & Vandervorst, W. Three-dimensional doping and diffusion in nano scaled devices as studied by atom probe tomography. Nanotechnology 24, 275705 (2013).
Koelling, S. et al. Direct Imaging of 3D Atomic-Scale Dopant-Defect Clustering Processes in Ion-Implanted Silicon. Nano Lett. 13, 2458–2462 (2013).
Norris, D. J., Efros, A. L. & Erwin, S. C. Doped Nanocrystals. Science 319, 1776–1779 (2008).
Pereira, R. N., Almeida, A. J., Stegner, A. R., Brandt M. S. & Wiggers, H. Exchange-Coupled Donor Dimers in Nanocrystal Quantum Dots. Phys. Rev. Lett. 108, 126806 (2012).
Wagner, C. D., Davis, L. E., Riggs, W. M. The energy dependence of the electron mean free path. Surf. Interface Anal. 2, 53–55 (1980).
Brown, G. W. et al. Observation of substitutional and interstitial phosphorus on clean Si(100)-(2×1) with scanning tunneling microscopy. Phys. Rev. B 72, 195323 (2005).
Ossicini, S. et al. Simultaneously B- and P-doped silicon nanoclusters: Formation energies and electronic properties. Appl. Phys. Lett. 87 173120 (2005).
Dalpian, G. M. & Chelikowsky, J. R. Self-Purification in Semiconductor Nanocrystals. Phys. Rev. Lett. 96, 226802 (2006).
Dalpian, G. M. & Chelikowsky, J. R. Dalpian and Chelikowsky Reply: Phys. Rev. Lett. 100, 179703 (2008).
Chan, T.-L., Tiago, M. L., Kaxiras, E. & Chelikowsky, J. R. Size Limits on Doping Phosphorus into Silicon Nanocrystals. Nano Lett. 8, 596–600 (2008).
Nicollian, E. H. & Brews, J. R. MOS (Metal Oxide Semiconductor) Physics and Technology. Wiley-Interscience: New York, 1982).
Holleman, A. F., Wiberg, E. & Wiberg, N. Lehrbuch der Anorganischen Chemie. [101st Ed] Walter deGruyter: Berlin, 1995). in German.
Zimina, A., Eisebitt, S., Eberhardt, W., Heitmann, J. & Zacharias, M. Electronic structure and chemical environment of silicon nanoclusters embedded in a silicon dioxide matrix. Appl. Phys. Lett. 88, 163103 (2006).
Gutsch, S. et al. Doping efficiency of phosphorus doped silicon nanocrystals embedded in a SiO2 matrix. Appl. Phys. Lett. 100, 233115 (2012)
Singh, A. & Davis, E. A. THE SiOx:Hy THIN FILM SYSTEM II. Optical bandgap behavior. J. of Non-Cryst. Sol. 122, 233–240 (1990).
König, D., Rudd, J., Green, M. A. & Conibeer, G. Role of the Interface for the Electronic Structure of Silicon Quantum Dots. Phys. Rev. B 78, 035339 (2008).
Gnaser, H. et al. Phosphorus doping of Si nanocrystals embedded in silicon oxynitride determined by atom probe tomography. J. Appl. Phys. 115, 034304 (2014).
Lannoo, M., Delerue, C. & Allan, G. Theory of radiative and nonradiative transitions for semiconductor nanocrystals. J. Luminescence 70, 170–184 (1996).
Sze, S. M. & Ng, K. K. Physics of Semiconductor Devices, 3rd Ed. Wiley 2007).
Trupke, T. et al. Temperature dependence of the radiative recombination coefficient of intrinsic crystalline silicon. J. Appl. Phys. 94, 4930–4937 (2003).
Richter, A., Glunz, S. W., Werner, F., Schmidt, J. & Cuevas, A. Improved quantitative description of Auger recombination in crystalline silicon. Phys. Rev. B 86, 165202 (2012).
Fujii, M., Yamaguchi, Y., Takase, Y., Ninomiya, K. & Hayashi, S. Control of photoluminescence properties of Si nanocrystals by simultaneously doping n- and p-type impurities. Appl. Phys. Lett. 85, 1158–1160 (2004).
Faulkner, R .A. Higher donor excited states for prolate-spheroid conduction bands: A re-evaluation of silicon and germanium. Phys. Rev. 184, 713–721 (1969).
König, D., Hiller, D., Gutsch, S. & Zacharias, M. Energy Offset Between Silicon Quantum Structures: Interface Impact of Embedding Dielectrics as Doping Alternative. Adv. Mater. Interfaces 1, 1400359 (2014).
Becke, D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C., Yang, W. & Parr, R. G. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. B 37, 785–789 (1988).
Hehre, W. J., Ditchfield, R. & Pople, J. A. Self-Consistent Molecular Orbital Methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 56, 2257–2261 (1972).
Francl, M. M. et al. Self-Consistent Molecular Orbital Methods. 23. A polarization-type basis set for 2nd-row elements. J. Chem. Phys. 77, 3654–3666 (1982).
Rassolov, V. A., Ratner, M. A., Pople, J. A., Redfern, P. C. & Curtiss, L. A. 6-31G* Basis Set for Third-Row Atoms. J. Comp. Chem. 22, 976–984 (2001).
Frisch, M.J. et al. GAUSSIAN03, Revision D.02, Gaussian Inc.: Wallingford, CT, (2004), Date of access: 28/11/2014; http://www.gaussian.com/g_misc/g03/citation_g03.htm.
Frisch, M.J. et al. GAUSSIAN09, Revision A.02, Gaussian Inc., Wallingford, CT (2010), Date of access: 28/11/2014; http://www.gaussian.com/g_tech/g_ur/m_citation.htm
König, D., Rudd, J., Green, M. A. & Conibeer, G. Critical Discussion of Computation Accuracy. supplemental material to Phys. Rev. B 78, 035339 (2008), Date of access: 28/11/2014; http://journals.aps.org/prb/abstract/10.1103/PhysRevB.78.035339.
Krukau, A. V., Vydrov, O. A., Izmaylov, A. F. & Scuseria, G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 125, 224106 (2006).
Becke, A. D. & Johnson, E. R. A simple effective potential for exchange. J. Chem. Phys. 124, 221101 (2006).
Koller, D., Tran, F. & Blaha, P. Improving the modified Becke-Johnson exchange potential. Phys. Rev. B 85, 155109 (2012).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865–3868 (1996).
Ravel, B. & Neville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption Spectroscopy using IFEFFIT. J. Synchrotron Radiation 12, 537–541 (2005).
Franke, R. & Hormes, J. The P K-near edge absorption spectra of phosphates. Physica B 216, 85–95 (1995).
Acknowledgements
D.K. acknowledges use of compute cluster Leonardi, engineering faculty, UNSW and financial support by Australian Government via Australian Renewable Energy Agency (ARENA). Australian government does not accept responsibility for any information expressed herein. D.H. acknowledges funding by the German Research Foundation (DFG) under grant number HI 1779/3-1. D.K., S.G. and D.H. acknowledge DAAD-Go8 joint research cooperation schemes (54385434 and 57060437). The ANKA Synchrotron Radiation Facility is acknowledged for providing beamtime.
Author information
Authors and Affiliations
Contributions
D.K. developed concepts, carried out h-DFT calculations, participated in characterisation data post processing and drafted manuscript. S.G. and D.H. processed samples and participated in sample characterisation and data post processing. H.G., M.K., M.W., J.G. and R.S. characterized samples and processed associated data. D.H., D.K. and M.Z. guided the project. All authors discussed and revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
König, D., Gutsch, S., Gnaser, H. et al. Location and Electronic Nature of Phosphorus in the Si Nanocrystal − SiO2 System. Sci Rep 5, 9702 (2015). https://doi.org/10.1038/srep09702
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09702
This article is cited by
-
Grain-boundary segregation of magnesium in doped cuprous oxide and impact on electrical transport properties
Scientific Reports (2021)
-
Boron-Incorporating Silicon Nanocrystals Embedded in SiO2: Absence of Free Carriers vs. B-Induced Defects
Scientific Reports (2017)
-
Defect-Induced Luminescence Quenching vs. Charge Carrier Generation of Phosphorus Incorporated in Silicon Nanocrystals as Function of Size
Scientific Reports (2017)
-
Modulation Doping of Silicon using Aluminium-induced Acceptor States in Silicon Dioxide
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
