
Abstract
Epidemiological evidence indicates that thyrotropin (TSH) is positively correlated with the severity of obesity. However, the mechanism remains unclear. Here, we show that TSH promoted triglyceride (TG) synthesis in differentiated adipocytes in a thyroid hormone-independent manner. Mice with subclinical hypothyroidism, which is characterized by elevated serum TSH but not thyroid hormone levels, demonstrated a 35% increase in the total white adipose mass compared with their wild-type littermates. Interestingly, Tshr KO mice, which had normal thyroid hormone levels after thyroid hormone supplementation, resisted high-fat diet-induced obesity. TSH could directly induce the activity of glycerol-3-phosphate-acyltransferase 3 (GPAT3), the rate-limiting enzyme in TG synthesis, in differentiated 3T3-L1 adipocytes. However, following either the knockdown of Tshr and PPARγ or the constitutive activation of AMPK, the changes to TSH-triggered GPAT3 activity and adipogenesis disappeared. The over-expression of PPARγ or the expression of an AMPK dominant negative mutant reversed the TSH-induced changes. Thus, TSH acted as a previously unrecognized master regulator of adipogenesis, indicating that modification of the AMPK/PPARγ/GPAT3 axis via the TSH receptor might serve as a potential therapeutic target for obesity.
Similar content being viewed by others


Introduction
Obesity, the excess accumulation of white adipose tissue (WAT), is a major health hazard worldwide and the epidemic incidence of obesity is increasing. Obesity is an established risk factor for metabolic diseases, including type 2 diabetes, hypertension and nonalcoholic fatty liver disease1,2. Obesity-related metabolic diseases are accompanied by abnormal serum lipid parameters3, which are usually caused by higher TG levels and ectopic TG accumulation. WAT contains white adipocytes, which specialize in the storage of energy as triglycerides (TGs) and in the secretion of numerous adipokines that affect numerous aspects of metabolism4. The etiology of obesity is complicated, with both genetic and environmental factors influencing its development and susceptibility5. In this study, we identified a hormone involved in obesity.
Thyrotropin (thyroid stimulating hormone, TSH) is a hypophyseal hormone, the major role of which is to stimulate thyrocyte proliferation, iodide uptake, hormonogenesis and the release of thyroid hormones (T4 and T3). Numerous studies have confirmed the association between serum TSH levels and obesity. Epidemiological evidence has indicated a positive correlation between elevated serum TSH concentrations and body mass index (BMI) in euthyroid subjects6,7,8,9,10,11. In patients with metabolic syndrome and/or subclinical hypothyroidism (SCH, a type of thyroid disease accompanied only by elevated serum TSH levels), serum TSH levels are correlated with the severity of obesity12,13. Furthermore, obese people exhibit higher serum TSH levels than non-obese people14,15,16. However, the molecular mechanism by which TSH affects obesity has not been fully elucidated.
Expression of the TSH receptor (TSHR), once thought to be limited to thyrocytes, has been detected in numerous extrathyroidal tissues, including liver17 and adipose tissues18,19. Our previous study indicated that TSH promotes 3T3-L1 preadipocyte differentiation. Additionally, knocking down Tshr blocked the effects of TSH on preadipocyte differentiation20. Similar results were observed in rat preadipocytes18, human orbital preadipocyte fibroblasts21,22 and mouse embryonic stem cells23.
Obesity is a hypertrophic disease resulting from an increase in the number or size of individual adipocytes. We previously demonstrated that TSH could increase the number of adipocytes by promoting preadipocytes to differentiate into mature adipocytes20. The number of adipocytes is set during childhood and adolescence24. Adipocyte hypertrophy due to increased TG synthesis was recently shown to be the determinant of the development of adult obesity25. Brook showed an increase in adipose cell size among all obese subjects, but the total number of adipose cells was only increased in obese children and in adults who developed obesity during childhood26. Thus, adipocyte size is a major determinant of obesity in adults. Still, the role of TSH in regulating TG synthesis among differentiated adipocytes has not been completely established.
Glycerol-3-phosphate acyltransferase (GPAT) is the rate-limiting enzyme involved in TG synthesis27. GPAT3 is the major GPAT isoform expressed in adipocytes and plays a crucial role in adipogenesis28. The over-expression of GPAT3 in mammalian cells resulted in increased TG formation29, whereas the targeted knockdown of GPAT3 in 3T3-L1 cells significantly impaired adipogenesis28. Recent studies have indicated that the expression and activity of GPAT3 are regulated by insulin29, alcohol30, the glucocorticoid receptor31 and thiazolidinediones (peroxisome proliferator-activated receptor γ agonists)29. However, whether TSH affects the adipogenesis of differentiated adipocytes through GPAT3 has not been previously investigated. In the present study, we tested the hypothesis that TSH might upregulate GPAT3 expression, resulting in adipogenesis and obesity. This study provides a more comprehensive understanding of the pathophysiological effects of TSH on adipogenesis and suggests a novel interpretation of the mechanism that causes metabolic syndrome (MS) or SCH patients to develop obesity and obesity-related metabolic diseases.
Results
TSH regulates triglyceride content in adipose tissue
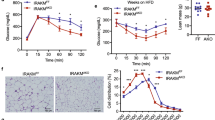
Based on the characteristic elevated serum TSH levels only in SCH, we first established the SCH mouse model. The SCH mice exhibited equal serum fT4 levels and higher TSH levels compared with their littermate controls (Fig. 1A). Meanwhile, higher body, total fat pad weight and BMI were found in the SCH mice (Fig. 1B). We found an apparent increase in total fat pad mass and adipocyte size in SCH mice compared with those in control mice (Fig. 1C). Epididymal WAT (Epi-WAT), a typical visceral adipose tissue, has been frequently used to study the relationship between adipose tissue and obesity32. As shown in Fig. 1D, the Epi-WAT mass to body weight ratio revealed a 25% relative increase in SCH mice compared with their littermate controls. In addition, both the Epi-WAT TG content and serum TG levels were increased in SCH mice. The serum concentration of adiponectin, an adipokine specifically secreted by adipocytes that is known to regulate glucolipid metabolism, was also reduced.

TSH-stimulated mature adipocyte TG synthesis.
Male C57/BL6 mice were given methimazole (MMI, 0.04 mg/kg BW·d, SCH group, n = 8) or a corresponding volume of vehicle (control group, n = 8) for 12 weeks. (A) The plasma fT4 and TSH levels were assayed, respectively. (B) The body weight, total fat pad weight and BMI levels were assayed, respectively.(C) Representative images and H&E staining of total white adipose tissue (Epi-WAT). (D) Effect of TSH on white adipose tissue metabolism. Epi-WAT index, Epi-WAT triglyceride (TG) contents, plasma levels of TG and adiponectin were measured, respectively. (E) Intracellular TG contents were determined with the indicated concentrations of TSH for indicated time in differentiated 3T3-L1 cells and were normalized by the total proteins in same a sample. The results are expressed as the mean ± SD. #p < 0.05 compared with control; ★p < 0.05 compared with 1 μM TSH at 24 h; ˙p < 0.05 compared with 2 μM TSH at 24 h; ♦p < 0.05 compared with 1 μM TSH at 48 h. (F) Oil red O staining differentiated 3T3-L1 cells treated with 2 μM TSH for 48 h.
To confirm the direct effect of TSH on adipocytes, we established the differentiated 3T3-L1 adipocyte model. TSH stimulation significantly increased the number of lipid droplets (Fig. 1E) and the intracellular TG contents increased in a dose- and time-dependent manner in vitro (Fig. 1F). These results are consistent with the increase in TG caused by TSH treatment.
Regulation of TG synthesis by TSH through GPAT3
TSH binds to the TSH receptor (TSHR) to perform its actions. To identify the molecular mechanism underlying the TSH-induced effect on TG synthesis, we generated Tshr knockout mice (Tshr−/− mice). Tshr−/− mice fed with the T4-supplemented diet exhibited equal levels of serum TSH and fT4 compared to the littermate control Tshr+/+ mice33. We conducted a systematic and quantitative analysis of a panel of genes involved in adipogenesis in WAT. The expression of most of the genes involved in adipogenesis was downregulated in the Epi-WAT of Tshr−/− mice (Fig. 2A), specifically including GPAT3 and diacylglyceride acyltransferase-2 (DGAT2), which control the initial and final steps of TG synthesis, respectively. The levels of GPAT3 and DGAT2 decreased by 88% and 62%, respectively, compared with those of the Tshr+/+ mice. The levels of peroxisome proliferator-activated receptor γ (PPARγ), a key molecule involved in adipogenesis, decreased by 55%. The expression of genes involved in fatty acid synthesis, uptake and TG storage, including sterol regulatory element-binding protein 1c (SREBP-1c), fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), stearoyl-CoA desaturase (SCD), fatty acid-binding protein 4 (FABP4), CD36 and FSP27, were downregulated in Tshr−/− mice. Meanwhile, the levels of CPT-1 and Dio2, which promote fatty acid oxidation and accelerate thyroid hormone conversion, respectively, did not change in Tshr−/− mice. These results imply that Tshr deletion in vivo results in an altered gene expression profile, leading to thyroid hormone-independent inhibition of the anabolism of triglycerides.

TSH promotes adipocyte GPAT3 activity, leading to elevated TG synthesis.
Homozygous Tshr-knockout mice (Tshr−/−) were supplemented with T4 (serum T4 levles were similar to that in the wild type mice, p > 0.05) to avoid the effects of the thyroid hormones. Tshr+/+ mice were used as controls. (A) RT-PCR analysis of adipogenesis-related gene expression in the Epi-WAT. #p < 0.05 compared with Tshr+/+ mice. (B) The expression of GPAT3 protein was detected by western blot. (C) RT-PCR analysis of GPAT3 mRNA in TSH (2 μM, for 48 h)-treated differentiated 3T3-L1 cells. #p < 0.05 compared with control. (D) The time-dependent and does-dependent effects of TSH on GPAT3 expression in differentiated 3T3-L1 cells. (E) Immunofluorescence staining of GPAT3 in differentiated 3T3-L1 cells treated with the indicated concentrations of TSH for 24 h. The blue color depicts the DAPI-stained nuclei and the red shows the GPAT3 protein (TRITC-conjugated). (F) HEK293 cells were transfected with GPAT3 luciferase reporter construct vectors for 48 h, then treated with vehicle or TSH (1 or 2 μM) for an additional 24 h. GPAT3 activity was detected by dual luciferase reporters. #p < 0.05 compared with control; ★p < 0.05 compared with 1 μM TSH at 24 h. (G–H) RNAi of GPAT3 inhibited TSH-induced TG synthesis. After transfection with non-targeting or GPAT3 siRNA for 48 h, the differentiated 3T3-L1 cells were incubated with TSH (2 μM) or vehicle for another 24 h. (G) Lipid droplet contents and (H) intracellular TG contents were assayed. The results are expressed as the mean ± SD. #p < 0.05 compared with the non-targeting siRNA group; ★p < 0.05 compared with TSH group. All panels above are representative of 3 independent experiments. All the gels were run under the same experimental conditions and key data cropped blots are used here. The full-length gel images are available in Supplementary Fig. 6.
Through molecular screening, we noted that the level of GPAT3, the rate-limiting enzyme of TG synthesis in adipocytes34, was significantly decreased in Tshr−/− mice. Consistently, the GPAT3 protein levels were significantly decreased in the Epi-WAT of Tshr−/− mice (Fig. 2B).
However, after treating differentiated 3T3-L1 adipocytes with TSH, the GPAT3 mRNA levels were enhanced (Fig. 2C) and the protein levels increased in a dose- and time-dependent manner (Fig. 2D). Similar changes were observed in the primary adipocytes of mice and humans (Figs. S1A and B). Immunofluorescence analysis indicated that after TSH treatment, the increased GPAT3 protein was mainly located in the cytoplasm in a dose-dependent manner (Fig. 2E).
We next determined the response of the promoter region of GPAT3 to TSH. When GPAT3 promoter constructs linked to a luciferase reporter gene were transfected into HEK293 cells, the luciferase activity of the GPAT3 promoter sharply increased following TSH challenge in a dose-dependent manner (Fig. 2F).
To assess whether TSH induces adipogenesis through GPAT3, we transfected GPAT3 siRNA into differentiated 3T3-L1 adipocytes for 48 hours, followed by an additional incubation with TSH for 24 hours. GPAT3 silencing markedly attenuated TSH-induced adipogenesis. The Oil Red O staining was weak (Fig. 2G) and the TG content significantly dropped (Fig. 2H) relative to the group treated with only TSH.
PPARγ is indispensable in TSH-induced adipogenesis
We observed that the PPARγ mRNA and protein levels were decreased in the Tshr−/− mice in vivo (Fig. 2A and Fig. 3A). Consistently, in differentiated adipocytes in vitro, TSH promoted the expression and nuclear accumulation of PPARγ protein (Fig. 3B) in a dose- and time-dependent manner (Fig. 3C; Figs. S1A and B). Additionally, TSH promoted PPARγ mRNA expression (Fig. 3D).

PPARγ is an essential requirement for the regulation of TSH-induced adipogenesis.
(A) PPARγ nuclear proteins were detected by western blot in the Epi-WAT of Tshr−/− and Tshr+/+ mice, respectively. (B) Immunofluorescence staining of PPARγ in differentiated 3T3-L1 cells treated with the indicated concentrations of TSH for 24 h. The blue color depicts the DAPI-stained nuclei and the green shows the PPARγ protein (FITC conjugated). (C) The time-dependent and does-dependent effects of TSH on PPARγ expression in differentiated 3T3-L1 cells. (D) RT-PCR analysis of PPARγ mRNA in TSH (2 μM, for 48 h)-treated differentiated 3T3-L1 cells. #p < 0.05 compared with control. (E) HEK293 cells were transfected with PPRE-luciferase reporter constructs, along with PPARγ expression vectors. Cells were treated with the vehicle, GW9662 (20 μM), or TSH (1 or 2 μM) for an additional 24 h. PPARγ activity was detected by dual luciferase reporters. #p < 0.05 compared with PPARγ plasmid group, ★p < 0.05 compared with PPARγ plasmid + GW9662 group. (F–G) Differentiation 3T3-L1 cells were infected with the PPARγ siRNA or non-targeting siRNA to silence PPARγ expression. After transfection with non-targeting or PPARγ siRNA for 48 h, differentiated 3T3-L1 cells were incubated with TSH (2 μM) or vehicle for another 24 h. (F)Intracellular TG contents were assayed, the results are expressed as the mean ± SD. #p < 0.05 compared with the non-targeting siRNA group; ★p < 0.05 compared with TSH group; ˙p < 0.05 compared with PPARγ siRNA group. (G) GPAT3 protein levels were analyzed using western blots. (H) After transfection with or without the PPARγ S112A plasmid for 48 h, differentiated 3T3-L1 cells were incubated with TSH (2 μM) or vehicle for another 24 h. GPAT3 protein was analyzed by western blot. All of the above panels are representative of 3 independent experiments. All the gels were run under the same experimental conditions and key data cropped blots are used here. The full-length gel images are available in Supplementary Fig. 6.
To confirm the effect of TSH on PPARγ activity, HEK293 cells were cotransfected with a luciferase reporter gene construct (PPREx3-tk-luc; PPRE) and a PPARγ over-expression plasmid. As expected, the PPARγ inhibitor (GW9662) reduced the luciferase reporter activity to a greater extent than that observed in the control. In contrast, TSH alone elevated the reporter activity in a dose-dependent manner. However, preincubating the cells with GW9662 prevented this increase in activity (Fig. 3E).
To reconfirm that TSH-induced adipogenesis is mediated by PPARγ, we used RNA interference to block PPARγ expression in differentiated 3T3-L1 adipocytes. PPARγ siRNA significantly knocked down PPARγ mRNA expression (p < 0.05) (data not shown). TSH-stimulated adipogenesis and GPAT3 upregulation were significantly attenuated by PPARγ siRNA (Figs. 3F and G). In addition, van Beekum reported that the phosphorylation of PPARγ at Ser112 resulted in decreased transcriptional activity in the reporter assays35. After the cells were transfected with the PPARγ S112A plasmid (Ser112 mutated to alanine), the regulation of GPAT3 by TSH was enhanced (Fig. 3H).
PPARγ inhibitor ameliorates high-fat diet (HFD)-induced obesity in vivo
To determine whether PPARγ is the upstream factor involved in the effects of GPAT3 on HFD-induced obesity in vivo, GW9662-administered mice were used. HFD-fed C57/BL6 mice (13 weeks old) were supplemented with or without GW9662 for 5 weeks. Compared with vehicle, GW9662 ameliorated the HFD-induced increase in weight, Epi-WAT index and adipocyte size (Figs. 4A and B). Furthermore, GW9662 inhibited GPAT3 expression (Fig. 4C), decreased intracellular and plasma TG levels (Fig. 4D and Fig. S2A) and suppressed adipose TG synthesis in vivo. In addition, the function of the adipose tissue was changed due to the suppression of adiponectin release by GW9662 (Fig. 4D). These results indicated that PPARγ is essential for the regulation of TG synthesis via TSH.

Antagonism of PPARγ prevents high-fat-diet-induced obesity in vivo.
High-fat-diet (HFD) chowed C57/BL6 mice were subjected to GW9662 at a dose of 0.35 mg/kg per day in their drinking water for 5 weeks (GW9662 group, n = 8) or to a corresponding volume of vehicle (control group, n = 8). (A) Increases in body fat were suppressed by GW9662 treatment and the adipose mass of Epi fat, retroperitoneal fat (RP fat) and adipocyte size were diminished in the GW9662 group. (B) Epi-WAT weight, WAT index, weight at 18 weeks and weight changes were measured. #p < 0.05 compared with control group. (C) The protein expression of GPAT3 was measured using western blots. The gels were run under the same experimental conditions and cropped blots are used here. The full-length gel images are available in Supplementary Fig. 6. (D) Intracellular TG levels and plasma adiponectin were detected, respectively. The results are expressed as the mean ± SD. #p < 0.05 compared with control group. All of the above panels are representative of 3 independent experiments.
TSH-stimulated adipogenesis in differentiated adipocytes is dependent on AMPK
As an energy sensor in metabolic tissues, AMP-activated protein kinase (AMPK) plays an important role in the regulation of PPARγ activity in adipocytes36. In Tshr−/− mice, AMPK Thr172 phosphorylation was increased compared with that in Tshr+/+ mice (Fig. 5A): these effects have been reported to correlate with kinase activity. Meanwhile, the key downstream targets of AMPK (i.e., SREBP-1c, FAS, SCD and ACC)37 were reduced in Tshr−/− mice (Fig. 2A). Based on these changes, we hypothesized that the functions of TSH are activated in association with AMPK.

TSH-stimulated adipogenesis was dependent on AMPK.
(A) Phosphorylated protein at Thr172 of AMPKα subtype (p-AMPK) was detected by western blot in the Epi-WAT of Tshr−/− and Tshr+/+ mice. (B) The time-dependent and dose-dependent effects of TSH on p-AMPK protein expression in differentiated 3T3-L1 cells. (C) After cells were incubated with TSH (1 μM), AICAR (1 mM) or vehicle for 24 h, the differentiated 3T3-L1 cells underwent AMPK activity assays with SAMS peptides. #p < 0.05 compared with control; ★p < 0.05 compared with TSH group. (D–F) Differentiated 3T3-L1 cells were transfected with constitutively activated AMPK (CA-AMPK) or dominant negative mutant of AMPK (DN-AMPK) plasmids for 48 h and then stimulated with or without 1 μM TSH for another 24 h. (D) Intracellular TG contents were detected. #p < 0.05 compared with control; ★p < 0.05 compared with TSH group; ˙p < 0.05 compared with CA-AMPK group; ♦p < 0.05 compared with CA-AMPK + TSH group. GPAT3 and PPARγ expression and location were detected by (E) western blotting or (F) immunofluorescent staining, respectively. (G–H) Tshr+/+ or Tshr−/− mice were injected intraperitoneally with AICAR (n = 5, respectviely) or PBS (n = 5, respectviely) for 3 weeks. (G) The p-AMPK, total AMPK, GPAT3 and PPARγ protein were detected by western blot. (H) GPAT3 and PPARγ mRNA levels were detected by RT- PCR. #p < 0.05 compared with Tshr−/− + AICAR group; ★p < 0.05 compared with Tshr−/− + Vehicle group. All of the panels above are representative of 3 independent experiments. All the gels were run under the same experimental conditions and key data cropped blots are used here. The full-length gel images are available in Supplementary Fig. 6.
As shown in Fig. 5B (Figs. S1A and B), the expression of phosphorylated AMPK decreased in mature adipocytes in vitro in a dose- and time-dependent manner following TSH treatment. Similarly, AMPK activity was inhibited by TSH according to a SAMS peptide phosphorylation assay (Fig. 5C).
To determine whether TSH functions by modulating AMPK activity, we transfected differentiated 3T3-L1 adipocytes with either a constitutively active form of AMPK (CA-AMPK) to increase their AMPK activity or a dominant negative mutant of AMPK (DN-AMPK) to reduce their AMPK activity. Transfection with CA-AMPK significantly ameliorated TSH-stimulated TG and lipid droplet production and transfection with DN-AMPK enhanced the effects of TSH (Fig. 5D and Fig. S3).
In addition, TSH-stimulated PPARγ and GPAT3 upregulation were significantly inhibited by transfection with CA-AMPK but were enhanced by transfection with DN-AMPK (Figs. 5E and F). This finding suggests that TSH stimulates PPARγ and GPAT3 expression via AMPK in vitro.
Thus, we analyzed whether TSH induces adipogenesis through AMPK in vivo by chronically treating mice with AICAR. As shown in Fig. 5G, chronic exposure to AICAR increased AMPK phosphorylation. Moreover, a significant decrease in PPARγ, GPAT3 mRNA and protein expression occurred in the Epi-WAT of AICAR-treated Tshr+/+ and Tshr−/− mice (Figs. 5G and H).
Tshr ablation ameliorates HFD-induced obesity in vivo
To address whether TSH-stimulated adipogenesis depended on TSHR, we transfected 3T3-L1 adipocytes with Tshr siRNA and found that Tshr mRNA expression was significantly reduced38. Compared with the non-targeting siRNA, Tshr siRNA inhibited the effects of TSH on the regulation of TG levels and lipid droplet content (Figs. 6A and B). Furthermore, Tshr siRNA inhibited the effects of TSH on the upregulation of PPARγ and GPAT3 expression, although AMPK phosphorylation increased (Fig. 6C).

The effects of TSH on adipogenesis were reversed when the expression of Tshr was silenced in vivo and in vitro.
(A–C) Differentiated 3T3-L1 cells were transfected with non-targeting or Tshr siRNA. After 48 h, the cells were incubated with or without TSH (2 μM), for 24 h. (A) TG contents and (B) lipid contents were measured, respectively. #p < 0.05 compared with non-targeting siRNA group; ★p < 0.05 compared with TSH group. (C) The p-AMPK, total AMPK, GPAT3 and PPARγ protein expression were analyzed by western blot. (D–H) Tshr−/− (n = 8) and Tshr+/+ (n = 8) mice were fed a high-fat diet for 10 weeks. (D) Weight gain was decreased in Tshr−/− mice. Body weight was monitored twice a week. (E) Adipose mass of Epi fat, RP fat and the size of the adipocytes were diminished in the Tshr−/− group. (F) Epi-WAT weight and the WAT index were assayed. #p < 0.05 compared with the Tshr+/+ group. (G) The protein expression levels of GPAT3 were detected by western blot. (H) Intracellular TG levels and plasma adiponectin were detected. The results are expressed as the mean ± SD. #p < 0.05 compared with Tshr+/+ group. All of the above panels are representative of 3 independent experiments. All the gels were run under the same experimental conditions.
For the in vivo study, we fed Tshr−/− and Tshr+/+ mice (8 weeks old) a HFD for 10 weeks and measured relevant obesity-related parameters. Although the body lengths and food intake of Tshr−/− mice were similar to those of Tshr+/+ mice (data not shown), the body weights of mice on the HFD (30.86 ± 1.03 g) were 23% less than those of the Tshr+/+ mice (39.41 ± 1.99 g) at 18 weeks of age (Fig. 6D). In Tshr−/− mice, due to the absence of TSHR expression, the Epi-, retroperitoneal-adipose mass and adipocyte size were significantly decreased compared with those of the littermate control mice (Figs. 6E and F). As shown in Figure 6G, decreased GPAT3 protein levels were observed in the adipose tissue of Tshr−/− mice. As a result, the intracellular and plasma TG contents were downregulated (Fig. 6H and Fig. S2B). The plasma adiponectin levels were higher in the Tshr−/− mice (Fig. 6H), consistent with their reduced fat mass, indicating that the function of the adipose tissue was changed. These various observations showed that the deletion of TSHR protected the mice from HFD-induced adipocyte hypertrophy and obesity.
Discussion
Here, we discovered a novel role of TSH in regulating adipogenesis and obesity. TSH increased TG accumulation via TSHR, in which GPAT3 activation is induced by the AMPK/PPARγ signaling pathway. SCH mice manifested obesity, whereas the ablation of Tshr in mice caused resistance to high fat diet-induced obesity. Our findings reveal a novel effect of TSH on TG metabolism in adipose tissue and might partially explain the pathogenesis of obesity in SCH patients.
Adipose tissue serves as a regulator of various physiological pathways and is the key tissue involved in the regulation of energy balance and lipid homeostasis39,40,41,42,43. Previous studies have reported that human, mouse and rat preadipocytes differentiation is accompanied by TSHR expression18,23,44. Toyoshi cloned Tshr cDNA from the white adipose tissue of rats and determined that the Tshr cDNA from adipose tissue was almost identical to that from the thyroid glands. The expression levels of TSHR in the epididymal adipose tissue and the thyroid were similar45. We previously demonstrated that TSHR expression increases as 3T3-L1 preadipocytes differentiate and that TSHR knockdown delays adipocyte differentiation20. All of these studies have focused on the effects of TSH on the adipocyte differentiation. However, whether TSH regulates TG synthesis in differentiated adipocyte had never been explored.
The functions of differentiated adipocytes include lipid storage, energy flow and the production of adipokines46. The hypertrophy of differentiated adipocytes results in dysfunction and induces an inflammatory state and abnormal glucolipid metabolism, in which altered adipokine secretions occur47. Moreover, mature adipocytes are the most important form in human adipose tissues. In the present study, we observed that the expression of a panel of genes known to play a critical role in adipogenesis was decreased in Tshr−/− mice. Therefore, we believe that deciphering the regulation of this process could provide new insight into the regulation of conventional adipogenesis.
The expression of TSHR in adipocytes is not constant20,48. Thus, it is inappropriate to use the differentiation process within the 3T3-L1 preadipocytes model to study the effect of TSH on adipogenesis. To determine the functional role of TSH in adipocytes, we studied 3T3-L1 adipocytes that were cultured for 16 days following the induction of differentiation. 3T3-L1 cells were the first non-thyroidal cell line reported to express TSHR and TSHR expression can also be induced in these cells48. 3T3-L1 cells subjected to long-term culture are considered to be representative of mature adipocytes, in which the expression of proteins linked to adipocyte differentiation, including TSHR (Fig. S4A), does not significantly change49. Furthermore, TSHR expression remains unchanged in TSH-stimulated differentiated adipocytes (Fig. S4B). In addition, using primary fat cells provides a suitable system for exploring fat cell biology and numerous regulatory mechanisms at the cellular level50.
GPAT has been suggested to be the rate-limiting enzyme in the TG biosynthesis pathway because of its low specific activity and its position as the initiating enzyme34. Although GPAT catalyzes the conversion of glycerol-3-phosphate to lysophosphatidic acid, Igal reported that GPAT directed the incorporation of fatty acids into TGs51. GPAT3 is the major GPAT isoform in adipocytes and plays a crucial role in adipogenesis28 and TG storage52. Because TGs are primarily stored in adipose tissue, we evaluated the TG contents and adipocyte sizes in vivo, as well as intracellular TG and lipid droplets contents in vitro, to estimate TG synthesis. These results illustrated that the changes in TG content were consistent with the shifts in GPAT3 expression and activity. Furthermore, we used TSH stimulation, siRNA-mediated depletion of TSHR, SCH and Tshr−/− mice models to show that TSH specifically promoted GPAT3 expression and TG synthesis. In the present study, we demonstrated for the first time that TSH regulates GPAT3 expression and activity. Based on our previous study20 and this current work, our results strongly suggest that TSH promotes TG accumulation, both by increasing adipocyte differentiation and by augmenting TG synthesis in differentiated adipocytes, resulting in the development of obesity and metabolic-related disorders.
In this study, we found that TSH promotes adipocyte hypertrophy by counteracting the inhibitory effect of AMPK on PPARγ/GPAT3 signaling. AMPK is directly regulated by cAMP/PKA and functions by inhibiting fatty acid synthesis, adipogenesis and activating fatty acid oxidation53. Numerous studies have established that TSH is involved in lipid metabolism through the cAMP/PKA or PI3K/Akt pathway38,54, but the role of TSH in the AMPK pathway was not fully elucidated. A better understanding of the molecular mechanisms by which TSH regulates AMPK activity in adipose tissues may provide new insight into the global modulation of adipogenesis. Our present data indicate that AMPK acts as the pivot in TSH-induced adipogenesis, as shown by the following results: 1) TSH functions as a potent suppressor of AMPK activity according to the SAMs peptide phosphorylation assay; 2) the selective PKA inhibitor, H89, activated AMPK in a dose- and time-dependent manner in C57/BL6 mice (Fig. S5); 3) CA-AMPK inhibited the nuclear accumulation of PPARγ, suppressed GPAT3 expression and resulted in reduced intracellular TG contents; 4) transfection with DN-AMPK resulted in diametrically opposed effects were observed; and 5) the potency of the proadipogenic effect of TSH was shown to be highly dependent on AMPK.
The PPARs are transcription factors that regulate diverse physiological and pathological processes, especially glucolipid metabolism. PPARγ is the most important PPAR species in adipose tissue; it is essential for the differentiation and function of white adipocytes and promotes lipid accumulation55. Previous studies consider PPARγ to mainly regulate adipocyte differentiation and development56. However, recent studies have shown that PPARγ is essential for the survival of mature adipocytes49,57. In addition, adipose-specific PPARγ-knockout mice displayed diminished TG stores in WAT58. Our results showed that PPARγ could increase GPAT3 expression through its involvement in TSH-stimulated TG synthesis. Meanwhile, the inhibition of PPARγ promotes the expression of the adipokine adiponectin. Therefore, we believe that PPARγ plays an important role in regulating the function and adipogenesis of mature adipocytes, not only in adipocyte differentiation. The characterization of PPARγ, a potential regulator in mature adipocytes, could open the door for novel approaches for the treatment of obesity and other disorders of mature adipocytes.
Thyroid hormones should be considered as important confounding factors when discussing the relationship between TSH and lipid metabolism. In our study, we used Tshr−/− mice to evaluate the effects of TSH on lipid profiles, independent of the thyroid hormone levels. Because the deletion of TSHR can cause TSH malfunction, Tshr−/− mice were compared with Tshr+/+ mice to investigate the function of TSH in vivo. Meanwhile, to study the function of TSH in vitro, we stimulated differentiated 3T3-L1 adipocytes with TSH or the vehicle, further used RNAi Tshr. Through the above-mentioned approaches, we excluded the role of thyroid hormones both in vivo and in vitro, confirming the relationship between TSH and TG synthesis in adipocytes.
Although strong clinical and experimental evidence supports the pivotal role of TSH in the regulation of metabolism, the molecular mechanisms involved in these actions remain largely unknown. In this study, we demonstrated that TSH, by binding to TSHR, inhibits AMPK Thr172 phosphorylation, activates the PPARγ transcriptional program and subsequently increases GPAT3 expression in mature adipocytes. In summary, our data indicate that TSH is linked with the promotion of adipocyte hypertrophy, thus contributing to the pathogenesis of obesity. The modification of the AMPK/PPARγ/GPAT3 axis via TSH might therefore serve as a potential therapeutic strategy for obesity-associated metabolic syndrome and SCH.
Methods
Primary adipocytes culture
The procedure to obtain primary normal human and mice adipocytes has been carefully standardized59. Human primary adipocytes were obtained by surgical resection of benign omental adipose tissue from male at Shandong Provincial Hospital Affiliated to Shandong University. Mice primary adipocytes were isolated from the epididymal fat pads of normal male C57/BL6 mice. In brief, excised adipose tissues from three to four mice were pooled together and visible blood vessels were removed. Then the minced adipose tissues were digested in D-hanks' buffer with 1 mg/ml collagenase type I, 100 mg/ml penicillin and 100 mg/ml streptomycin for 30 min at 37.8°C. Subsequently, the cell suspension was filtered sequentially through 100 mm nylon mesh and centrifuged at 200 g for 10 min. In the laminar flow hood, remove and discard the underlying pellet (containing preadipocytes, fibroblasts and erythrocytes) and media. Resuspend the remaining fat layer in 20 ml D-hanks' and centrifuge for 10 min at 200 g. Repeat this step two additional times. After the last centrifugation step, transfer the fatty layer (containing the mature adipocytes) to 35 mm cell culture dished. Fill the dishes with DMEM/F12 + 10% FBS. The culture medium was replaced with fresh medium every 48 h59. All the patients were properly informed and written informed consent was obtained from each individual. Our research was approved by the Ethics Committee of Shandong Provincial Hospital (Jinan, China). All animal experiments were performed according to the Shandong Provincial Hospital Animal Care and Use Committee (Jinan, China).
Animals and treatment
All the mice were kept in a SPF room in which lighting was controlled (12 hours on, 12 hours off) and the temperature was maintained at 23°C.
SCH mice
Male C57/BL6 mice (8 weeks old) were obtained from Vital River Corporation (Beijing, China). Mice were divided into two groups: one group were given methimazole, a drug that inhibits thyroid hormone synthesis [MMI, 0.04 mg/kg BW·d] in the drinking water, another group were given a corresponding volume of vehicle (control group, n = 8) for 12 weeks. We weighed the mice each week and adjusted the MMI dose according to the body weight. After MMI was administered for 12 weeks, the mice were fasted for 6 h and then euthanized using pentobarbital sodium. Serum samples were collected immediately prior to sacrificing the mice and were tested for fT4 and TSH levels.
GW9662 treated mice
High-fat-diet (HFD) chowed male C57/BL6 mice (13 weeks old) were subjected to GW9662 at a dose of 0.35 mg/kg BW·d in their drinking water for 5 weeks (GW9662 group, n = 8) or to a corresponding volume of vehicle (control group, n = 8).
HFD chowed Tshr−/− and Tshr+/+ mice
Male Tshr−/− and Tshr+/+ mice were obtained from Jackson Laboratory (USA). All the information of these mice could get from our previous research33. Tshr−/− (n = 8) and Tshr+/+ (n = 8) mice (8 weeks old) fed HFD for 10 weeks.
H89 treated mice
Male C57/BL6 mice (8 weeks old) were gavaged with H89 at a dose of 20 nM for 3 or 5 days (n = 3, respectively), or 40 nM for 3 days (n = 3), or with a corresponding volume of vehicle (n = 3).
AICAR treated Tshr−/− and Tshr+/+ mice
Male Tshr−/− and Tshr+/+ (8 weeks old) mice were injected intraperitoneally with AICAR at a dose of 0.5 μg/kg BW·d for 3 weeks (n = 5, respectively), or with a corresponding volume of vehicle (n = 5, respectively).
For each animal experiment, Epi-WAT weight, Epi-WAT weight/body weight, Epi-WAT triglyceride contents were measured. Blood from animals was then obtained for analyses of serum TG and adiponectin (SCH mice also detected BMI, fT4 and TSH levels). In addition, adipose tissue from all animals were collected and immediately frozen for assay.
AMPK activity assay
AMPK activity was measured after immunoprecipitation with polyclonal rabbit AMPK α1/2 antibodies raised against synthetic peptides of AMPK. AMPK activity was measured using the SAMs peptide phosphorylation assay60. Briefly, the kinase assay was performed in 30 μl kinase buffer containing 200 μM SAMs peptide (HMRSAMSGLHLVKRR, Upstate Technology), 200 μmol/l AMP, 5 mM MgCl2, 200 μM ATP and 10 μCi [γ-32P]ATP (specific activity 3,000 Ci/mmol) at 30°C for 15 min with continuous shaking. 20 μl aliquots of the reaction mixture were spotted on half-cut P81 Whatman phosphocellular paper. The P81 paper was washed three times for 15 min in 1% phosphoric acid with gentle stirring to remove free ATP and washed once with acetone for 5 min. The papers air-dried and radioactivity was counted 32P-labeled incorporation into SAMs peptide in 4 ml of scintillation liquid by scintillation counting. Control immunoprecipitation that contained no AMPK antibody was performed to ensure that SAMs peptide phosphorylation was dependent on specific immunoprecipitation of AMPK. AMPK activity was calculated as picomoles per minute per milligram of protein.
Materials, 3T3-L1 cells culture, Oil Red O staining, Triglyceride assay, H&E staining, Adiponectin assay, Cell transfection, Knockdown of Tshr, PPARγ or GPAT3 by RNAi, PPRE-luciferase reporter assay, GPAT3-luciferase reporter assay, Western blot analysis, Real-time quantitative PCR, Immunofluorescence.
These methods are provided in the Supplemental Materials and Methods.
Statistical analysis
Results were presented as the means ± standard deviations (SD) for the number of experiments indicated. For statistical analysis, Differences between means were compared using either unpaired Student t tests for two-group comparisons or one-way analysis of variance (ANOVA) (Dunnett's t or LSD test) for multiple comparisons. Data were analyzed using SPSS 11.0 softwear. Differences were considered significant at a level of p < 0.05.
References
Hotamisligil, G. S. & Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8, 923–934 (2008).
Boucher, J. et al. Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat Commun 3, 902 (2012).
Steinberger, J., Moorehead, C., Katch, V. & Rocchini, A. P. Relationship between insulin resistance and abnormal lipid profile in obese adolescents. J Pediatr 126, 690–695 (1995).
Cawthorn, W. P., Scheller, E. L. & MacDougald, O. A. Adipose tissue stem cells: the great WAT hope. Trends Endocrinol Metab 23, 270–277 (2012).
Russo, P., Lauria, F. & Siani, A. Heritability of body weight: moving beyond genetics. Nutr Metab Cardiovasc Dis 20, 691–697 (2010).
Diez, J. J. & Iglesias, P. Relationship between thyrotropin and body mass index in euthyroid subjects. Exp Clin Endocrinol Diabetes 119, 144–150 (2011).
Kitahara, C. M. et al. Body fatness and markers of thyroid function among U.S. men and women. PLoS One 7, e34979 (2012).
Knudsen, N. et al. Small differences in thyroid function may be important for body mass index and the occurrence of obesity in the population. J Clin Endocrinol Metab 90, 4019–4024 (2005).
Reinehr, T. Thyroid function in the nutritionally obese child and adolescent. Curr Opin Pediatr 23, 415–420 (2011).
Ruhla, S. et al. A high normal TSH is associated with the metabolic syndrome. Clin Endocrinol (Oxf) 72, 696–701 (2010).
Solanki, A., Bansal, S., Jindal, S., Saxena, V. & Shukla, U. S. Relationship of serum thyroid stimulating hormone with body mass index in healthy adults. Indian J Endocrinol Metab 17, S167–169 (2013).
Lai, Y. et al. The relationship between serum thyrotropin and components of metabolic syndrome. Endocr J 58, 23–30 (2011).
Rapa, A. et al. Subclinical hypothyroidism in children and adolescents: a wide range of clinical, biochemical and genetic factors involved. J Clin Endocrinol Metab 94, 2414–2420 (2009).
Ambrosi, B. et al. Relationship of thyroid function with body mass index and insulin-resistance in euthyroid obese subjects. J Endocrinol Invest 33, 640–643 (2010).
Bastemir, M., Akin, F., Alkis, E. & Kaptanoglu, B. Obesity is associated with increased serum TSH level, independent of thyroid function. Swiss Med Wkly 137, 431–434 (2007).
Muscogiuri, G. et al. High-normal TSH values in obesity: is it insulin resistance or adipose tissue's guilt? Obesity (Silver Spring) 21, 101–106 (2013).
Zhang, W. et al. Presence of thyrotropin receptor in hepatocytes: not a case of illegitimate transcription. J Cell Mol Med 13, 4636–4642 (2009).
Haraguchi, K., Shimura, H., Lin, L., Endo, T. & Onaya, T. Differentiation of rat preadipocytes is accompanied by expression of thyrotropin receptors. Endocrinology 137, 3200–3205 (1996).
Sorisky, A., Bell, A. & Gagnon, A. TSH receptor in adipose cells. Horm Metab Res 32, 468–474 (2000).
Lu, S. et al. Role of extrathyroidal TSHR expression in adipocyte differentiation and its association with obesity. Lipids Health Dis 11, 17 (2012).
Valyasevi, R. W., Harteneck, D. A., Dutton, C. M. & Bahn, R. S. Stimulation of adipogenesis, peroxisome proliferator-activated receptor-gamma (PPARgamma) and thyrotropin receptor by PPARgamma agonist in human orbital preadipocyte fibroblasts. J Clin Endocrinol Metab 87, 2352–2358 (2002).
Zhang, L. et al. Biological effects of thyrotropin receptor activation on human orbital preadipocytes. Invest Ophthalmol Vis Sci 47, 5197–5203 (2006).
Lu, M. & Lin, R. Y. TSH stimulates adipogenesis in mouse embryonic stem cells. J Endocrinol 196, 159–169 (2008).
Bjorntorp, P. & Sjostrom, L. +SJOSTROM. Number and size of adipose tissue fat cells in relation to metabolism in human obesity. Metabolism 20, 703–713 (1971).
Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 27, 163–177 (2013).
Brook, C. G., Lloyd, J. K. & Wolf, O. H. Relation between age of onset of obesity and size and number of adipose cells. Br Med J 2, 25–27 (1972).
Takeuchi, K. & Reue, K. Biochemistry, physiology and genetics of GPAT, AGPAT and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab 296, E1195–1209 (2009).
Shan, D. et al. GPAT3 and GPAT4 are regulated by insulin-stimulated phosphorylation and play distinct roles in adipogenesis. J Lipid Res 51, 1971–1981 (2010).
Cao, J., Li, J. L., Li, D., Tobin, J. F. & Gimeno, R. E. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc Natl Acad Sci U S A 103, 19695–19700 (2006).
Li, Q. et al. Preservation of hepatocyte nuclear factor-4alpha contributes to the beneficial effect of dietary medium chain triglyceride on alcohol-induced hepatic lipid dyshomeostasis in rats. Alcohol Clin Exp Res 37, 587–598 (2013).
Yu, C. Y. et al. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One 5, e15188 (2010).
Wronska, A. & Kmiec, Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol (Oxf) 205, 194–208 (2012).
Wang, T. et al. Decreased fasting blood glucose is associated with impaired hepatic glucose production in thyroid-stimulating hormone receptor knockout mice. Endocr J 60, 941–950 (2013).
Wendel, A. A., Lewin, T. M. & Coleman, R. A. Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim Biophys Acta 1791, 501–506 (2009).
van Beekum, O., Fleskens, V. & Kalkhoven, E. Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator. Obesity (Silver Spring) 17, 213–219 (2009).
Lee, Y. S. et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55, 2256–2264 (2006).
Hardie, D. G. Hot stuff: thyroid hormones and AMPK. Cell Res 20, 1282–1284 (2010).
Tian, L. et al. A novel role for thyroid-stimulating hormone: up-regulation of hepatic 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase expression through the cyclic adenosine monophosphate/protein kinase A/cyclic adenosine monophosphate-responsive element binding protein pathway. Hepatology 52, 1401–1409 (2010).
Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep 10, 306–315 (2010).
Goossens, G. H. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav 94, 206–218 (2008).
Halberg, N., Wernstedt-Asterholm, I. & Scherer, P. E. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am 37, 753–768, x–xi (2008).
Qatanani, M. & Lazar, M. A. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 21, 1443–1455 (2007).
Duteil, D. et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nat Commun 5, 4093 (2014).
Bell, A. et al. Functional TSH receptor in human abdominal preadipocytes and orbital fibroblasts. Am J Physiol Cell Physiol 279, C335–340 (2000).
Endo, T., Ohta, K., Haraguchi, K. & Onaya, T. Cloning and functional expression of a thyrotropin receptor cDNA from rat fat cells. J Biol Chem 270, 10833–10837 (1995).
Dodson, M. V. & Fernyhough, M. E. Mature adipocytes: are there still novel things that we can learn from them? Tissue Cell 40, 307–308 (2008).
Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab 40, 16–28 (2014).
Haraguchi, K. et al. Functional expression of thyrotropin receptor in differentiated 3T3-L1 cells: a possible model cell line of extrathyroidal expression of thyrotropin receptor. Biochem Biophys Res Commun 223, 193–198 (1996).
Tamori, Y., Masugi, J., Nishino, N. & Kasuga, M. Role of peroxisome proliferator-activated receptor-gamma in maintenance of the characteristics of mature 3T3-L1 adipocytes. Diabetes 51, 2045–2055 (2002).
Rodbell, M. Metabolism of isolated fat cells. I. Effects of hormones on glucose metabolism and lipolysis. J Biol Chem 239, 375–380 (1964).
Igal, R. A., Wang, S., Gonzalez-Baro, M. & Coleman, R. A. Mitochondrial glycerol phosphate acyltransferase directs the incorporation of exogenous fatty acids into triacylglycerol. J Biol Chem 276, 42205–42212 (2001).
Gimeno, R. E. & Cao, J. Thematic review series: glycerolipids. Mammalian glycerol-3-phosphate acyltransferases: new genes for an old activity. J Lipid Res 49, 2079–2088 (2008).
Daval, M., Foufelle, F. & Ferre, P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol 574, 55–62 (2006).
Morshed, S. A., Latif, R. & Davies, T. F. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology 150, 519–529 (2009).
Ahmadian, M. et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med 19, 557–566 (2013).
Floyd, Z. E. & Stephens, J. M. Controlling a master switch of adipocyte development and insulin sensitivity: covalent modifications of PPARgamma. Biochim Biophys Acta 1822, 1090–1095 (2012).
Imai, T. et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci U S A 101, 4543–4547 (2004).
Jones, J. R. et al. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A 102, 6207–6212 (2005).
Feng, L. et al. Long-term ethanol exposure inhibits glucose transporter 4 expression via an AMPK-dependent pathway in adipocytes. Acta Pharmacol Sin 31, 329–340 (2010).
Yan, H., Zhang, D., Zhang, Q., Wang, P. & Huang, Y. The activation of AMPK in cardiomyocytes at the very early stage of hypoxia relies on an adenine nucleotide-independent mechanism. Int J Clin Exp Pathol 5, 770–776 (2012).
Acknowledgements
We thank Prof. Xu Haiyan (Hallett Center for Diabetes and Endocrinology, Brown Medical School, Providence, RI, USA.) for providing generous gift the 3T3-L1 cells; Prof. Bruce Spiegelman (Department of Cell Biology, Harvard Medical School, Boston, MA, USA.) for providing the PPARγ S112A plasmid; Prof. David Carling (MRC Clinical Sciences Centre, Cellular Stress Group, Hammersmith Hospital Campus, London, UK.) for providing the CA-AMPK and DN-AMPK plasmids. This work was supported by grants from the National Basic Research Program (2012CB524900), the National Natural Science Foundation (81230018, 81170794, 81300644, 81370891,81400828) and the Chinese Society of Endocrinology (13050830468).
Author information
Authors and Affiliations
Contributions
The work presented here was carried out in collaboration between all authors. S.M. performed the experiments, S.M. and F.J. wrote the main manuscript text, J.Z., Q.G. and L.G. designed the experiments, L.Z. prepared all the animals, C.X. and Y.L. prepared figures 1–3, Y.S. and C.Y. prepared figures 4–6. D.J. prepared supplymental figures. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFO
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ma, S., Jing, F., Xu, C. et al. Thyrotropin and Obesity: Increased Adipose Triglyceride Content Through Glycerol-3-Phosphate Acyltransferase 3. Sci Rep 5, 7633 (2015). https://doi.org/10.1038/srep07633
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07633
This article is cited by
-
The association between dyslipidaemia in the first trimester and adverse pregnancy outcomes in pregnant women with subclinical hypothyroidism: a cohort study
Lipids in Health and Disease (2024)
-
Atypical pituitary hormone-target tissue axis
Frontiers of Medicine (2023)
-
Significance of levothyroxine treatment on serum lipid in pregnant women with subclinical hypothyroidism
BMC Pregnancy and Childbirth (2022)
-
RETRACTED ARTICLE: Relationship between the development of hyperlipidemia in hypothyroidism patients
Molecular Biology Reports (2022)
-
A preliminary study: proteomic analysis of exosomes derived from thyroid-stimulating hormone-stimulated HepG2 cells
Journal of Endocrinological Investigation (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
