
Abstract
Human rhinovirus-C (HRV-C) has been increasingly detected in patients with acute respiratory diseases (ARDs). Prolonged surveillance was performed on children with ARD to investigate the molecular epidemiology and clinical characteristics of HRV in Chongqing, China. Nasopharyngeal aspirates (NPA) were collected from hospitalized children with ARD during 2009–2012. HRV-C was genotyped by sequencing the VP4/VP2 coding region. Among the 1,567 NPAs obtained, 223 (14.2%) were HRV positive and 75.3% of these 223 NPAs were co-infected with other viruses. HRV-A (54.7%) and HRV-C (39.9%) accounted for the majority of HRV infections. Logistic regression models demonstrated significant associations between HRV-A, HRV-C and asthma attacks, as well as between HRV-C and wheezing. A phylogenetic tree showed that HRV-C2 was the predominant type of HRV-C, followed by HRV-C43, HRV-C1 and HRV-C17. Three novel genotypes were proposed on the basis of a low identity with the known HRVs. Our results showed that HRV-A and HRV-C were the predominant types of HRV infection and HRV-C showed a high genetic variation in Chongqing, China. HRV infection was associated with asthma attacks and wheezing; furthermore, HRV infections played a minor role in causing severe pneumonia. This knowledge provides information for the prevention and control of HRV associated with ARDs.
Similar content being viewed by others
Introduction
Human rhinoviruses (HRVs) are a group of small, single-stranded, positive-sense RNA viruses of the genus Enterovirus that belong to the family Picornaviridae. HRV is one of the most frequent causes of acute respiratory diseases (ARDs) and highly affects the morbidity and mortality of children and infants1,2,3. HRV has been known for decades; however, no effective vaccine or drug is available to prevent and cure this infection.
HRV exhibits a high genetic diversity and has been re-classified into three species, namely, HRV-A, HRV-B and HRV-C. HRV-C is a novel species with a unique phylogenetic position and distinct genomic characteristics4. Previous studies have shown that HRV-C exhibits a significantly higher genetic diversity than the HRV-A and HRV-B serotypes5,6,7. The complete genome set of this virus also confirmed interspecies recombination events in HRV-C evolution8,9. These findings provide evidence that the recently identified HRV-C is not an emerging strain; instead, HRV-C has been present for a long time without being detected10,11.
Studies have revealed that HRV-C has been detected in patients with respiratory tract infections worldwide1,12,13,14,15,16,17,18,19. Although the clinical manifestations of HRV-C infections vary in different areas4,20, this infection is mainly associated with cough and asthma exacerbations2,21, acute lower respiratory tract infections, bronchiolitis and pneumonia, particularly in children4,22. A few studies have shown that HRV-C is possibly more virulent and associated with more severe diseases than HRV-A and HRV-B2,4,21,23,24; however, controversial results have been obtained from other studies8,25. Seasonal patterns of HRV-C infection have been revealed, in which HRV-C incidence reaches its peak in the early fall and late spring in many temperate or subtropical countries and in the rainy season in tropical countries4,19,25,26.
Cross-sectional studies in China have suggested that HRV-C, in combination with HRV-A, is a predominant species associated with acute respiratory illnesses in hospitalized children and adults27,28. However, previous studies were limited by short observation periods, small sample sizes and inadequate genetic analyses25,27,29. These studies were performed before the classification of HRV species as HRV-A and HRV-B was proposed in 2010 and before the later publication of the proposal for HRV-C in 201330,31. Furthermore, the presence of more than 100 genetically and serologically distinct HRVs and their roles in human illnesses remain unexplored. To provide a timely estimation of HRV circulation and to acquire the exact genetic characteristics of HRVs in China, researchers should continuously monitor HRV infection by using new classification methods.
In the current study, the molecular epidemiology and clinical characteristics of HRV, particularly HRV-C, were investigated by a three-year surveillance study on children with ARD.
Methods
The retrospective study was conducted at the Children's Hospital, Chongqing Medical University (CHCMU), Chongqing, China, as a part of a viral surveillance of ARD cases from June 2009 to June 2012. Chongqing is located in a subtropical region; CHCMU is the largest children's hospital in southeastern China and serves the population of Sichuan Province and two neighboring provinces. ARD was determined on the basis of an acute disease onset with symptoms of cough, rhinorrhea, dyspnea and a fever of more than 37.5°C. Children aged < 14 years who were admitted in respiratory clinics for ARD were recruited in this study. Nasopharyngeal aspirates (NPAs) were collected and subjected to a laboratory analysis. Severe pneumonia was defined as pneumonia plus hypoxemia (at a constant SaO2 < 92% in the air) or an increase in respiratory and pulse rates with clinical evidence of respiratory distress and exhaustion with or without an increase in PaCO2 concentrations.
RNA was extracted from each specimen by using QIAamp® MinElute virus spin kits (Qiagen, Hilden, Germany). The cDNA was synthesized using a SuperScript® III first-strand synthesis system for the reverse transcription polymerase chain reaction (RT-PCR; Invitrogen, USA). HRV was detected by PCR and the amplicons were further sequenced using the primers of 5′-NTR sequences to classify the subtypes of HRV, as described previously32. The samples were also screened to determine the presence of the influenza virus (A, B and C), metapneumovirus, respiratory syncytial virus (RSV), parainfluenza virus (types 1–4) and coronaviruses (229E and OC43) by using molecular methods, as described previously33.
The VP4/VP2 coding regions of the HRV-C-positive samples were further amplified and sequenced to identify the HRV type and genetic characteristics, as described in a previous study31. Primers targeting the VP4/VP2 coding region of HRV (forward, 5′-GGGACCAACTACTTTGGGTGT CCGTGT-3′; reverse, 5′-CACGGACACCCAAAGTAGT-3′) were used. PCR was performed at 95°C for 5 min followed by 40 cycles at 95°C for 30 s, at 60°C for 45 s, at 72°C for 45 s and at 72°C for 10 min for a final extension.
The genomic sequences were assembled using Lasergene's DNA SeqMan software (version 7.1.0, DNA Star Inc., Madison, WI, USA). Available partial HRV-C VP4/VP2 coding region sequences were obtained from GenBank. The alignments were compared and a phylogenetic tree was constructed using a neighbor-joining method with 1,000 bootstrap pseudoreplicates (MEGA 5). Similarities between the strains were calculated using BioEdit (version 7.13, www.mbio.ncsu.edu/bioedit/bioedit.html).
A standardized questionnaire was designed to obtain the patients' information from medical records, including demographic characteristics, underlying medical conditions, symptoms and signs, laboratory tests, radiographic findings, sputum culture results and the disease outcome. Consent was obtained from the guardians of the children. The study protocol was reviewed and approved by the Ethics Review Committee of CHCMU. The study was conducted in accordance with the approved guidelines.
The variables were subjected to descriptive statistics: the continuous variables were summarized as the means and standard deviations or medians and ranges and the categorical variables were summarized as frequencies and proportions. To determine the differences between groups, we performed chi-square tests, Fisher's exact tests and non-parametric tests or ANOVAs whenever appropriate. A logistic regression analysis was performed to identify the associations between HRV subtypes and severe pneumonia. Odds ratios (ORs) and their 95% confidence intervals (CIs) were estimated using maximum-likelihood methods. A two-sided P value of less than 0.05 was considered statistically significant. The statistical analyses were conducted using SAS 9.1.3 (SAS Institute, Cary, North Carolina, USA).
Results
A total of 1,567 NPAs (145 from 2009, 446 from 2010, 698 from 2011 and 278 from 2012) were collected from children aged 1 month to 14 years (median = 9 months). Approximately 66.1% (1036) of these children were male. Furthermore, 223 (14.2%) were HRV positive and aged 1 month to 12 years (median = 8 months), from which 159 (71.3%) were male. The age and gender distributions of the HRV-positive patients were not significantly different from the other patients. According to the 5′-UTR sequences, 54.7% (122/223) of the HRVs detected were HRV-A, 5.4% (12/223) were HRV-B and 39.9% (89/223) were HRV-C (P < 0.001).
Among the HRV-positive patients, 24.7% (55/223) were affected by single infections and 75.3% (168/223) were co-infected with other viruses, such as RSV (74/223, 33.2%), parainfluenza virus (60/223, 26.9%) and influenza virus (58/223, 26.0%). Co-infection occurred evenly among the three genotypes.
The clinical and epidemiological characteristics of the HRV-positive patients are shown in Table 1. The age and gender distributions were comparable between groups with single infections and co-infection. Cough, expectoration and moist rales were the main clinical manifestations. The clinical manifestations of expectoration and diarrhea were significantly overrepresented in the patients with HRV co-infections than in the patients with HRV single infections (Table 1). Severe pneumonia was identified in 31 (13.9%) HRV-infected patients; among these patients, nine exhibited single infections. A similar frequency was obtained in the patients with co-infection. Except for the higher frequencies of cough and moist rales in the patients with HRV-A and HRV-C, all the other clinical manifestations were evenly distributed among the three HRV genotype groups.
The occurrences of wheezing, asthma and severe pneumonia were further analyzed using multivariate-logistic regression models. In general, HRV infection was significantly associated with wheezing (OR = 1.37, 95% CI = 1.02–1.84) and asthma attacks (OR = 1.68, 95% CI = 1.15–2.45) after considering the effects of age, sex and the presence of other respiratory viruses. These associations were not observed in the patients with single HRV infections (Table 2). The effects of the HRV subtypes on clinical outcomes revealed that HRV-A and HRV-C were more likely associated with asthma using the multivariate logistic regression analysis (OR = 1.68, 95% CI = 1.03–2.74 and OR = 1.89, 95% CI = 1.10–3.27, respectively). Additionally, HRV-C was associated with cough (OR = 1.71, 95% CI = 1.08–2.69). No effects on severe pneumonia were observed from either the single HRV or the co-infected HRV genotypes.
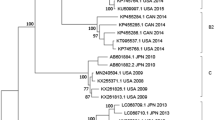
The identified HRV-C was further characterized by constructing a phylogenetic tree using the sequences of the VP4/VP2 coding region from representative subtypes in GenBank. Based on the criteria of the HRV-C subtypes presented by Simmonds et al.30, the 89 HRV-C samples from the study were grouped into 32 HRV-C subtypes (Figure 1). HRV-C2 was the predominant subtype, followed by HRV-C43, HRV-C1 and HRV-C17 (Figure 2). Six HRV-Cs demonstrated less than 90% similarities with 51 HRV gene sequences available in GenBank (Table 3, detailed in Supplementary Table S1). Among these six, CQ4290 (maximum identity, 77.7%), CQ3144-CQ4245 (maximum identity, 83.5%) and CQ4260 (maximum identity, 84.5%) could be designated into novel genotypes and were temporarily named HRV-Cpat52, HRV-Cpat53 and HRV-Cpat54, respectively. Another two strains (CQ5641 and CQ2026) had homologies close to 90% and were accordingly classified as HRV-C2 and HRV-C16, respectively.

Phylogenetic analysis of the HRV-C VP4/VP2 coding region using the neighbor-joining method with 1000 bootstrap replicates with MEGA 5.1.
The branches under the same taxa are labeled by color. The strains in this study are marked with CQ (Chongqing).

Composition of HRV-C subtypes in children with ARD.
The temporally named new subtypes are labeled in red.
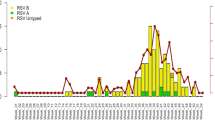
Temporally, HRV occurred throughout the whole year and displayed a characteristic biennial outbreak pattern with alternating high and low peaks during the winter and spring seasons, respectively (Figure 3). Independently, HRV-A showed comparable magnitudes of winter and spring peaks, whereas HRV-C demonstrated distinct peaks during the winter, spring and early summer. Notably, the HRV-C peaks constantly appeared behind the HRV-A peaks at 1–2 month intervals, regardless of the year or season. The seasonal distribution of HRV-B could not be clearly defined because of the low number of positive detections.

Monthly detection rate of HRV and subtypes from 2009 to 2012.
The lines for the HRV, HRV-A, HRV-B and HRV-C detection rates use the primary y-axis and the histograms for the sample number in every month use the secondary y-axis.
Discussion
In this study, HRV-A and HRV-C were identified as the predominant species present in pediatric patients in China for the most recent three years. HRV-C is genetically closer to HRV-A than to HRV-B. The three new lineages of HRV-C that were identified possibly indicate the rapid evolution of HRV-C in China. These findings further substantiate the necessity of continuous surveillance.
In China, HRV was found to circulate among patients with ARDs by retrospective investigation. Jin et al.29 reported a 13.1% HRV detection rate from 406 NPA samples collected from children in Lanzhou from 2006 to 2007, which consisted of 42% HRV-A, 23% HRV-B and 36% HRV-C species. A study in Hong Kong, China, conducted in 2004–2005 revealed a significantly higher frequency of HRV (29.7%) in pediatric patients with ARDs with the HRV-genotype group composition comparable to the results of the current study (50.4% HRV-A, 41.4% HRV-C and 8.2% HRV-B)25. The detection rate of HRV or its subtypes appears to be influenced by detection methods as well as by the spatial and temporal characteristics of detection in various studies.
The seasonality of HRV circulation has been studied previously, with seasonal patterns varying geographically in different countries or regions. The highest HRV-C occurrences are observed during October in America34, fall and winter in Hong Kong, December and April in Gansu, China29 and between September and December in Spain3. In the current study, a biennial HRV outbreak pattern that occurred during the winter and spring seasons was identified. Chongqing is a subtropical region in China and the identified seasonal pattern was largely similar to the pattern of influenza that has been identified in other subtropical regions in China (unpublished data). The high circulation pattern in the winter and spring may be explained by meteorological variables, such as temperature, humidity and precipitation. Further study is warranted to investigate the relationship between meteorological parameters and HRV circulation in other regions. Different but interactive patterns of HRV-A and HRV-C circulation were identified. This temporal pattern of a high HRV-A peak followed by a low HRV-C peak could be attributed to the variation in magnitude of the susceptible population, which considerably diminished following a large outbreak of HRV-A, assuming that a cross protection exists among HRV genotypes.
HRV-C causes significant febrile cough and asthmatic exacerbations in children, thereby requiring hospitalization. Previous studies have reported that HRV-C causes more severe clinical manifestations compared with the other HRV types2,18,22. However, in this current study, we only identified an association between HRV-C and an increased development of cough and asthma attacks; no correlation was observed with severe pneumonia. This discrepancy from previous studies could be attributed to the varied recruitment criteria for patients and the impact of a high frequency of co-infection. Considering that the patients in the current study were all hospitalized, they only represented a subset of HRV infections that were more severe than the average. This representation may have suppressed the association between HRV and severe diseases. Nevertheless, the significant associations in this study were inferred after excluding the effects of co-infection by multivariate logistic regression models. This approach presents an advantage over previous studies that lack multivariate analyses for the consideration of the effects from co-infection or other confounding factors1,3.
Simmonds et al.30 proposed 13% and 10% divergence thresholds on VP1 nucleotide sequences for type assignment and for identifying different HRV-C types using VP4/VP2. Subsequently, they proposed that the equivalent system for the assignment of new HRV types can be extended to HRV-A and HRV-B31. In the proposed criteria, the VP4/VP2 sequences are predictive of the HRV species and type, which can be used for type identifications in epidemiological studies. Results from previous studies suggest that most or all HRV-C types circulate freely worldwide, among which HRV-C2, C6, C16 and C43 have higher prevalences than other types31. VP4/VP2 sequences were used to study the genetic characteristics of HRV-C and the HRV-C2 genotype was found to be predominant in Chongqing, China. Additionally, some new sequences divergent from the VP4/VP2 sequences were identified in mainland China. Although lacking the complete gene sequences, the available VP4/VP2 sequences logically support the classification of a novel HRV-C2 lineage, confirming the high genetic diversity of HRV-C in the region. Nonetheless, further characterization of the VP1 sequences is necessary for a comprehensive analysis of the novel HRV-C lineage.
In summary, HRV-A and HRV-C were shown to be predominant in HRV infections, with HRV-C displaying a high genetic variation in Chongqing, China. HRV-C infection has been associated with asthma attacks and wheezing occurrence and, to a lesser extent, with severe pneumonia. This knowledge provides information for the prevention and control of HRV-associated ARDs.
References
Iwane, M. K. et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J Infect Dis 204, 1702–1710 (2011).
Khetsuriani, N. et al. Novel human rhinoviruses and exacerbation of asthma in children. Emerg Infect Dis 14, 1793–1796 (2008).
Calvo, C. et al. Role of rhinovirus C respiratory infections in sick and healthy children in Spain. Pediatr Infect Dis J 29, 717–720 (2010).
Lau, S. K. et al. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J Clin Microbiol 45, 3655–3664 (2007).
Kiang, D. et al. Molecular characterization of a variant rhinovirus from an outbreak associated with uncommonly high mortality. J Clin Virol 38, 227–237 (2007).
Cordey, S. et al. Rhinovirus genome evolution during experimental human infection. PLoS One 5, e10588 (2010).
Kistler, A. L. et al. Genome-wide diversity and selective pressure in the human rhinovirus. Virol J 4, 40 (2007).
Huang, T. et al. Evidence of recombination and genetic diversity in human rhinoviruses in children with acute respiratory infection. PLoS One 4, e6355 (2009).
Bochkov, Y. A. & Gern, J. E. Clinical and molecular features of human rhinovirus C. Microbes Infect. 14, 485–494 (2012).
Briese, T. et al. Global distribution of novel rhinovirus genotype. Emerg Infect Dis 14, 944–947 (2008).
Arden, K. E. & Mackay, I. M. Newly identified human rhinoviruses: molecular methods heat up the cold viruses. Rev Med Virol 20, 156–176 (2010).
Fuji, N. et al. Detection of human rhinovirus C viral genome in blood among children with severe respiratory infections in the Philippines. PLoS One 6, e27247 (2011).
Arakawa, M. et al. Molecular epidemiological study of human rhinovirus species A, B and C from patients with acute respiratory illnesses in Japan. J Med Microbiol 61, 410–419 (2012).
Chan, Y. F. et al. Diverse human rhinoviruses A and C from children with respiratory infections in Kuala Lumpur, Malaysia. J Infect 64, 633–636 (2012).
Esposito, S. et al. Circulation of different rhinovirus groups among children with lower respiratory tract infection in Kiremba, Burundi. Eur J Clin Microbiol Infect Dis 31, 3251–3256 (2012).
Landa-Cardena, A. et al. Clinical characteristics and genetic variability of human rhinovirus in Mexico. Viruses 4, 200–210 (2012).
Onyango, C. O. et al. Molecular epidemiology of human rhinovirus infections in Kilifi, coastal Kenya. J Med Virol 84, 823–831 (2012).
Pierangeli, A. et al. Molecular epidemiology and genetic diversity of human rhinovirus affecting hospitalized children in Rome. Med Microbiol Immunol 202, 303–311 (2013).
Xiang, Z. et al. Human rhinovirus group C infection in children with lower respiratory tract infection. Emerg Infect Dis 14, 1665–1667 (2008).
Lee, W. M. et al. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS One 2, e966 (2007).
Miller, E. K. et al. Human rhinovirus C associated with wheezing in hospitalised children in the Middle East. J Clin Virol 46, 85–89 (2009).
Cox, D. W. et al. HRV-C Infection in Young Children with Acute Wheeze is Associated with Increased Acute Respiratory Hospital Admissions. Am J Respir Crit Care Med 188, 1358–1364 (2013).
Tapparel, C. et al. Pneumonia and pericarditis in a child with HRV-C infection: a case report. J Clin Virol 45, 157–160 (2009).
Wisdom, A. et al. Genetics, recombination and clinical features of human rhinovirus species C (HRV-C) infections; interactions of HRV-C with other respiratory viruses. PLoS One 4, e8518 (2009).
Lau, S. K. et al. Clinical and molecular epidemiology of human rhinovirus C in children and adults in Hong Kong reveals a possible distinct human rhinovirus C subgroup. J Infect Dis 200, 1096–1103 (2009).
Watanabe, A. et al. Rhinovirus species and their clinical presentation among different risk groups of non-hospitalized patients. J Med Virol 82, 2110–2115 (2010).
Xiang, Z. et al. Human rhinovirus C infections mirror those of human rhinovirus A in children with community-acquired pneumonia. J Clin Virol 49, 94–99 (2010).
Piralla, A., Baldanti, F. & Gerna, G. Phylogenetic patterns of human respiratory picornavirus species, including the newly identified group C rhinoviruses, during a 1-year surveillance of a hospitalized patient population in Italy. J Clin Microbiol 49, 373–376 (2011).
Jin, Y. et al. Prevalence and clinical characterization of a newly identified human rhinovirus C species in children with acute respiratory tract infections. J Clin Microbiol 47, 2895–2900 (2009).
Simmonds, P. et al. Proposals for the classification of human rhinovirus species C into genotypically assigned types. J Gen Virol 91, 2409–2419 (2010).
McIntyre, C. L., Knowles, N. J. & Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J Gen Virol 94, 1791–1806 (2013).
Imamura, T. et al. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 17, 1430–1435 (2011).
Tiveljung-Lindell, A. et al. Development and implementation of a molecular diagnostic platform for daily rapid detection of 15 respiratory viruses. J Med Virol 81, 167–175 (2009).
Miller, E. K. et al. A novel group of rhinoviruses is associated with asthma hospitalizations. J Allergy Clin Immunol 123, 98–104 e101 (2009).
Acknowledgements
This study is supported by the China Mega-Project on Infectious Disease Prevention (No. 2013ZX10004202) and the National Natural Science Foundation (No. 81222037).
Author information
Authors and Affiliations
Contributions
W.C.C. and W.L. conceived and designed this study and revised the manuscripts. Q.B.L., Y.W., L.Y.W., H.Y.W., D.D.H. and X.A.Z. collected the samples and performed the experiments. Q.B.L. and Y.W. analyzed the data and wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Table 1 Sequence identity matrix between the sequences from our study and from GenBank
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lu, QB., Wo, Y., Wang, LY. et al. Molecular Epidemiology of Human Rhinovirus in Children with Acute Respiratory Diseases in Chongqing, China. Sci Rep 4, 6686 (2014). https://doi.org/10.1038/srep06686
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06686
This article is cited by
-
Molecular epidemiology and clinical characterization of human rhinoviruses circulating in Shanghai, 2012-2020
Archives of Virology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
