
Abstract
Enrichment of spermatogonial stem cells (SSCs) from the mammalian adult testis faces several limitations owing to their relatively low numbers among many types of advanced germ cells and somatic cells. The aim of the present study was to improve the isolation efficiency of SSCs using a simple tissue grafting method to eliminate the existing advanced germ cells. Sliced testis parenchyma obtained from adult ICR or EGFP-expressing transgenic mice were grafted heterotropically under the dorsal skin of nude mice. The most advanced germ cells disappeared in the grafted tissues after 2–4 weeks. Grafted tissues were dissociated enzymatically and plated in culture dishes. During in vitro culture, significantly more SSCs were obtained from the grafted testes than from non-grafted controls and the isolated SSCs had proliferative potential and were successfully maintained. Additionally, EGFP-expressing SSCs derived from graft parenchyma were transplanted into bulsufan-treated recipient mice testes. Finally, we obtained EGFP-expressing pups after in vitro fertilization using spermatozoa derived from transplanted SSCs. These results suggest that subcutaneous grafting of testis parenchyma and the subsequent culture methods provide a simple and efficient isolation method to enrich for SSCs in adult testis without specific cell sorting methods and may be useful tools for clinical applications.
Similar content being viewed by others
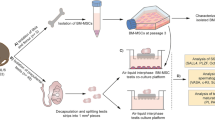


Introduction
Spermatogenesis is the process by which undifferentiated germ cells divide and mature, resulting in the sustainment of male fertility via the daily production of millions of spermatozoa in the testis. The foundation of this process lies in spermatogonial stem cells (SSCs), which undergo self-renewal and produce daughter cells by undergoing complicated differentiation processes1,2,3,4. In the past few decades, techniques for long-term, in vitro maintenance of SSCs have been greatly improved by co-culturing on feeder cells and/or in defined medium containing various growth factors, such as glial cell line-derived neurotrophic factor (GDNF), glial cell line-derived neurotrophic factor receptor-α1 (GFR-α1), basic fibroblast growth factor (bFGF), epidermal growth factor (EGF) and leukemia inhibitory factor (LIF)5,6,7. Similarly, other researchers have reported that the selection of testicular cells expressing integrin α6 (CD49f), integrin β1, CD9, Thy-1 or GFR-α1 resulted in significantly increased efficiency in SSC colonization8,9,10,11. These studies indicated that a relatively high ratio of SSCs might be required for the efficient isolation of cell lines with biological potential. Indeed, the testes of neonatal or transgenic animals that are amenable to various cell sorting methods have usually been used as starting materials for the establishment of SSC lines12,13. Meanwhile, because of the small ratio of SSCs in the human adult testis14,15, conventional methods for isolating adult derived-human SSCs may limit clinical trials16,17.
Recently, some groups have applied various methods to develop an enrichment procedure for adult-derived SSCs. For example, SSCs can bind to laminin but not to fibronectin or collagen type IV18. When laminin-binding (lamB) and laminin-non-binding (lamNB) GFP+ germ cell populations selected from 19-day-old mice were transferred to the testes of recipient males, only the lamB fraction was substantially colonized. This finding suggested that SSCs were greatly enriched in the lamB fraction, which represented approximately 5% of the total germ cell population. However, unlike the pre- or peri-pubertal testis, this protocol was not applicable to the isolation of SSCs from adult testes. Other researchers have taken advantage of the sensitivity of advanced germ cells to high core body temperature. When the testis of most mammals is retained in the body cavity, spermatogenesis fails to occur. Additionally, if the mature testis is surgically fixed in the abdomen, the mature stages of germ cells are lost. This condition is known as cryptorchidism. An experimental procedure for cryptorchidism has been used in a variety of laboratory studies19,20, but this procedure is not appropriate for human clinical trials. In the third model, a testis tissue graft in an immunodeficient host mouse has been shown to model the structural integrity of the testicular tissue; this model therefore facilitated the accessibility and controlled manipulation of testicular function21,22. However, early studies using this procedure were focused on the completion of spermatogenesis in vivo following long-term transplantation (6 ~ 12 months) of immature testis23,24 and not on the isolation or maintenance of SSCs.
We hypothesized that short-term (1–4 weeks), ectopic adult testis tissue grafting would lead to a new method for isolating or maintaining SSCs and could serve as a model system that would be applicable in human clinical trials via the simple grafting of the patient's own tissue to eliminate the advanced germ cells present in the adult testis. To verify this possibility, in the present study, SSCs were isolated and cultured from grafting tissues using a collagen type IV-coated dish and then evaluated their self-renewal capacity. The biological potential of the SSCs was confirmed by the production of advanced germ cells and offspring after transplantation to busulfan-treated, infertile recipient mice.
Results
Evaluation of grafted testicular parenchyma survival and morphology
Fig. 1A shows the typical morphology of the tissue sections of non-grafts and whole or slice grafts. At the time of grafting, the seminiferous tubules included all types of cells: Sertoli cells, SSCs and differentiated germ cells. Two weeks after grafting, 14/20 (70.0%) and 17/20 (85.0%) of the grafted tissues (whole tissue and slice transplantation, respectively) contained germ cells and Sertoli cells in the dorsal skin. However, round spermatids and sperm were not observed in the seminiferous tubules. In the whole tissue and slice groups, 4/20 (20.0%) and 3/20 (15.0%) of the grafted tissues, respectively, were observed to have fully sclerotic tubules. To determine whether SSCs were included in the grafting tubules, we used immunohistochemical staining to detect the SSC markers GFR-α1 and Thy-1. The results showed that 14/16 (87.5%) and 14/17 (82.4%) of the whole and slice grafted tissues, respectively, contained SSC marker-positive cells in the dorsal skin. Four weeks after grafting, the numbers of sclerotic tubules increased to 7/10 (70%) and 6/10 (60%) in the whole tissue and slice groups, respectively. Interestingly, no seminiferous tubules exhibited GFR-α1/Thy-1-positive staining (Table 1). Therefore, the 2-week grafting groups were selected for subsequent experiments.

Grafting of testis tissue from an adult mouse under the dorsal skin of recipient nude mice.
(A) Histological appearance of adult mouse testis tissue before and 2 weeks after grafting into recipient nude mice. (B) Apoptosis of cells was detected by a TUNEL assay (TMR-red staining, red staining of cells indicates apoptotic cells). The yellow dotted box indicates the magnified area. (C) Graphical representation of the results. The left and right panels show the number of total cells (DAPI) and the percentage of apoptotic cells (TUNEL/DAPI) in adult mouse testis tissue after grafting into recipients. Note: Non-graft: testis at the time of grafting; Graft (whole): tissue 2 weeks after grafting into the dorsal skin of recipient nude mice; Graft (slice): tissue 2 weeks after grafting into the dorsal skin of recipient nude mice. The size of the slice testis grafts was 4–5 slices (approximately 0.2–0.5 mm in diameter). The data are presented as the mean ± SEM. a vs b: significantly different (P < 0.05). Scale bars, 100 μm.
To analyze the viability of cells within the seminiferous tubules, twelve slides, four from each of the three groups, were stained using TUNEL methods. In the non-grafts (control), the percentage of apoptotic cells (TMR-red stained) was 4.76 ± 4.97%. Although the total number of cells (DAPI-stained) within the seminiferous tubules was significantly decreased compared with the control, the percentages of apoptotic cells were not different between the whole or slice graft tissues (8.89 ± 8.22% vs.9.48 ± 8.83%) (Fig. 1B,C).
In vitro culture of SSCs derived from grafted testicular parenchyma
Some of the dissociated cells from the testicular parenchyma (cells unattached to the collagen-coated dish) were cultured in StemPro-34 SFM supplemented with 1% KSR and with four growth factors (LIF, EGF, FGF2 and GDNF). Owing to the gelatin coating of the culture plate, some somatic cells attached and proliferated significantly by 7 days of culture. Additionally, SSC-enriched cells, which formed small clumps (>30 cells), grew in size during the first 7 days of culture (Fig. 2A). At first, AP staining could be used to distinguish SSCs from somatic cells in the clumps and small clumps were strongly positive for AP staining (Fig. 2B). To confirm that an AP staining-positive clump was an SSC clump, we performed co-staining for E-cadherin (a marker for SSCs) and 3β-HSD (a marker for interstitial cells). As shown in Fig. 2C, AP was co-expressed in both the E-cadherin-positive and some of the 3β-HSD-positive cells. However, 3β-HSD was not detected in SSC clumps showing AP- and E-cadherin-double positive staining. Attached somatic cells gradually decreased during the second 7 days of culture, but after passaging, they re-formed clumps that subsequently became loosely attached to dishes and continued to proliferate for more than 8 passages (>2 months).

Morphology and alkaline phosphatase staining of cultured SSCs from adult mouse testis tissues 1 week (upper panel) and 2 weeks (lower panel) after grafting into recipient nude mice.
(A) Typical morphology of the attached SSC colonies from the small number of remaining somatic cells in the SSC culture medium containing four growth factors. In the non-graft tissues, initial SSC clumps (1–5 cells) were formed on the culture plate and somatic cells were enriched after a 1-week culture. However, at the second passage, the SSC colonies disappeared after a 2-week culture. Photographs of the primary SSC colonies of the graft groups on the culture plate showing the small-sized (10–20 cells) clumps after a 1-week culture (upper panel). Photographs of the second-passage SSC colonies on the culture plate showing the increased-size colonies after a 2-week culture (lower panel). (B) Alkaline phosphatase (AP) activity of SSC colonies growing from the small number of remaining somatic cells. (C) Histological appearance (hematoxylin and eosin staining) of adult testis. (I) region of interstitial cells, (S) region of SSCs. Immunolocalization of E-cadherin or 3β-HSD in the adult testis and testis-derived SSCs after a 2-week culture. SSCs were characterized by AP staining. Note: Scale bars, 50 μm.
Localization of SSC markers and characterization of cultured cells
It is well known that Thy-1 and GFR-α1 are expressed on mouse and human SSCs and this was confirmed by co-staining with E-cadherin (a marker for SSCs) in seminiferous tubules (Fig. 3A and 3B). Thus, we hypothesized that the ratios of Thy-1 and GFR-α1-positive cells could be increased after dissolution of differentiated germ cells using the grafting method. Immunohistochemical analysis of normal adult mouse testis showed that Thy-1 and GFR-α1-positive cells (putative SSCs) were detected on the basement membrane, but their ratios were low. However, in the grafted testicular tissues, although their tubules appeared small in size, the ratio of cells showing Thy-1 and GFR-α1 signals were relatively higher than those in fresh tissue (Fig. 3A and 3A′). Observed under high magnification, as expected, Thy-1 and GFR-α1 were strongly expressed on the surface membrane of cultured SSCs. During in vitro culture, the colonization efficiency of the SSCs was higher in the grafted testis than in non-grafted controls (Fig. 3C and 3C′).

Immunolocalization of E-cadherin, GFR-α1 and Thy-1 in adult mouse testis after a 2-week grafting and in their cultured SSCs.
(A) Cross-sections of seminiferous tubules in an adult mouse testis stained for Thy-1 and GFR-α1. Thy-1 and GFR-α1, which indicate an SSC-positive signal, are shown in red and green (Thy-1; CY3 and GFR-α1; FITC) and were found in cells located at the basement membrane. (A′) Graphical representation of the results of the graft group. The ratios of SSC-marker-positive cells were significantly increased when compared with the non-graft group. (B) Cross-sections of seminiferous tubules in an adult mouse testis stained for GFR-α1 and E-cadherin. GFR-α1 and E-cadherin, which indicate an SSC-positive signal, are shown in green and red (GFR-α1; FITC and GFR-α1; CY3) and were found in cells located at the basement membrane. The right panel shows a high magnification image of the rectangled area. (C) Cultured SSC colonies from graft tissues at 2 weeks were fixed and stained with Thy-1 and GFR-α1. Thy-1 and GFR-α1 were abundantly expressed on the membrane and in some of the cytoplasm of the SSC colonies. C′. During culture (60 days), SSCs proliferated continuously and maintained similar size morphology. Note: Mock primary Ab/DAPI: DAPI counter-stained SSCs without a primary antibody. The data are presented as the mean ± SEM. a vs b: significantly different (P < 0.05). Scale bars, 20 μm.
Flow cytometrical characterization and RT-PCR of cultured cells
After 2 weeks of culture, the relative ratios of the SSCs were determined using FACS with Thy-1 and GFR-α1. The ratios of CY3-labeled (Thy-1) cells in testicular cells that were obtained from whole and slice grafted testes were 85.00% and 83.40%, respectively, but the ratio from non-grafted testis was 7.33%. Similarly, the ratios of FITC-labeled (GFR-α1) cells in testicular cells that were obtained from whole and slice grafted testes were 78.92% and 68.26%, respectively, but the ratio of those cells from non-grafted testis was 3.93% (Fig. 4A).

Flow-cytometric analysis and RT-PCR of cultured SSCs from adult mouse testis tissues 2 weeks after grafting into recipient nude mice.
(A) The percentage of Thy-1 and GFR-α1-positive cells in the non-graft group was only 7.33 and 3.93%, respectively. The percentage of Thy-1 and GFR-α1-positive cells in the graft (whole) group was 85.00 and 78.92%, respectively, which was significantly higher than that in the non-graft group. The percentage of Thy-1 and GFR-α1-positive cells in the graft (slice) group was 83.40 and 68.26%, respectively, which was also significantly higher than that in the non-graft. (B) Germ cell-specific mRNA expression patterns by real time RT-PCR. Gfr-α1, lin 28 and, ngn3 mRNA were used as undifferentiated spermatogonia marker genes. c-kit, piwil2 and stra8 were used for differentiating spermatogonia and spermatocyts. TH2B was used for spermatocytes. TP-1 mRNA was used as a spermatid marker gene, MVh was used for all germ cells and 18S ribosomal RNA was used as a control gene.
Additionally, to characterize the cultured cells that were derived from the non-grafted, whole and sliced testis grafted groups, we analyzed the expression of spermatogenesis-specific genes by RT-PCR. First, we analyzed the expression of Gfr-α1, lin28 and ngn3, which are specifically expressed in undifferentiated spermatogonia. Cultured cells from the whole grafted group demonstrated high expression of the mRNAs of Gfr-α1 and lin28 compared with cells from the non-grafted and slice grafted groups. Interestingly, the expression of ngn3 was not different in any of the three groups. However, stra8 and piwil2, which are specifically expressed in differentiated spermatogonia and spermatocytes, were highly expressed in cultured cells of the non-grafted group and were weakly expressed in both grafted groups. However, the expression of c-kit was similar across the groups. Additionally, the TH2B and TP-1 genes, which are specifically expressed in spermatocytes and spermatids, were decreased in cells from the grafted groups. Mvh, which is expressed in all stages of germ cell development, was slightly decreased in cells of the grafted groups, but not significantly. These results indicated that the majority of cultured cells included SSCs but did not include advanced-stage germ cells, such as spermatocytes and spermatids (Fig. 4B).
Generation of functional spermatogenic cells by transplantation of cultured cells into the testis of busulfan-treated males
To determine the function of SSCs that had been isolated in vivo and proliferated in vitro, we next transplanted them into the testis of infertile recipient mice. For these experiments, SSCs isolated from EGFP-expressing (C57BL/6-Tg (CAG-EGFP)) transgenic mice were used. The efficiency of isolating SSCs from the transgenic mice was similar to that of the previous experiment using wild type mice (Fig. 5A). Following a culture period of 4 weeks, isolated SSCs were harvested and transplanted into the seminiferous tubules of busulfan-treated adult mice via an efferent duct. Some EGFP-positive germ cells in recipient testes were observed under UV light or in paraffin sections 4 weeks after transplantation (Fig. 4B).

Procedure for SSC transplantation and offspring production.
Graft tissues were prepared from the testis of a fertile male (EGFP-expressing C57BL/6-Tg). The dissociated cells were cultured under the same SSC culture conditions. (B) In vitro cultured SSCs were microinjected into the seminiferous tubules of bulsufan-treated recipient mice (upper panel; 0.1% trypan blue-stained cells). Four weeks after transplantation, the recipient seminiferous tubules that had received EGFP-labeled donor SSCs and their cross sections were observed by fluorescence under UV light (middle and lower panels). Scale bars, 50 μm. (C) After ICSI, cultured blasotocysts emitted fluorescence under UV light. (D) An offspring derived from EGFP-labeled SSCs emitted fluorescence under UV light. Note: Arrows indicate the fluorescence signal.
To produce embryos, spermatozoa or elongated spermatids were collected from the recipient testes 4–8 weeks after transplantation and injected into BDF1 oocytes. After ICSI, 80/101 (79.2%) eggs survived and 71/80 (88.7%) developed to the 2-cell stage within 24 hours in culture. In some preimplantation embryos produced by ICSI, a strong EGFP signal (6/10) was detected at the blastocyst stage. However, some cultured blastocysts did not express EGFP (4/10), which seems to have been due to the production of embryos by fertilization from the recipient's own sperm that had re-initiated spermatogenesis after busulfan treatment (Fig. 4C). To generate offspring, the embryos (30 blastocysts) were transferred into the oviducts of two pseudopregnant females, which resulted in the delivery of 7 live pups (four EGFP-expressing pups and three non-EGFP-expressing pups, Supplementary Table 1 and Fig. 4D). As expected, genotyping analysis by PCR revealed that among the seven pups, four expressed EGFP (Supplementary Fig. 1). These results indicated that SSCs isolated in vivo can be differentiated into functionally advanced germ cells and are capable of producing normal offspring.
Discussion
Although successful isolation and proliferation of SSCs has recently been reported5,10,13,25,26,27, these methods are still limited because high efficiency is only accomplished in SSC-enriched, prepubertal animals or by cell sorting-based strategies using specific antibodies. However, for clinical purposes, the isolation of SSCs from adult testes is more important than isolation from prepubertal testes. Moreover, a high ratio of advanced germ cells in the adult testis may reduce the isolation efficiency of cell sorting using specific antibodies. Therefore, the development of a novel, efficient method for the enrichment and isolation of SSCs from adult testis is a prerequisite for the treatment of male infertility and the study of spermatogenesis in vitro. Many studies have focused on germ cell production of SSCs and several recent studies have reported that SSCs can be converted to pluripotent stem cells in specific culture conditions28,29. However, unlike neonatal testicular cells, the conversion or isolation of pluripotent stem cells from adult testis has rarely been reported in mammalian and human systems. Thus, the difficulty of isolating sufficient numbers of SSCs has likely hindered the progression of their study and application15,30.
To enrich SSCs from the mammalian adult testis, the experimental cryptorchid procedure was applied and resulted in the death of most of the germ cells, leaving only undifferentiated spermatogonia 2 months after surgery in mice31,32, which led to a 5- to 25-fold enrichment of SSCs. McLean et al. reported that the colonization (an SSC property) of germ-line stem cells from hyperthermia-exposed testes was 6- to 19-fold higher than in controls when calculated based on the number of colonies and the area of colonization33.
Grafting of testis tissue was developed as a tool for androgen substitution in the 1950s and has been subsequently applied to the study of steroidogenesis and spermatogenesis. Rathi et al. and Schoenfeld et al. reported that testicular stem cells continuously proliferated in the aging testis after grafting34,35. In the present study, we introduced a testis tissue grafting method for the enrichment of SSCs from adult testis connected with the simultaneous cultivation of these cells because tissue grafting is potentially be a better strategy for germ cell preservation and transplantation in humans than the cryptorchid or hyperthermia-exposed methods. In fact, as a model for application, testis grafting into a recipient mouse enriched the SSC population and culture using collagen selection further increased the concentration of SSCs by decreasing the number of advanced germ cells and somatic cells (Fig. 2). Additionally, isolated SSCs proliferated continuously in in vitro culture conditions for 2–3 months and they kept their stem-cell characteristics. To confirm that the cultured cells were SSC colonies, we performed an immunocytochemical analysis to detect GFR-α1 and Thy-1 expression in the cultured germ cell clumps. As expected, GFR-α1 and Thy-1 were strongly expressed in the cultured SSCs (Fig. 3 and Fig. 4). The purity of the SSC population was increased during passaging; this finding suggested that advanced germ cells and somatic cells in the peripheral area of the colony were gradually decreased (Fig. 4B). Additionally, these observations explain why the specific sorting procedure for germ cells could be skipped in our graft/culture system using one type of ECM (collagen type IV).
The technique for the transplantation of cultured cells into the seminiferous tubules of recipient males is well established in mice36,37. In the present study, SSCs were isolated from the testes of EGFP-expressing transgenic mice and propagated (Fig. 5A) and harvested cell suspensions were microinjected into seminiferous tubules via rete testis38. Transplanted SSCs colonized the basement membrane of the seminiferous tubules and begin proliferating (Fig. 5B). After 4–6 weeks, differentiated germ cells begin to fill the tubule from the basement membrane toward the lumen and a few spermatids or spermatozoa appeared in the lumen39. Using the ICSI technique, the donor-derived, differentiated germ cells produced embryos and offspring (Fig. 5C and 5D), suggesting that the biological potential of SSCs isolated by our grafting/culture method can be well maintained.
Recent advances in chemotherapy and radiotherapy have significantly improved the remission and complete recovery rates of cancer patients. However, because germ cells are highly susceptible to cytotoxic treatments, iatrogenic loss of fertility has emerged as a major side effect of successful treatment40. In males, sperm freezing is an established method to preserve germline cells and is performed routinely in clinics for patients who wish to preserve their fertility before undergoing treatment for malignancy41. Unfortunately, the technique cannot be applied to prepubertal patients, who do not have sperm but have different types of germ cells. Therefore, the tissue grafting/germ cell culture method may provide a new avenue for SSC-based cell therapy37,42 for the preservation of an individual male's SSCs and re-introduction of those cells into the testis after patient recovery while avoiding re-introduction of cancer cells during tissue transplantation. Additionally, this technology may increase the efficiency of studies on SSCs for germ cell and multipotent stem cell production from adult male patients.
Methods
Approvals of animal experiment
The protocols for the use of animals in these studies were approved by the Institutional Animal Care and Use Committee (IACUC) of CHA University (Project No. IACUC-120013) and all experiments were carried out in accordance with the approved protocols.
Animals
All mice were housed under a 12 hr/12 hr light/dark cycle in a temperature- and humidity-controlled room. Donor testes for transplantation were obtained from 6- to 8-week-old adult, outbred (wild-type) ICR male mice (Samtaco Co., Ltd., Seoul, Korea) or EGFP-expressing (C57BL/6-Tg(CAG-EGFP)) transgenic mice (SLC, Inc., Hamamatsu, Japan). Six- to 8-week-old adult BALB/c-Nu (Samtaco Co., Ltd.) male mice were used as the testis recipients.
Adult testis transplantation
Donor adult mice were killed by cervical dislocation and their testes were collected and/or cut transversally into 3–5 slices. The tissues were maintained in ice-cold buffered medium (Dulbecco's modified Eagle's medium), with added antibiotics and 10% fetal bovine serum (FBS; Invitrogen) at room temperature for up to 30 min until the recipient was surgically prepared for grafting. Recipient nude mice were anesthetized using ketamine (80 mg/kg body weight) and xylazine (6 mg/kg body weight) in saline and castrated through two scrotal incisions. The spermatic cord was ligatured and the testes were removed. The scrotal skin was then closed using sutures and two dorsal sideline incisions were made in the skin and body wall. Donor testes or testicular tissues were placed under the dorsal skin using fine forceps and closed by sutures. The mice were maintained in groups of four per cage with food and water available ad libitum for 2–4 weeks.
Isolation of grafted parenchyma and in vitro culture of SSCs
The testis parenchyma were harvested at 2 weeks (n = 20) and 4 weeks (n = 10) after grafting. The dorsal skin of each sacrificed mouse was removed, the grafts were excised and 1/4 of the tissue was fixed in Bouin's solution overnight at room temperature for histological and immunohistochemical analyses. The remaining graft parenchyma were dissociated enzymatically in buffer containing 5 mg/ml collagenase (Type IV; Sigma-Aldrich, St Louis, MO), 10 μg/ml DNase I and 1 mg/ml hyaluronidase (Sigma-Aldrich) for 5 min with agitation at 37°C. Dissociated cell suspensions were then plated and incubated on collagen-coated dishes in selection medium composed of Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) containing 20% FBS, 10 μM 2-mercaptoethanol (Invitrogen) and 1% non-essential amino acids (Invitrogen) at 37°C under humidified conditions of 5% CO2 in air. Over the next 12–24 h, unattached cells were harvested and plated on 0.1% gelatin-coated dishes (1–1.5 × 104 cells/cm2) in serum-free germline-stem cell (GS) medium with modifications as previously described6. The composition of the GS medium was as follows: StemPro-34 SFM (Invitrogen) supplemented with 6 mg/ml D(+)-glucose, 5 × 10−5 M β-mercaptoethanol, 1 μM d(L)-lactic acid, 2 mM L-glutamine, 30 μM pyruvic acid, 10−4 M ascorbic acid, 60 ng/ml progesterone, 30 ng/ml β-estradiol (Sigma-Aldrich), 0.2% BSA (ICN Chemicals, Costa Mesa, CA), 100 U/ml penicillin, 100 μg/ml streptomycin, 1× Insulin-Transferrin-Selenium supplement, 1× MEM vitamin solution, 1× MEM non-essential amino acids, 20 ng/ml mouse EGF (Invitrogen), 10 ng/ml human bFGF (Invitrogen), 1% Knockout™ serum replacement (KSR, Invitrogen), 10 ng/ml rat GDNF (R&D systems, Inc., Minneapolis, MN) and 103 U/ml ESGRO (Chemicon, Temecula, CA). During culture, some remaining somatic testicular cells attached to the bottom of the dish. Adult testicular cell-derived SSCs proliferated slowly and then formed clump-like structures on the somatic cells 2 to 4 weeks after seeding. Cultured cells were dissociated by trypsin treatment and re-plated every 7 to 10 days. Cultured cells were maintained at 37°C in 5% CO2 and the medium was changed every 2 days.
Histological and immunocytochemical characterization of graft tissue and cultured SSCs
After fixation, tissue samples were embedded in paraffin and cut into 5- to 7-μm-thick sections. Each section was stained with hematoxylin and eosin for morphological evaluation. For the detection of apoptotic cells, sections were permeabilized by incubation in 0.5% Triton X-100 for 1 h at room temperature and a TUNEL assay was performed using an In Situ Cell Death Detection Kit as described by the manufacturer (Roche Diagnostics, Indianapolis, IN). Sections were washed twice with PBS and incubated with the TUNEL reaction mixture (10 μl of terminal deoxynucleotidyl transferase and 90 μl of nucleotide mixture) for 60 min at 37°C in a humidified atmosphere in the dark. To detect alkaline phosphatase (AP) activity, cultured cells were fixed in 4% paraformaldehyde at room temperature for 1 min, washed twice with PBS and stained with an alkaline phosphatase substrate solution (10 ml FRV-Alkaline Solution, 10 ml Naphthol AS-BI Alkaline Solution; Alkaline Phosphatase Assay Kit, Sigma-Aldrich) for 30 min at room temperature. AP activity was detected colorimetrically (red) by light microscopy. For the detection of SSC-specific marker proteins, tissue or cell colonies were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) for 30 min at 4°C. Slides were then stained with primary antibodies (1:100–500 dilutions) against E-cadherin (Santa Cruz), Thy-1 (BD Biosciences), GFR-α1 (Chemocon) or 3β-HSD (Santa Cruz). The primary antibodies were detected using Cy3- or fluorescein isothiocyanate (FITC)-conjugated secondary antibodies (Zymed) and all slides were washed and transferred to a solution containing 1 μg/ml 4,6-diamidino 2-phenyindiol (DAPI; Sigma-Aldrich) and incubated for 30 min at room temperature in the dark. After being washed three times, the slides were mounted and optical images were obtained using an epifluorescence microscope (Nikon Corp., Tokyo, Japan).
Flow cytometric analysis
Flow cytometric analyses were performed using a standard protocol. SSC colonies were dissociated in trypsin-EDTA and aliquots of 106 cells were suspended in 0.1 ml of PBS containing 2% fetal bovine serum (PBS/FBS) and incubated with primary antibodies. To detect SSC-specific cells, the cells were incubated with 10 mg/ml anti-mouse Thy-1 antibody (BD Biosciences) or anti-rabbit GFR α-1 antibody (Abcam). The primary antibodies were then detected using 5 mg/ml of Cy3-conjugated (BD Biosciences) or FITC-conjugated antibody (BD Biosciences). Control cells were not treated with primary antibodies. The cells were kept in the dark on ice until analysis using a Becton Dickinson FACS IV Calibur (Becton Dickinson, San Jose, CA). At least, 5,000 or 10,000 events were acquired for each sample.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Graft parenchyma or 2-week cultured samples were analyzed by RT-PCR to assess the expression of the following stage-specific marker genes: Gfr-α1, lin28 and ngn3 for spermatogonial stem cells (undifferentiated spermatogonia); c-kit, piwil2 and stra8 for differentiating spermatogonia and spermatocytes; TH2B for spermatocytes; TP-1 for spermatids; and MVh for germ cells. Total RNA was isolated from tissues (~10 mg) and cell clumps (50–100 clumps) using the TRIzol method (Invitrogen). Total RNA (1 μg) was reverse transcribed using a reverse transcription kit (Superscript, Invitrogen) and amplifications of the PCR reaction mixtures (Invitrogen) were performed in a total volume of 20 μl. The following targets were amplified from cDNA using the primers indicated in parentheses: Gfr-α1 (F: 5′-TGCGTATCTACTGGAGCATGT -3′, R: 5′-CATCGAGGCAGTTGTTCCCTT -3′; 171 bp; Gen Bank accession number NM010279); lin28 (F: 5′-CCTTTGCCTCCGGACTTCTC-3′, R: 5′-GGGCGGTCATAGACAGGAAG-3′; 256 bp; Gen Bank accession number NM145833); ngn3 (F: 5′-TCTCGCCTCTTCTGGCTTTC-3′, R: 5′-CGGCAGATTTGAATGAGGGC-3′; 234 bp; Gen Bank accession number NM009719.6); c-kit (F: 5′-TGTGGAGGAGTTGTGGAGGA-3′, R: 5′-CCCACTGCTGAAACAAAGTCA-3′; 492 bp; Gen Bank accession number NM013598.2); piwil2 (F: 5′-CATTATGGTCAAGTATCTGTT-3′, R: 5′-AGAGGTTGGCGAGGAATAAGG-3′; 250 bp; Gen Bank accession number NM021308); stra8 (F: 5′-AAGGACTTGAGGTTTATTCCT-3′, R: 5′-CTGACGTTCATAATTGAAGTC-3′; 345 bp; Gen Bank accession number NM009292); TH2B (F: 5′-AAGGCCTTAAATACCCAGAC-3′, R: 5′-AGCAATGTGTGCCTAAGTTT-3; 254 bp; Gen Bank accession number NM59924); TP-1 (F:5′-AAGGCCTTAAATACCCAGAC-3′; R:5′-AGCAATGTGTGCCTAAGTTT-3′; 254 bp; Gen Bank accession number NM59924); MVh (F: 5′-AGCACAGCCTCTAGTTAAAGT-3′, R: 5′-ATATCAGGTTTCACACTTCTGT-3′; 328 bp; Gen Bank accession number NM010029) and 18S ribosomal RNA (F: 5′-TACCTACCTGGTTGATCCTG-3′, R: 5′-GGGTTGGTTTTGATCTGATA-3′; 255 bp; GenBank accession number NC000083). The PCRs were initiated by denaturation at 94°C for 5 min, followed by 30–35 cycles of 30 s at 94°C, 30 s at 55–60°C and 30 s at 72°C. After a final extension step for 10 min at 72°C, the products were separated by 1.5% agarose-gel electrophoresis. Negative controls included mock transcription without mRNA.
Transplantation of cultured SSCs
Cultured SSC colonies (EGFP-expressing C57BL/6-Tg) were transplanted into busulfan-treated recipient mice in the first (3 mice), second (3 mice) and third experiments (4 mice) to test their in vivo biological activities. To transfer the cells into the seminiferous tubules of a recipient testis, we used a transfer technique via the efferent duct38. Cultured SSC colonies were dissociated and suspended in DPBS medium containing 0.4% trypan blue (Sigma-Aldrich). The recipient mice were anesthetized and the testes were exteriorized through a midline abdominal incision and immobilized. The injection pipette was constructed from a three-inch length of borosilicate glass with an internal diameter of 0.75 mm and an external diameter of 1 mm (World Precision Instruments # TW100-3). The glass was drawn on a Kopf pipette puller (Model 750), which created two potential injection pipettes (40–60 μm outside diameter (o.d.)). The glass pipette was inserted into the efferent duct using a dissecting microscope and the pressure in the injection tubing was raised until the cell suspension flowed into the seminiferous tubule. The flow was monitored based on the color change. A cell suspension of approximately 3–5 μl (less than 1 × 105 cells) was injected into each recipient testis, which filled ~50% of the tubules in each testis. Four to 8 weeks after transplantation, the recipient testes were collected and analyzed for EGFP expression using a fluorescent microscope and then collected haploid germ cells were used for embryo production.
Preparation of gametes, intracytoplasmic sperm injection (ICSI), embryo culture and embryo transfer (ET)
BDF1 female mice were superovulated by injection of 5 IU of pregnant mare serum gonadotropin (PMSG, Folligon, Intervet Co., Holland) followed 48 hr later by injection of human chorionic gonadotropin (hCG, Chorulon, Intervet Co). At 14 hr post-hCG, the oviducts were removed from the female mice and placed in a Petri dish containing modified human tubal fluid medium (HTF, Irvine Scientific, Santa Ana, CA) at room temperature. After being washed, the oviducts were placed in fresh medium and cumulus-oocyte complexes were released from the ampulla using Dumont forceps. In the ICSI procedure, the cumulus cells were dispersed by incubation for 3–5 min in HTF medium containing 0.1% hyaluronidase (Sigma-Aldrich); after being washed, the oocytes were maintained in KSOM medium at 37°C in 5% CO2 in air until ICSI. Testicular sperm from the recipients were collected in M2 medium by excision with a pair of fine scissors and forceps, followed by squeezing in a 50-μl drop of M2 in mineral oil. The sperm were allowed to disperse at 37°C for 30 min and each suspension was transferred to a 50-μl Eppendorf tube for centrifugation at 1500 rpm for 5 min. Sperm were mixed with 40 μl of 10% polyvinylpyrrolidone (PVP-360) in M2, followed by placement in a culture dish for microinjection. Injections were performed using a PMM-150 FU piezo-impact unit (Prime Tech) and micromanipulators using a blunt-ended, mercury-containing pipette (inner volume, 6–7 μl). After 15 min of recovery at room temperature in M2 medium, the surviving oocytes were returned to mineral oil-covered KSOM and cultured at 37°C in an atmosphere of 5% CO2 in air. For embryo culture, 50 μl of KSOM covered with mineral oil were equilibrated overnight at 37°C in a humidified atmosphere of 5% CO2. After 92–96 hr of culture, in vitro grown blastocysts were transferred into the uterus of a 2.5-day pseudopregnant female that had been mated with the vasectomized male mice. The newborns were observed using a UV illuminator.
Statistical analysis
Unless otherwise specified, each experiment was carried out using at least three replicates. Data are expressed as the means ± SEM. The statistical significances of the differences between treated groups were evaluated by one-way analysis of variance (ANOVA) using a log-linear model in the Statistical Analysis System (SAS, Cary, NC, USA). P values < 0.05 were considered statistically significant.
References
Aponte, P. M., van Bragt, M. P., de Rooij, D. G. & van Pelt, A. M. Spermatogonial stem cells: characteristics and experimental possibilities. APMIS 113, 727–742, APMapm_302 (2005).
de Rooij, D. G. Spermatogonial stem cell renewal in the mouse. I. Normal situation. Cell Tissue Kinet 6, 281–287 (1973).
Oakberg, E. F. Spermatogonial stem-cell renewal in the mouse. Anat Rec 169, 515–531, 10.1002/ar.1091690305 (1971).
Russell, L. D. et al. Spermatogenesis in Bclw-deficient mice. Biol Reprod 65, 318–332 (2001).
Kanatsu-Shinohara, M. et al. Long-term culture of mouse male germline stem cells under serum-or feeder-free conditions. Biol Reprod 72, 985–991, biolreprod.104.036400 (2005).
Kanatsu-Shinohara, M. et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 69, 612–616, 10.1095/biolreprod.103.017012 (2003).
Kubota, H., Avarbock, M. R. & Brinster, R. L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc Natl Acad Sci U S A 101, 16489–16494, 0407063101 (2004).
Buageaw, A. et al. GDNF family receptor alpha1 phenotype of spermatogonial stem cells in immature mouse testes. Biol Reprod 73, 1011–1016, biolreprod.105.043810 (2005).
Kanatsu-Shinohara, M., Toyokuni, S. & Shinohara, T. CD9 is a surface marker on mouse and rat male germline stem cells. Biol Reprod 70, 70–75, 10.1095/biolreprod.103.020867 (2004).
Shinohara, T., Avarbock, M. R. & Brinster, R. L. beta1- and alpha6-integrin are surface markers on mouse spermatogonial stem cells. Proc Natl Acad Sci U S A 96, 5504–5509 (1999).
Shinohara, T., Orwig, K. E., Avarbock, M. R. & Brinster, R. L. Spermatogonial stem cell enrichment by multiparameter selection of mouse testis cells. Proc Natl Acad Sci U S A 97, 8346–8351, 97/15/8346 (2000).
Kanatsu-Shinohara, M. et al. Genetic and epigenetic properties of mouse male germline stem cells during long-term culture. Development 132, 4155–4163, dev.02004 (2005).
Kubota, H. & Brinster, R. L. Culture of rodent spermatogonial stem cells, male germline stem cells of the postnatal animal. Methods Cell Biol 86, 59–84, S0091-679X(08)00004-6 (2008).
Goharbakhsh, L. et al. Isolation and culture of human spermatogonial stem cells derived from testis biopsy. Avicenna J Med Biotechnol 5, 54–61 (2013).
He, Z., Kokkinaki, M., Jiang, J., Dobrinski, I. & Dym, M. Isolation, characterization and culture of human spermatogonia. Biol Reprod 82, 363–372, biolreprod.109.078550 (2010).
Lee, D. R. et al. Isolation of male germ stem cell-like cells from testicular tissue of non-obstructive azoospermic patients and differentiation into haploid male germ cells in vitro. Hum Reprod 21, 471–476, dei319 (2006).
Lim, J. J. et al. Long-term proliferation and characterization of human spermatogonial stem cells obtained from obstructive and non-obstructive azoospermia under exogenous feeder-free culture conditions. Cell Prolif 43, 405–417, CPR691 (2010).
Hamra, F. K. et al. Self renewal, expansion and transfection of rat spermatogonial stem cells in culture. Proc Natl Acad Sci U S A 102, 17430–17435, 0508780102 (2005).
Alpert, P. F. & Klein, R. S. Spermatogenesis in the unilateral cryptorchid testis after orchiopexy. J Urol 129, 301–302 (1983).
Skinner, J. D. & Rowson, L. E. Some effects of unilateral cryptorchism and vasectomy on sexual development of the pubescent ram and bull. J Endocrinol 42, 311–321 (1968).
Honaramooz, A. et al. Sperm from neonatal mammalian testes grafted in mice. Nature 418, 778–781, 10.1038/nature00918 (2002).
Goossens, E., Frederickx, V., De Block, G., Van Steirteghem, A. C. & Tournaye, H. Reproductive capacity of sperm obtained after germ cell transplantation in a mouse model. Hum Reprod 18, 1874–1880 (2003).
Schlatt, S., Kim, S. S. & Gosden, R. Spermatogenesis and steroidogenesis in mouse, hamster and monkey testicular tissue after cryopreservation and heterotopic grafting to castrated hosts. Reproduction 124, 339–346 (2002).
Shinohara, T. et al. Birth of offspring following transplantation of cryopreserved immature testicular pieces and in-vitro microinsemination. Hum Reprod 17, 3039–3045 (2002).
Nagano, M., Ryu, B. Y., Brinster, C. J., Avarbock, M. R. & Brinster, R. L. Maintenance of mouse male germ line stem cells in vitro. Biol Reprod 68, 2207–2214, 10.1095/biolreprod.102.014050 (2003).
Naughton, C. K., Jain, S., Strickland, A. M., Gupta, A. & Milbrandt, J. Glial cell-line derived neurotrophic factor-mediated RET signaling regulates spermatogonial stem cell fate. Biol Reprod 74, 314–321, biolreprod.105.047365 (2006).
Shen, F. et al. Long-term culture and transplantation of spermatogonial stem cells from BALB/c mice. Cells Tissues Organs 191, 372–381, 000276586 (2010).
Guan, K. et al. Generation of functional cardiomyocytes from adult mouse spermatogonial stem cells. Circ Res 100, 1615–1625, 01.RES.0000269182.22798.d9 (2007).
Kanatsu-Shinohara, M. et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell 119, 1001–1012, S0092867404010578 (2004).
Ehmcke, J. & Schlatt, S. A revised model for spermatogonial expansion in man: lessons from non-human primates. Reproduction 132, 673–680, 132/5/673 (2006).
de Rooij, D. G., Okabe, M. & Nishimune, Y. Arrest of spermatogonial differentiation in jsd/jsd, Sl17H/Sl17H and cryptorchid mice. Biol Reprod 61, 842–847 (1999).
Shinohara, T., Avarbock, M. R. & Brinster, R. L. Functional analysis of spermatogonial stem cells in Steel and cryptorchid infertile mouse models. Dev Biol 220, 401–411, 10.1006/dbio.2000.9655 (2000).
McLean, D. J., Russell, L. D. & Griswold, M. D. Biological activity and enrichment of spermatogonial stem cells in vitamin A-deficient and hyperthermia-exposed testes from mice based on colonization following germ cell transplantation. Biol Reprod 66, 1374–1379 (2002).
Rathi, R., Honaramooz, A., Zeng, W., Turner, R. & Dobrinski, I. Germ cell development in equine testis tissue xenografted into mice. Reproduction 131, 1091–1098, 131/6/1091 (2006).
Schoenfeld, H. A., Hall, S. J. & Boekelheide, K. Continuously proliferative stem germ cells partially repopulate the aged, atrophic rat testis after gonadotropin-releasing hormone agonist therapy. Biol Reprod 64, 1273–1282 (2001).
Brinster, R. L. Germline stem cell transplantation and transgenesis. Science 296, 2174–2176, 10.1126/science. (2002).
Brinster, R. L. & Avarbock, M. R. Germline transmission of donor haplotype following spermatogonial transplantation. Proc Natl Acad Sci U S A 91, 11303–11307 (1994).
Ogawa, T., Arechaga, J. M., Avarbock, M. R. & Brinster, R. L. Transplantation of testis germinal cells into mouse seminiferous tubules. Int J Dev Biol 41, 111–122 (1997).
Ogawa, T., Dobrinski, I., Avarbock, M. R. & Brinster, R. L. Transplantation of male germ line stem cells restores fertility in infertile mice. Nat Med 6, 29–34, 10.1038/71496 (2000).
Aslam, I., Fishel, S., Moore, H., Dowell, K. & Thornton, S. Fertility preservation of boys undergoing anti-cancer therapy: a review of the existing situation and prospects for the future. Hum Reprod 15, 2154–2159 (2000).
Lass, A., Akagbosu, F. & Brinsden, P. Sperm banking and assisted reproduction treatment for couples following cancer treatment of the male partner. Hum Reprod Update 7, 370–377 (2001).
McLean, D. J., Johnston, D. S., Russell, L. D. & Griswold, M. D. Germ cell transplantation and the study of testicular function. Trends Endocrinol Metab 12, 16–21, S1043-2760(00)00330-1 (2001).
Acknowledgements
This study was supported by a grant from the Stem Cell Research Program (NRF-2006-2004127) of the Ministry of Science, ICT & Future Planning and the Basic Science Research (2009-0093821) and the BK21 PLUS Program (22A20130012640) through NRF funded by the Ministry of Education, Republic of Korea.
Author information
Authors and Affiliations
Contributions
J.J.L. and D.R.L. conceived and designed the research. J.J.L., D.W.S., K.H.C. and D.H.S. performed the experiments. J.J.L., H.J.K., S.H.S. and D.R.L. analyzed the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lim, J., Seol, D., Choi, K. et al. Spermatogonial stem cell enrichment using simple grafting of testis and in vitro cultivation. Sci Rep 4, 5923 (2014). https://doi.org/10.1038/srep05923
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05923
This article is cited by
-
Relative safety of various spermatogenic stem cell purification methods for application in spermatogenic stem cell transplantation
Stem Cell Research & Therapy (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
