
Abstract
Serine/threonine kinase 39 (STK39) gene has been reported to be a hypertension-susceptibility gene by a recent genome-wide association study in Western populations. To validate this finding in Chinese, we focused on five well-characterized common polymorphisms in STK39 gene to examine their potential association with hypertension in a large northeastern Han population. This is a hospital-based case-control study involving 1009 hypertensive patients and 756 normotensive controls. Data were analyzed by the Haplo.Stats and multifactor dimensionality reduction (MDR) softwares. The genotype and allele distributions of rs6749447, rs3754777 and rs6433027 differed significantly between patients and controls (P < 0.001) even after the Bonferroni correction. The majority of derived haplotypes also showed remarkable differences between the two groups (P ≤ 0.001). As indicated by MDR analysis, a three-locus model including rs6749447, rs35929607 and rs3754777 was selected as the overall best with a larger testing accuracy of 0.7309 and a maximum cross-validation consistency of 10 (P < 0.001). The utility of this model was reinforced by a Logistic regression analysis. Taken together, our findings suggest the potential interactive role of STK39 gene multiple polymorphisms in the development of hypertension among northeastern Han Chinese.
Similar content being viewed by others

Introduction
Hypertension is a common complex disease that causes significant morbidity and mortality worldwide. Evidence is mounting suggesting that arterial hypertension is a leading risk factor for global disease burden with 16.5% of all deaths attributable to high blood pressure (BP)1. Also hypertension is a polygenic disease with a considerable genetic component2. Considering the fact that some individuals are more susceptible to hypertension than others3,4, it is therefore of timely importance to explain this inter-individual difference in disease susceptibility in order to improve the existing therapy and prevention strategies.
Not until a recent genome-wide association study by Wang et al in Western populations5 has the potential contributory role of serine/threonine kinase 39 (STK39) gene in the pathogenesis of hypertension has drawn global attention6. More recently, Xi et al performed a meta-analysis of 10 studies on STK39 gene rs3754777 and found significant association in Europeans and East Asians, but not in Africans7. There exists a common drawback for these two studies, that is, the genetic defects of STK39 gene were examined individually without allowing for their potential interactions. Such interaction cannot be overlooked because polymorphisms do not exist in isolation and it may be the combination of base changes at several loci that influences gene function8,9.
STK39 is a sterile 20-like-related proline-alanine-rich kinase that not only mediates the phosphorylation of several cation-chloride-coupled cotransporters to maintain salt-water homeostasis but also interacts with the components of p38 MAP kinase pathway to attenuate the injury of cellular stress10,11. The gene encoding STK39 consists of 18 exons and spans about 300 kb on chromosome 2q24.3. The genomic sequence of STK39 gene is polymorphic and so far 34 polymorphisms have been validated by SNPbrowser™. Several genetic polymorphisms in STK39 gene have been reported to be associated with hypertension; however, the results are often not reproducible12,13,14,15,16. To generate more information and unveil an epistatic picture, we in this study focused on five well-characterized common polymorphisms of STK39 gene, aiming to examine their individual and interactive association with the risk of hypertension among northeastern Han Chinese.
Methods
Study population
This was a hospital-based case-control study. Altogether, 1765 unrelated subjects of Han nationality were enrolled from the local residents of Qiqihar city, Heilongjiang province in northeast China. The Ethics Committee of Qiqihar Medical University approved this study that was conducted in accordance with the guidelines outlined in Declaration of Helsinki. All subjects signed informed consent at the time of enrollment.
All study subjects were assigned to the hypertensive group or normotensive group based on clinical examinations and laboratory measurements. Essential hypertension, with an unknown etiology, is the cause of 95% of all cases of diagnosed hypertension.
The hypertensive group consisted of inpatients or outpatients of the Second Affiliated Hospital of Qiqihar Medical University. Hypertensive patients with clinical evidence of secondary hypertension and renal disease were excluded. The normotensive controls who underwent a medical examination at the same hospital were clinically confirmed to be free of hypertension and had a negative family history of hypertension in their first-degree relatives. All study subjects were genetically unrelated local residents of Han Chinese nationality who were recruited consecutively between June 2008 and December 2012.
Diagnosis
Hypertension is defined as a mean systolic BP of at least 140 mm Hg or a mean diastolic BP of at least 90 mm Hg or the current intake of antihypertensive drugs. BP was measured using a calibrated mercury sphygmomanometer with an appropriate adult cuff size by certified examiners.
As recommended by Tobin et al17, for subjects under antihypertensive treatment, BP was imputed by respectively adding 15 and 10 mmHg for systolic and diastolic BP and this imputation was also adopted by Newton-Cheh et al18. Unless otherwise indicated, the imputed systolic and diastolic BP was used in the analysis.
Sample size
The hypertensive group included 1009 sporadic patients with a mean age of 64.48 (standard deviation: 8.53) years. Males comprised 54.31% of the hypertensive group. The remaining subjects (n = 756) whose blood pressure was normal formed the age-, gender- and ethnicity-matched control group.
Demographic and clinical measurement
At the time of enrollment, the data on age, gender, body weight and height were recorded. Body weight and height were measured when subjects in light clothes and bare feet to the nearest 0.1 kg and 0.1 cm, respectively. Body mass index (BMI) was computed as the weight in kilograms divided by height in meters squared.
Fasting venous blood was drawn from each subject and the upper sera were isolated immediately by centrifugation. The plasma levels of triglycerides, total cholesterol, high-density lipoprotein cholesterol (HDL-C), blood urea nitrogen, creatinine and urea acid were determined enzymatically using available kits and auto-analyzers. High-sensitivity C-reactive protein (hsCRP) was measured using a particle-enhanced immunoturbidimetric method.
Polymorphism selection
With the use of PubMed and Online Mendelian Inheritance in Man datasets, five intronic common polymorphisms in STK39 were selected, including rs6749447, rs12692877, rs35929607, rs3754777 and rs6433027. These polymorphisms have been reported to be associated with hypertension or BP by many studies5,7,16,19,20. The genomic organization of human STK39 gene and the localization of five examined polymorphisms are provided in Supplementary material online, Figure S1.
Genotyping
Genomic DNA was isolated from peripheral blood leukocytes according to a standard phenol-chloroform method and stored at −40°C until required for batch genotyping. Genotypes of all examined polymorphisms were determined by a polymerase chain reaction-ligase detection reaction (PCR-LDR) method as previously described9.
The PCR reaction was conducted in an EDC-810 Amplifier (Dongsheng Innovation Biotech Co., Ltd., Beijing, China). The cycling parameters were as follows: 94°C for 2 min; 35 cycles of 94°C for 20 s, 60°C for 20 s and 72°C for 20 s; and a final extension step at 72°C for 3 min.
Two specific probes were synthesized for each polymorphism to discriminate specific bases with one common probe labeled by 6-carboxy-fluorescein (FAM) at the 3′ end and by phosphorylated at the 5′ end. The multiplex ligation reaction was carried out in a reaction volume of 10 μl containing 2 μl of PCR product, 1 μl 10 × Taq DNA ligase buffer, 1 μM of each discriminating probe, 5 U Taq DNA ligase with the ligation parameters of 30 cycles of 94°C for 30 s and 56°C for 3 min. After the reaction, 1 μl LDR reaction product was mixed with 1 μl ROX passive reference and 1 μl loading buffer and then denatured at 95°C for 3 min and chilled rapidly in ice water. The fluorescent products of LDR were differentiated using the ABI 3730XL sequencer (Applied Biosystems, USA).
To test the accuracy of this PCR-LDR method, 96 DNA samples were randomly selected and run in duplicates with 100% concordance.
Statistical analysis
Statistical analyses were completed with the STATA software (version 11.0) for Windows (StataCorp LP, College Station, TX, USA). Study power was computed by the Power and Sample Size Calculations (PS) software (v3.0.7).
Demographic indexes and clinical biomarkers were compared between patients and controls by the unpaired t-test or the Mann-Whitney U test or the χ2 test where appropriate. The deviation from Hardy-Weinberg equilibrium was assessed by a Pearson goodness-of-fit test. The genotype and allele distributions of five examined polymorphisms between the two groups were compared by the χ2 test.
Each genotype of examined polymorphisms was regressed in a Logistic model, assuming the additive (major homozygotes versus heterozygotes versus minor homozygotes), the dominant (major homozygotes versus heterozygotes plus minor homozygotes) and the recessive (major homozygotes plus heterozygotes versus minor homozygotes) models of inheritance with covariates of age, gender and BMI. Risk estimates are expressed as the odds ratio (OR) and its 95% confidence interval (95% CI).
The frequencies of haplotypes derived from five examined polymorphisms in STK39 gene were estimated by the haplo.em program. This program computes the maximum likelihood estimates of haplotype probabilities using the progressive insertion algorithm which progressively inserts batches of loci into haplotypes of growing lengths. In this study, only haplotype with estimated frequency of greater than 1% was considered. P value was simulated on 1000 replicates. In addition, the haplo.score program was used to model a subject's intermediate phenotype as a function of each inferred haplotype, weighted by their estimated probability, to account for the haplotype ambiguity. This program provides the global and haplotype-specific test21. The haplo.em and haplo.score programs are implemented in the Haplo.Stats software (version 1.4.0) operated in the R language (version 2.14, available at the website http://www.r-project.org).
The open-source data-mining multifactor dimensionality reduction (MDR) approach22,23 (version 3.0, available at the website www.epistasis.org) was employed to identify and characterize the interaction of STK39 gene multiple polymorphisms. This approach aims to construct all possible combinations of examined polymorphisms and selects the overall best model. The accuracy of each model is evaluated by a Bayes classifier in the context of 10-fold cross validation. A single best model simultaneously has the maximum testing accuracy and cross-validation consistency (a measure of the number of times of 10 divisions of the dataset that the best model is extracted). Statistical significance was evaluated using a 1000-fold permutation test to compare the observed testing accuracy with the expected one under the null hypothesis of null association. The permutation test corrects for multiple testing by repeating the entire analysis on 1000 datasets that are consistent with the null hypothesis. Further to test the utility of MDR analysis, a classical Logistic regression analysis was undertaken.
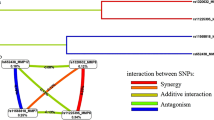
Furthermore, an interaction entropy graph was depicted to quantify the synergistic and non-synergistic interactions between examined polymorphisms. This graph is implemented in MDR approach. The information gain value expressed as the percentage in the node signifies the independent main effect of each examined polymorphism. For the information gain value on the connected line, the positive, negative and zero values indicate synergism (the red or the orange line), redundancy (the green or the blue line) and independence (the yellow line), respectively.
Results
Baseline characteristics
Demographic indexes and clinical biomarkers are compared between hypertensive patients and normotensive controls in Table 1. The distribution of age (P = 0.751) and gender (P = 0.843) did not differ significantly between the two groups. Relative to controls, the levels of BMI (P < 0.001), systolic and diastolic BP (both P < 0.001), fasting glucose (P < 0.001), triglycerides (P = 0.024), creatinine (P = 0.002) and hsCRP (P < 0.001) were significantly higher in patients, whereas the HDL-C level was significantly lower (P < 0.001). No significant difference was observed for the other clinical biomarkers (P > 0.05). The proportion of patients undergoing antihypertensive treatment was 14.17%.
Single-locus analysis
The genotype distributions and allele frequencies of five examined polymorphisms in STK39 gene are provided in Table 2. There was no deviation from the Hardy-Weinberg equilibrium for all polymorphisms (P > 0.05). Overall differences were significant for the genotype and allele distributions of rs6749447, rs3754777 and rs6433027 between patients and controls (P < 0.001 for all) even after the Bonferroni correction (Bonferroni significance threshold P = 0.05 divided by the total number of 5 examined polymorphisms: P = 0.01). The power to reject the null hypothesis of no differences in risk allele frequencies of rs6749447, rs3754777 and rs6433027 between the two groups was 100%, 100% and 98.4%, respectively.
Table 3 summarizes the risk prediction of five examined polymorphisms in STK39 gene for hypertension under three genetic models of inheritance. Compared with the additive model, the risk estimates of rs6749447 and rs3754777 were slightly increased under the dominant model, whereas this increase was obvious under the recessive model. Taking rs3754777 as an example, the odds of having hypertension was 3.44 (P < 0.001), 3.94 (P < 0.001) and 6.23 (P < 0.001) under the additive, dominant and recessive models, respectively. It is worth noting that this significant association was independent of the confounding factors including age, gender and BMI. In contrast to rs6433027, the crude highest risk estimate was seen under the dominant model (OR = 1.82; 95% CI: 1.45–2.28; P < 0.001) but attenuated after adjusting for the confounders (OR = 1.63; 95% CI: 1.26–2.10; P < 0.001).
Haplotype analysis
The haplotype frequencies of five examined polymorphisms in STK39 gene are compared between patients and controls in Table 4. To improve the power to detect an association, only haplotype with frequency of at least 1% in overall subjects was analyzed. All derived haplotypes accounted for 96.77%, 95.18% and 97.49% of the cumulative variance in overall subjects, patients only and controls only, respectively.
The frequencies of most derived haplotypes differed significantly (P ≤ 0.001) between the two groups, except for three low-penetrance haplotypes C-C-G-G-C (alleles in order of rs6749447, rs12692877, rs35929607, rs3754777 and rs6433027) (P = 0.115), A-G-A-G-C (P = 0.393) and C-C-G-G-T (P = 0.460) even after the Bonferroni correction (Bonferroni significance threshold P = 0.05 divided by the total number of comparisons (n = 17): P = 0.0029).
Haplotype-phenotype analysis
When treating all derived haplotypes as a whole, the omnibus association with demographic indexes and clinical biomarkers is presented in Table 5. In hypertensive patients, the marginally significant association was observed for SBP (global simulated P = 0.019), HDL-C (global simulated P = 0.017) and BUN (global simulated P = 0.047). There was no observable significance between haplotypes and phenotypes in controls.
Interaction analysis
In view of the significant findings in the haplotype analysis, it is of great interest to explore the potential interaction of five examined polymorphisms in STK39 gene. To achieve this goal, a promising data-mining analytical approach MDR was employed (Table 6). Each best model across all possible combinations is assessed by the testing accuracy, cross-validation consistency and significance level.
Compared with the other models, a four-locus model incorporating rs6749447, rs35929607, rs3754777 and rs6433027 was the best with the maximum testing accuracy of 0.7329 and cross-validation consistency of 10 out of 10. However, the best three-locus model including rs6749447, rs35929607 and rs3754777 also had a maximum cross-validation consistency of 10 and higher testing accuracy of 0.7309, secondary to the best four-locus model. The best three- and four-locus models were both significant at P < 0.001, indicating that a model this good or better was observed less than 1 out of 1000 permutations and thus unlikely hinged on the null hypothesis of null association. In view of this subtle difference, to pinpoint the polymorphisms that are of particular interest, the three-locus model was regarded as the overall best MDR model in this study.
Additionally to test the utility of MDR analysis, a classical Logistic model was conducted with the outcome variable of hypertension status, predictor variable of the product of three polymorphisms (rs6749447, rs35929607, rs3754777) in the best three-locus model and covariates of age, gender and BMI. Consistently, the interaction of these three polymorphisms was found to be significantly associated with a 1.44-fold (95% CI: 1.36–1.53; P < 0.001) increased risk of hypertension under the additive model. After adjusting for the confounding factors, this interaction was associated with a 1.40-fold increased risk (95% CI: 1.32–1.49; P < 0.001).
Interaction graph
Supplementary Figure S2 depicts the interaction graph of five examined polymorphisms in STK39 gene. The independent main effect was largest for rs3754777 with the information gain value of 6.65%, whereas this largest effect was remarkably attenuated by interacting with the other polymorphisms. Notably, the interaction of rs3754777 with rs6749447 was antagonistic with the information gain value of 2.12%. What's more, the interaction of rs6749447 with rs6433027 resulted in a negative information gain value of −1.25%.
Discussion
With a relatively large study population from northeast China, we examined the association of five well-characterized common polymorphisms in STK39 gene with the risk of hypertension and found that three of them exhibited independent significant association in the single-locus analysis. To the authors' knowledge, this is the first report assessing the interactive contribution of STK39 gene multiple polymorphisms in susceptibility to hypertension. These findings not only deepen our understanding that complex genetic interactions account for disease risk but also provide evidence supporting the logical functional role of STK39 in the pathogenesis of hypertension.
Recently, we have genotyped four top polymorphisms in STK39 gene with genome-wide significance among 1108 southern Han Chinese from Shanghai. Unfortunately none of these polymorphisms were successfully replicated for hypertension risk16. Moreover, another replication in a British Caucasian cohort also failed to detect any relationship between these polymorphisms and BP variation19. However, in a recent meta-analysis by Xi et al, STK39 gene rs3754777 was observed to be significantly associated with hypertension risk in Europeans and East Asians7, in agreement with the results of this study among northeastern Han Chinese. Besides, we additionally identified another two polymorphisms in STK39 gene that individually showed independent association with hypertension even after the stringent Bonferroni correction. A possible explanation is genetic heterogeneity across races and ethnicities. For example, the frequency of rs6749447 minor A allele was 23% in this study population, which was slightly lower than that (28%) observed in a Caucasian cohort from HapMap dataset. Contrastingly, the identification in a South African cohort yielded an exceedingly high frequency at 53%19. It is not uncommon to encounter genetic heterogeneity in any disease identification strategy24, which can be somewhat avoided by studying homogeneous populations25. In fact, our study population is characterized by genetic homogeneity and geographic stability. Moreover, the subjects are most likely uniform in terms of environmental exposures, including habitual dietary intake of high amount of salt and fat26 and a lower rate of hypertension recognition and treatment. These factors render this population more appropriate for enhancing the understanding of genetic predisposition to hypertension. Furthermore, it cannot be fully ruled out that the evolutionary history of linkage disequilibrium patterns varies significantly across different ethnic populations. Usually a locus is in close linkage with another nearby causal locus in one ethnic group but not in another27. As a consequence, there is a need to construct a database of hypertension-susceptibility genes or polymorphisms in each racial/ethnic group.
Another possibility for these inconsistent findings is the potential modifiable impact of environmental or lifestyle factors on genetic predisposition. Genetic factors may increase an individual's vulnerability to develop a disease; however its occurrence largely depends on the exposure to a certain environment or lifestyle. For example, a high-salt diet is known to induce or aggravate the development of hypertension in both animal models and human beings28,29. In China, the amount of salt consumption differs remarkably between northern and southern Chinese. According to a multi-center observation in China, the average urinary sodium excretion in northern Chinese (271 mmol per day) is nearly double the amount of that in southern Chinese (139 mmol per day), leading to average 7.4 and 6.9 mmHg increases of systolic and diastolic BP, respectively30. In view of the divergent STK39-hypertension association between northeastern (the present study) and southern Chinese16, it seems reasonable to speculate that the high-salt intake may modify an individual's genetic predisposition to hypertension. In fact, STK39 encodes a protein kinase more commonly known as SPAK, which participates in a signaling pathway that activates thiazide-sensitive NaCl cotransport in the kidney. An emerging body of evidence demonstrates that genetic defects that increase STK39 expression can alter renal salt handling and the BP set point5,31,32. The exact mechanism underlying the significant STK39-hypertension association in this study is so far unclear and thus if involved, these intronic polymorphisms might be in linkage disequilibrium with other flanking functional loci on STK39 gene that affect the final bioavailability of STK39.
Extending the previous observations, our findings revealed strong evidence that STK39 gene multiple polymorphisms may act interactively to increase hypertension risk. It is generally believed that the relative risk attributable to a single locus is small. To make up for this flaw, we first applied a haplotype analysis by pooling all examined polymorphisms together. Haplotype is composed of different alleles and so haplotype analysis could be more informative regarding the effect of genetic interaction on phenotype than an analysis with a single polymorphic marker33. The possible reason of this effect may owe to the synergistic action between rs6749447-C and rs3754777-G alleles, as the haplotypes simultaneously possessing these two alleles were overrepresented in hypertensive patients, albeit their protective tendency in a single-locus analysis, or to the antagonist action between rs6749447-A and rs6433027-T alleles, as the haplotypes with simultaneous presence of these two alleles were frequently observed in normotensive controls. In view of the complex allelic interaction in a haplotype, we then adopted a promising data-mining approach MDR. This approach is nonparametric and model-free in design and has been successfully applied to detect and characterize high-order gene-gene and gene-environment interactions in studies with relatively small samples3,34. By using MDR approach, we identified a significant interaction between rs6749447, rs35929607 and rs3754777. Further in interaction entropy investigation, despite the predominant role of rs6749447 and rs3754777, their joint effect was strongly attenuated as reflected by the information gain value. Moreover, the predictive value of re6437027 was totally offset by rs6749447 with negative entropy value. These interactions further reinforced the results of our haplotype analysis. Although the independent main effect of rs35929607 was extremely low, its potential role was fully embodied by interacting with rs3754777. Considering the ubiquity of epistasis in determining hypertension susceptibility, it is a high priority to examine the interaction of more candidate genes or pathways.
This study has several possible limitations. First, the cross-sectional nature of our study did not allow us to make inference about the causality for the effects35. Second, this study of 1765 subjects may be underpowered to discern a valid, medically important effect. However, a sample size calculation ensured us with more than 98% statistical power to detect significance. Third, this study had a limited coverage of genetic markers in STK39 gene and it is highly encouraged to incorporate other polymorphisms, especially some low-penetrance mutations or other logical hypertension-susceptibility genes, such as the genes encoding CaMK436 and GRKs37,38. Fourth, due to our design flaw, some clinical and lifestyle details on the study population were not available, as well as other reproducible risk factors for hypertension, which prevented further adjustment in risk estimates and may have overestimated the true effect size. Fifth, the MDR approach used in this study has some underlying drawbacks, including computational intensiveness, indistinct interpretation, lack of sensitivity and heterogeneity-free assumption39,40. Last but not the least, the fact that our study subjects were of northeastern Han Chinese descent limits the generalizability of our findings and necessitates further confirmation in other ethnic populations.
Despite these limitations, our findings suggest the potential interactive role of STK39 gene multiple polymorphisms in the development of hypertension among northeastern Han Chinese. As the development of hypertension is believed to be largely under genetic control, more emphases should be placed on the detection and characterization of multiple genetic interactions to predict high-risk individuals for prevention and personalized treatment of hypertension.
References
Santulli, G. Epidemiology of Cardiovascular Disease in the 21st Century: Updated Numbers and Updated Facts. JCvD 1, 1–2 (2013).
Niu, W. et al. Validation of genetic association in apelin-AGTRL1 system with hypertension in a larger Han Chinese population. J Hypertens 28, 1854–1861 (2010).
Niu, W. et al. Haplotype-based association of the renin-angiotensin-aldosterone system genes polymorphisms with essential hypertension among Han Chinese: the Fangshan study. J Hypertens 27, 1384–1391 (2009).
Wang, Y. L. et al. Tag polymorphisms of solute carrier family 12 member 3 gene modify the risk of hypertension in northeastern Han Chinese. J Hum Hypertens (2014).
Wang, Y. et al. From the Cover: Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A 106, 226–231 (2009).
Maatta, K. M., Nikkari, S. T., Lahteela, K. H., Palmroos, P. B. & Kunnas, T. A. A functional variant in the serine-threonine kinase coding gene is associated with hypertension: a case-control study in a Finnish population, the Tampere adult population cardiovascular risk study. J Hypertens 31, 516–520; discussion 520 (2013).
Xi, B. et al. STK39 polymorphism is associated with essential hypertension: a systematic review and meta-analysis. PloS one 8, e59584 (2013).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Niu, W. et al. Confirmation of top polymorphisms in hypertension genome wide association study among Han Chinese. Clin Chim Acta 411, 1491–1495 (2010).
Piechotta, K., Garbarini, N., England, R. & Delpire, E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl- cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem 278, 52848–52856 (2003).
Johnston, A. M. et al. SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene 19, 4290–4297 (2000).
Rhee, M. Y. et al. Novel genetic variations associated with salt sensitivity in the Korean population. Hypertens Res 34, 606–611 (2011).
Xu, J. et al. Lack of association between STK39 and hypertension in the Chinese population. J Hum Hypertens 27, 294–297 (2013).
Xi, B. et al. Influence of obesity on association between genetic variants identified by genome-wide association studies and hypertension risk in Chinese children. Am J Hypertens 26, 990–996 (2013).
Shin, D. J., Lee, S. H., Park, S. & Jang, Y. Association between Serine/Threonine Kinase 39 Gene Polymorphism, Hypertension and Other Cardiovascular Risk Factors in Koreans. Korean Circ J 43, 13–22 (2013).
Niu, W. Q., Zhang, Y., Ji, K. D., Gao, P. J. & Zhu, D. L. Contribution of five top whole-genome association signals to hypertension in Han Chinese. J Hum Hypertens 25, 278–280 (2011).
Tobin, M. D., Sheehan, N. A., Scurrah, K. J. & Burton, P. R. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med 24, 2911–2935 (2005).
Newton-Cheh, C. et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 41, 666–676 (2009).
Cunnington, M. S. et al. STK39 polymorphisms and blood pressure: an association study in British Caucasians and assessment of cis-acting influences on gene expression. BMC Med Genet 10, 135 (2009).
Chen, L. Y. et al. STK39 is an independent risk factor for male hypertension in Han Chinese. Int J Cardiol 154, 122–127 (2012).
Schaid, D. J., Rowland, C. M., Tines, D. E., Jacobson, R. M. & Poland, G. A. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70, 425–434 (2002).
Pattin, K. A. et al. A computationally efficient hypothesis testing method for epistasis analysis using multifactor dimensionality reduction. Genet Epidemiol 33, 87–94 (2009).
Hahn, L. W., Ritchie, M. D. & Moore, J. H. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics 19, 376–382 (2003).
Kato, N. Ethnic differences in genetic predisposition to hypertension. Hypertens Res 35, 574–581 (2012).
Pineda-Krch, M. & Lehtila, K. Costs and benefits of genetic heterogeneity within organisms. J Evol Biol 17, 1167–1177 (2004).
Niu, W. & Qi, Y. Association of alpha-adducin and G-protein beta3 genetic polymorphisms with hypertension: a meta-analysis of Chinese populations. PLoS One 6, e17052 (2011).
Niu, W. et al. A meta-analysis of receptor for advanced glycation end products gene: four well-evaluated polymorphisms with diabetes mellitus. Mol Cell Endocrinol 358, 9–17 (2012).
Gu, J. W. et al. Long-term high salt diet causes hypertension and alters renal cytokine gene expression profiles in Sprague-Dawley rats. Beijing Da Xue Xue Bao 41, 505–515 (2009).
Stolarz-Skrzypek, K., Bednarski, A., Czarnecka, D., Kawecka-Jaszcz, K. & Staessen, J. A. Sodium and potassium and the pathogenesis of hypertension. Curr Hypertens Rep 15, 122–130 (2013).
Zhao, L. et al. Blood pressure differences between northern and southern Chinese: role of dietary factors: the International Study on Macronutrients and Blood Pressure. Hypertension 43, 1332–1337 (2004).
Welling, P. A., Chang, Y. P., Delpire, E. & Wade, J. B. Multigene kinase network, kidney transport and salt in essential hypertension. Kidney Int 77, 1063–1069 (2010).
Donner, K. M., Hiltunen, T. P., Hannila-Handelberg, T., Suonsyrja, T. & Kontula, K. STK39 variation predicts the ambulatory blood pressure response to losartan in hypertensive men. Hypertens Res 35, 107–114 (2012).
Qi, Y., Niu, W., Zhou, W., Hou, S. & Qiu, C. Correlation between angiotensinogen gene polymorphisms and essential hypertension in Chinese population. J Hum Hypertens 22, 147–150 (2008).
Qi, Y., Niu, W., Zhu, T., Zhou, W. & Qiu, C. Synergistic effect of the genetic polymorphisms of the renin-angiotensin-aldosterone system on high-altitude pulmonary edema: a study from Qinghai-Tibet altitude. Eur J Epidemiol 23, 143–152 (2008).
Gu, M. et al. Strong association between two polymorphisms on 15q25.1 and lung cancer risk: a meta-analysis. PLoS One 7, e37970 (2012).
Santulli, G. et al. CaMK4 Gene Deletion Induces Hypertension. J Am Heart Assoc 1, e001081 (2012).
Santulli, G., Trimarco, B. & Iaccarino, G. G-protein-coupled receptor kinase 2 and hypertension: molecular insights and pathophysiological mechanisms. High Blood Press Cardiovasc Prev 20, 5–12 (2013).
Lobmeyer, M. T. et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharmacogenet Genomics 21, 42–49 (2011).
Moore, J. H. & Ritchie, M. D. STUDENTJAMA. The challenges of whole-genome approaches to common diseases. JAMA 291, 1642–1643 (2004).
Gui, J. et al. A simple and computationally efficient sampling approach to covariate adjustment for multifactor dimensionality reduction analysis of epistasis. Hum Hered 70, 219–225 (2010).
Acknowledgements
This study received the grants from the Department of Education Science and Technology Research Project in Heilongjiang Province (12521621), the Shanghai Rising Star Program (11QA1405500) and the Beijing New Star Program (Z111107054511072).
Author information
Authors and Affiliations
Contributions
W.N. and B.W. conceived and designed the experiments; H.Z., Y.Q. and Yuefei W. performed the experiments; W.N. analyzed the data; Yanli W., C.L. and Y.X. contributed materials/analysis tools; W.N. and B.W. wrote and revised the manuscript. All authors reviewed and approved the manuscript prior to submission.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhao, H., Qi, Y., Wang, Y. et al. Interactive contribution of serine/threonine kinase 39 gene multiple polymorphisms to hypertension among northeastern Han Chinese. Sci Rep 4, 5116 (2014). https://doi.org/10.1038/srep05116
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05116
This article is cited by
-
MiR-223-3p-loaded exosomes from bronchoalveolar lavage fluid promote alveolar macrophage autophagy and reduce acute lung injury by inhibiting the expression of STK39
Human Cell (2022)
-
WNK signalling pathways in blood pressure regulation
Cellular and Molecular Life Sciences (2017)
-
A meta-analytical assessment of STK39 three well-defined polymorphisms in susceptibility to hypertension
Scientific Reports (2016)
-
Female genetic distribution bias in mitochondrial genome observed in Parkinson’s Disease patients in northern China
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
