
Abstract
Ultrasmall SnO2 nanocrystals as anode materials for lithium-ion batteries (LIBs) have been synthesized by bubbling an oxidizing gas into hot surfactant solutions containing Sn-oleate complexes. Annealing of the particles in N2 carbonifies the densely packed surface capping ligands resulting in carbon encapsulated SnO2 nanoparticles (SnO2/C). Carbon encapsulation can effectively buffer the volume changes during the lithiation/delithiation process. The assembled SnO2/C thus deliver extraordinarily high reversible capacity of 908 mA·h·g−1 at 0.5 C as well as excellent cycling performance in the LIBs. This method demonstrates the great potential of SnO2/C nanoparticles for the design of high power LIBs.
Similar content being viewed by others

Introduction
SnO2 is a very appealing candidate as a substitute for conventional anodes in lithium-ion batteries (LIBs) because of its high theoretical capacity, high electric conductivity (~21.1 Ω·cm), low potential of lithium ion intercalation, as well as superior electron mobility (100–200 cm2·V−1·s−1) compared to other n-type oxide semiconductors1,2. Nanostructured SnO2 materials such as 0D nanocrystals (NCs) or quantum dots (QDs)3,4, 1D nanowires5 and/or nanorods6, 2D nanosheets/nanofilms4 and 3D-mesoporous SnO27 have attracted wide interest due to their potential for use in a wide variety of applications such as gas sensors4, photocatalysts8, transparent electrodes for energy conversion9 and energy storage devices2,5. The wide applicability of nanostructured SnO2 materials arises from their quantum size effect, large surface area and high surface activity10. In terms of applications in LIBs, 1D- and 3D-mesoporous SnO2 structures are of particular importance due to their use as anode materials capable of delivering improved energy and power densities2,5,11,12. For example, SnO2 nanowires have been demonstrated to deliver a discharge capacity of 468 mA·h·g−1 after 30 cycles and a current density of 100 mA·g−113. Also 3D-mesoporous SnO214 and hollow nanospheres15 were found to have a surprisingly high capacity of 773 mA·h·g−1 after 50 cycles. Unfortunately, SnO2 or Sn-based anodes suffer from volume changes of 200 ~ 300% during the lithiation/delithiation process, which leads to pulverization and loss of electrical contact between particles subsequently resulting in severe capacity fade and poor rate capability16. A number of strategies have been suggested to alleviate this problem including: interconnecting structures to accommodate volume change17, nano-sized crystals with dense stacking to reduce the volume variation18, hybrids with other inactive substances, e.g., SnO2/carbon19, in which the inactive carbon serves as a confining buffer for the volume variations. Despite significant progress already made using these methods, SnO2 electrodes are still far from commercialization. Therefore, it is urgent that new anode materials with novel architectures are investigated towards the development of commercially viable SnO2 electrodes with high energy and power densities. Self-assembled SnO2 hybrid nanoparticles can satisfy the second and third requirements mentioned above, making them an interesting anode material.
In this research, we present a general approach for the synthesis of SnO2 NCs both plain and hybridized with a carbon shell (SnO2/C) via the bubbling of an oxidizing gas through a hot surfactant solution and subsequent annealing. This approach to the synthesis of highly crystalline SnO2 NCs also has the commercial advantage of continuous operation in production of the material and high yield. This is in contrast to the low crystallinity, uncontrollable aggregation and restricted yields obtained using many of the previous and commonly employed hydrothermal or aqueous based methods. Other drawbacks of employing hydrothermal or aqueous solution based gas-liquid reactions to synthesize NCs are the inherent limitations imposed by a lower reaction temperature (<100°C), limited selection of the protecting agents in aqueous solution and/or the high pressure required during the synthesis20,21. These conditions tend to result in an uneven size distribution and less densely packed capping ligands on the surface resulting in large aggregates22. In contrast, gas-liquid reactions based on high boiling-point surfactants have unique merits such as high diffusion rates, ease of homogeneous nucleation, facile control of the reaction process (derived from controllable gas velocity), possibility of providing a ligand layer on the NCs and suitability for use in fixed fluidized bed reactors23,24,25. Here we report that SnO2 NCs synthesized using a gas-liquid reaction and subsequently annealed in an N2 atmosphere have proven to be a good substitute for use as anodic materials in LIBs. The SnO2 NCs, when encapsulated in carbon, exhibited an extraordinarily high reversible capacity of 908 mA·h·g−1 at 0.5 C as well as an excellent rate capability. The high surface area resulting from the assembly of SnO2/C assists in the storage of a greater number of lithium ions26. The porous carbon layers act as an elastic matrix to buffer the volume changes of SnO2, which afford brilliant cycling performances during the lithiation/delithiation process.
Results and discussion
Physical characterization
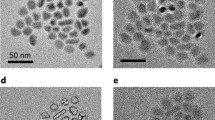
The as-synthesized SnO2 colloidal nanocrystals are greyish in color and can be readily dispersed in nonpolar solvents such as toluene, chloroform and tetrahydrofuran (Supporting Information, SI), which suggests that they have been well capped with the long alkyl chains ligands used in the synthesis. The typical transmission electron microscope (TEM) images of the sample prepared with Sn-oleate complex are presented in Figure 1a and 1b. The Sn-oleate was synthesized from freshly prepared precursor stannic hydroxide and oleic acid in high-boiling point solutions. Detailed procedures are presented in the method section. Monodisperse crystalline nanoparticles with an average size of ~3.0 nm were observed using high resolution TEM (HRTEM) as shown in Figure 1b. The XRD pattern of the as-synthesised colloidal particles is shown as a black curve in Figure 1c whose peaks can be indexed to that of tetragonal SnO2 (JCPDS no. 41-1445). The diffraction peaks (110), (101) and (211) are clearly seen in the XRD profiles. However, the peaks are obviously broadened, especially when compared with the corresponding samples which have subsequently been annealed at 600°C. The XRD profiles of SnO2 NCs annealed in O2 and N2 atmosphere are shown in Figure 1c. These broadened peaks manifest that crystallite domain effects are prominent in the as-synthesized colloidal NCs4. Based on the Scherrer equation, rutile SnO2 NCs have an average lattice plane separation of ~3.3 nm in the [110] direction, which is consistent with the TEM observations27. To study the growth kinetics, the absorption (Abs) spectrum of the colloidal NCs sampled at different stages was monitored. Although the NCs are quite monodisperse and small, the optical absorption of the NC solutions gave a scattering-type response. Our prepared SnO2 NCs are on the same size level as the exciton Bohr radius of SnO2 (2.7 nm), therefore they demonstrate weak quantum confinement effects28. The absorption spectra of the NCs synthesized with different growth times are shown in Figure S1 (see SI). We found a good trend of longer-wavelength shifting during the growth of our colloidal SnO2 NCs.

(a, b) TEM images of SnO2 NCs as-synthesized from Sn-oleate complex via a hot-bubbling route; (c) XRD profiles of the colloidal SnO2 NCs and those annealed in atmosphere of O2 or N2 at temperature of 600°C; (d and inset) TEM images of the colloidal SnO2 NCs synthesized from tin 2-ethylhexanoate capped with oleylamine (OLA).
The microstructure and phase transitions of the annealed samples were studied. As shown by the electronic microscopy images (Figure 2b ~ d in the main manuscript and Figure S2c in the SI), uniformly sized particles were obtained after annealing and the average diameter of the quasi-spheral particles lies between 3.0 and 10.0 nm depending on annealing procedures. The particle size was always found to be smaller than those obtained by hydrothermal reactions, which can be micron-sized19,29. The particles were observed by HRTEM to be composed of numerous single crystals. The two inter-planar observed were 0.26 and 0.33 nm which is consistent with the (101) and (110) of tetragonal SnO2 (Figure 1d in the main manuscript or S2b in the SI), respectively. The average size of SnO2 nanoparticles becomes larger after annealing, especially in an O2 atmosphere, which is consistent with previous findings where the crystal seeds tend to aggregate and merge into larger crystals during the annealing process. Other tin precursors such as tin 2-ethylhexanoate or di-n-octyltin oxide were also employed to synthesize colloidal SnO2 NCs by the same synthetic route. The TEM images of the as-synthesized NCs are displayed in Figure 1d and Figure S2d in the SI, respectively. Both images exhibit uniform particles of a size which is less than 3.0 nm. The inset of Figure 1d and S2d showing higher resolution images of a single particle assists in confirming the crystallinity of the particles obtained by the hot-bubbling method.

(a) TGA plots of SnO2 NCs with thermal treatment in atmosphere of O2 and N2; (b) SEM image of SnO2/C annealed in N2 at 600°C; (c, d) TEM and HRTEM images of SnO2/C and the circled white lines demonstrates amorphous carbon surrounding SnO2 nanoparticles. The inset of (c) is the SAED pattern corresponding to the selected area in figure (c).
The effects of ligand decomposition on the encapsulating carbon – critical for the lithiation/delithiation process – were investigated using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Figure 2a displays the TGA profiles of SnO2 colloidal particles capped with OLA measured in atmosphere of O2 and N2. The corresponding DSC plot at a heating rate of 5°C·min−1 is given in the SI (Figure S3). When the calcination temperature was increased from room temperature to 800°C, a mass loss of around 8 to 15% wt. was observed for the N2 thermal treatment samples. However, in the case of O2 annealing, the mass loss was between 38 and 42% wt. when the temperature was held at 600°C and further increases to 50–55% wt. as the temperature reached 800°C. Three distinct stages in the mass loss can be observed in the TG profile as presented in Figure 2a (black curve). The first stage is located below 200°C and can be explained by the removal of crystalline and adsorbed water. A second mass loss of 30% was found between 320 and 450°C and a prominent exothermic peak (at approx. 390°C) occurs in this region. This large mass loss is due to the oxidation and partial removal of capping agents. Compared to previously published SnO2/C spheres (450°C) synthesized by hydrothermal means30, we can therefore infer that part of the capping ligands (oleic acid (OA) or OLA as demonstrated by Raman and FTIR spectra, Figure S4 and S5 in the SI) are oxidized at these temperatures. The third stage of the mass loss may be related to a phase transition and further combustion of carbon species as is suggested by the exothermic peak at 793°C32. The carbon content in the SnO2 colloidal NCs presented here is close to that of Archer's SnO2 carbon hollow nanospheres which have been determined by TGA to be 32.3% by weight33. TGA together with IR and Raman analysis demonstrates that the colloidal SnO2 NCs prepared in this study were capped with a dense packing layer of long chained alkyl ligands which is similar to what was observed in the synthesis of other quantum dots23. The ligands can be thermally carbonized in N2 annealing, which provides a protecting layer for the SnO2 NCs. As seen from Figure 2b, the sample annealed in N2 atmosphere and which was visually black, was composed of numerous ultrasmall (size below the resolution of FESEM) particles as shown in the SEM panoramic overview and the edges of the particles are blurred compared to those calcined in O2 (SEM image shown in Figure S2a). The shaggy substance on the surface of the particles is amorphous carbon as determined by selected area electron diffraction (SAED) patterns. We thus name this N2-annealled sample as SnO2/C nanoparticles or hybrid NCs. The presence of amorphous carbon layers clearly increase the surface area compared to the sample calcined under O2. The N2 adsorption-desorption isotherms and BJH pore sized distribution both samples are presented in the SI (Figure S6). The specific surface area of the samples annealed in N2 and O2 are 90.78 and 74.65 m2·g−1, respectively. The average pore size lies within 2.0 nm in the SnO2/C sample. Figure 2c shows a typical TEM image of the N2 annealed sample where it can be seen that the uniform size distribution of the particles is still preserved. The SAED pattern in the inset demonstrates that the sample is highly crystalline. The observed diffraction rings can be roughly indexed to tetragonal SnO2. A closer inspection by the HRTEM image of the SnO2/C reveals that the crystalline domains are tightly encapsulated with an amorphous layer and some of the amorphous substance appears between the crystals which separate the crystalline particles as indicated by the white curves in Figure 2d or TEM images of Figure S2e and S2f in the SI. The crystalline domain can be attributed to rutile SnO2 and the amorphous layer, with a thickness of ~2 nm as demonstrated, is the carbon residue produced during the N2 annealing. The results therefore provide a good indication that a physical buffer to prevent the collapse (or fusion) of the SnO2 caused by the volume changes is present and thus a higher capacity for the storage of Li-ions could be obtained by the introduction of this amorphous carbon. To further analyze the chemical composition of the SnO2/C, energy dispersive X-ray analysis (EDXA) was conducted by scanning an area of 0.1 × 0.1 μm2 using TEM. The corresponding element mapping profiles are presented in Figure S7 (see SI). Sn and O were found to be the main elements in the annealed sample. A 1.84% by weight abundance of N was observed which originates from the OLA ligand (Table S1, SI). As previously reported by Zhou and Lou et al.33,34, the incorporation of nitrogen into the anodes could substantially improve the electrochemical performance of LIBs.
During the N2-annealing processes at ~600°C, the ligands can transform into encapsulating carbon layers as further shown by X-ray photoelectron spectroscopy (XPS). The surface chemical composition and the fine-scanning XPS spectra of Sn3d, O1s, C1s and N1s for both the SnO2 colloidal NCs and N2-annealed sample, are detailed in the SI (Table S3 and Figure S8). It was found that the peaks of Sn(IV) at 487.3 (Sn 3d3/2) and 495.9 eV (Sn 3d5/2) in the as-synthesized colloidal SnO2 NCs shift to 486.9 and 495.2 eV after N2-annealing, which reveals that a lower valence Sn species such as Sn(II) (3d5/2, 487.0 eV) or Sn(0) (3d5/2, 485.0 eV) was detected after decomposing the photoemission bands of Sn3d (see the deconvoluted peaks in Figure S8d of the SI)6. Also the O1s XPS spectra manifest that the surface is deficient in O with respect to the stoichiometry of SnO2. In the spectra of C1s, it is found that not only C1 (284.3 eV) and C2 (285.3 eV) peaks ascribed to C-C sp2 bonds in graphite and C-C sp3 diamond-like bonds are present in the amorphous carbon, but also an independent peak located at 289.5 eV is observed, which suggests the presence of C-Nx on the surface35. The N1s peak was also found in the survey scanning profile although it is quite weak (Figure S8f, SI). This suggests that carbon thermal reactions take place in the N2 annealing process, during which amorphous carbon and nitrogen residues are produced34. Although the relative carbon percentage in the annealed sample is lower than that of the as-synthesized colloidal NCs, there is still ~20% atomic percentage (Table S3). It is worth mentioning that the detected carbon content (19.24% at. or 5.0% wt.) is lower than that obtained from TG analysis (~30% wt.). This difference could be due to the micro-area of detection used by XPS where arbitrary errors can occur36. However both results demonstrate a high percentage of carbon content in the N2-annealled sample, due to the existence of densely packed capping ligands on the as-synthesized colloidal SnO2 NCs.
Electrochemical characterization
To assess the suitability of the prepared SnO2 NCs for their incorporation into LIBs devices as active centers, a mixed slurry composed of the annealed SnO2 NCs, acetylene black and poly-vinylidenefluoride (PVDF) was coated onto a copper foil and then the electrochemical performance was studied using two-electrode Swagelok-type half cells with lithium metal serving as both the counter and reference electrode. Figure 3a shows the cyclic voltammetry (CV) profiles of a SnO2/Li half-cell during the first three-cycles. As expected, a notable cathodic peak at 0.75 V can be attributed to the reduction of SnO2 to metallic Sn and the formation of the solid electrolyte interphase (SEI) layer in the first cycle ( , Eq.(1))33. This peak disappears in the following cycles. However, extra reduction peaks located at 0.12 and 0.01 V were found, which can be related to the following reactions: (
, Eq.(1))33. This peak disappears in the following cycles. However, extra reduction peaks located at 0.12 and 0.01 V were found, which can be related to the following reactions: ( , Eq.(2));
, Eq.(2));  (Eq.(3)) and
(Eq.(3)) and  (Eq.(4))37. Three new oxidation peaks appear at 0.13, 0.54 and 1.26 V corresponding to lithium extraction, de-alloying of LixSn and the partially reversible reaction of the SEI layer formation, respectively. Figure 3b shows the galvanostatic discharge/charge profiles up to 20 cycles of the SnO2/C at a density of 0.5 C. The initial discharge and charge capacities are 1590 and 908 mA·h·g−1, respectively. The capacities are calculated on basis of the mass of sample SnO2/C or bare SnO2 nanoparticles loaded in the electrodes. The extraordinarily high capacity corresponding to the first Li+ charge curve can be ascribed to the SEI layer and the formation of Li2O and/or electrolyte decomposition38. The pores in the sample SnO2/C as evidenced by the N2 adsorption-desorption, which have an average size that lies within the Li diffusion length39, would also contribute to a larger irreversible capacity of SEI. A sloping plateau at ~1.2 V due to the partially reversible reaction of SEI layer formation in the Li+ discharge curve was observed in the discharge/charge voltage profile (Figure S9 in the SI), which is consistent with the CV curves shown in Figure 3a. During the subsequent cycles, the Coulombic efficiency of the hybrid electrodes increases from 73% to 98% after 20 cycles, indicating a good trend of reversibility in the electrochemical reactions. Figure 3c shows the cycling performance up to 100 cycles at a current density of 0.5 C in the voltage range of 0.01–2.0 V vs. Li+/Li. Although there are a decline trend and temperature fluctuations observed during the discharge/charge cycles, e.g., two typical capacities of 1120 and 1034 mA·h·g−1 at the fifth and twentieth cycle, a relative stable capacity was achieved at ~910 mA·h·g−1 at the 100th cycle. The capacity is much higher than the bare SnO2 nanoparticles annealed in O2 (~610 mA·h·g−1 at the 50th cycle as shown in Figure S10,) or the previous reports in case of hybrid electrodes (~650 mA·h·g−1)15,17 and the stability is also better than that of bare SnO2 nanoparitles. The SnO2/C electrode demonstrates a capacity retention of about 60.5% over 100 cycles and the capacity loss rate of 0.05% per cycle is far less than those of bare SnO2 and SnO2 nanoparticles coated on carbon nanotubes40. After cycling 100 times, the rate is increased stepwise up to 8 C (Figure 3d). As can be seen, even at a high rate of 8 C, the SnO2/C sample can still deliver a stable capacity of about 450 mA·h·g−1, which is higher than the current commercialized graphite anode9. The high capacity for Li-ion storage of SnO2/C in this research is probably related to the small size of SnO2 as verified by TEM images (Figure 2b, 2c and Figure S2a–c in the SI), which offers a high reactive activity and shortens electronic and ionic transport lengths41. The thin carbon shells can act as minicurrent collectors for the encapsulated SnO2 particles that facilitate fast electron transport during the insertion/extraction processes42. Furthermore, the high degree of crystalline SnO2 particles that are deficient in O but doped with N and have a high specific area, can contribute to the efficient accessibility by the electrolyte thus enhance the electrochemical performance34. It is usually believed that the theoretical capacity of SnO2 is 782 mA·h·g−1 which is only based on the reversible reaction Eq.(2), however a number of investigations have revealed that the reaction of Eq.(1) can be reversible under certain conditions, which would lead to a theoretical capacity of 1494 mA·h·g−141,42. Herein, the as-prepared SnO2 NCs have an ultrasmall size (<3.0 nm) which is similar to the exciton Bohr radius. The diameter of SnO2/C particles remains at about 5.0 nm even after N2 annealing owing to the dense capping ligands after carbonization. As is known43,44,45, the reversibility of reaction Eq.(1) is mainly influenced by the intrinsic conductivity of SnO2, grain size of Li2O and Sn, their degree of dispersion and the contact degree with conductive additives. We thus indicate there can be reversible reaction or at least part of the Eq. (1) occurs in our system. Taking in consideration of SnO2 percentages (70.0%) and C (30.0%) in Sn2O/C, we obtain a data of 1154 mA·h·g−1 (1494 × 0.70 + 372 × 0.3 = 1154 mA·h·g−1). This evaluation is much closer to our experimental results. The high stability or cyclability is mainly attributable to the encapsulating carbon layers which are able to suppress the aggregation of the SnO2 nanoparticles and can preserve the electrochemical activity during the lithiation/delithiation process46. As stated in the literature, the main reason for the rapid fading of SnO2 electrodes is that a large volume expansion of the SnO2 material occurs during the battery cycling, leading to the pulverization of the electrode16. Here, in the case of SnO2/C as confirmed by HRTEM and N2 adsorption-desorption, the carbon layers homogeneously encapsulate the annealed SnO2 NCs separating them. The protecting layers are porous due to the pyrolysis of the ligands. Such an amorphous carbon layer could provide an elastic matrix which is helpful in tolerating the reversible volume change while avoiding the merging of adjacent SnO2 nanoparticles34. Furthermore, the porosity in SnO2/C offers lithium ions easier accessibility during the cycling process, which leads to increased accommodation behavior for lithium as well as improve the kinetics of the SnO2/C anode46. These results demonstrate that the carbon layer not only enhances the electrochemical activity of the SnO2 NCs, but also improve cycling performance significantly34.
(Eq.(4))37. Three new oxidation peaks appear at 0.13, 0.54 and 1.26 V corresponding to lithium extraction, de-alloying of LixSn and the partially reversible reaction of the SEI layer formation, respectively. Figure 3b shows the galvanostatic discharge/charge profiles up to 20 cycles of the SnO2/C at a density of 0.5 C. The initial discharge and charge capacities are 1590 and 908 mA·h·g−1, respectively. The capacities are calculated on basis of the mass of sample SnO2/C or bare SnO2 nanoparticles loaded in the electrodes. The extraordinarily high capacity corresponding to the first Li+ charge curve can be ascribed to the SEI layer and the formation of Li2O and/or electrolyte decomposition38. The pores in the sample SnO2/C as evidenced by the N2 adsorption-desorption, which have an average size that lies within the Li diffusion length39, would also contribute to a larger irreversible capacity of SEI. A sloping plateau at ~1.2 V due to the partially reversible reaction of SEI layer formation in the Li+ discharge curve was observed in the discharge/charge voltage profile (Figure S9 in the SI), which is consistent with the CV curves shown in Figure 3a. During the subsequent cycles, the Coulombic efficiency of the hybrid electrodes increases from 73% to 98% after 20 cycles, indicating a good trend of reversibility in the electrochemical reactions. Figure 3c shows the cycling performance up to 100 cycles at a current density of 0.5 C in the voltage range of 0.01–2.0 V vs. Li+/Li. Although there are a decline trend and temperature fluctuations observed during the discharge/charge cycles, e.g., two typical capacities of 1120 and 1034 mA·h·g−1 at the fifth and twentieth cycle, a relative stable capacity was achieved at ~910 mA·h·g−1 at the 100th cycle. The capacity is much higher than the bare SnO2 nanoparticles annealed in O2 (~610 mA·h·g−1 at the 50th cycle as shown in Figure S10,) or the previous reports in case of hybrid electrodes (~650 mA·h·g−1)15,17 and the stability is also better than that of bare SnO2 nanoparitles. The SnO2/C electrode demonstrates a capacity retention of about 60.5% over 100 cycles and the capacity loss rate of 0.05% per cycle is far less than those of bare SnO2 and SnO2 nanoparticles coated on carbon nanotubes40. After cycling 100 times, the rate is increased stepwise up to 8 C (Figure 3d). As can be seen, even at a high rate of 8 C, the SnO2/C sample can still deliver a stable capacity of about 450 mA·h·g−1, which is higher than the current commercialized graphite anode9. The high capacity for Li-ion storage of SnO2/C in this research is probably related to the small size of SnO2 as verified by TEM images (Figure 2b, 2c and Figure S2a–c in the SI), which offers a high reactive activity and shortens electronic and ionic transport lengths41. The thin carbon shells can act as minicurrent collectors for the encapsulated SnO2 particles that facilitate fast electron transport during the insertion/extraction processes42. Furthermore, the high degree of crystalline SnO2 particles that are deficient in O but doped with N and have a high specific area, can contribute to the efficient accessibility by the electrolyte thus enhance the electrochemical performance34. It is usually believed that the theoretical capacity of SnO2 is 782 mA·h·g−1 which is only based on the reversible reaction Eq.(2), however a number of investigations have revealed that the reaction of Eq.(1) can be reversible under certain conditions, which would lead to a theoretical capacity of 1494 mA·h·g−141,42. Herein, the as-prepared SnO2 NCs have an ultrasmall size (<3.0 nm) which is similar to the exciton Bohr radius. The diameter of SnO2/C particles remains at about 5.0 nm even after N2 annealing owing to the dense capping ligands after carbonization. As is known43,44,45, the reversibility of reaction Eq.(1) is mainly influenced by the intrinsic conductivity of SnO2, grain size of Li2O and Sn, their degree of dispersion and the contact degree with conductive additives. We thus indicate there can be reversible reaction or at least part of the Eq. (1) occurs in our system. Taking in consideration of SnO2 percentages (70.0%) and C (30.0%) in Sn2O/C, we obtain a data of 1154 mA·h·g−1 (1494 × 0.70 + 372 × 0.3 = 1154 mA·h·g−1). This evaluation is much closer to our experimental results. The high stability or cyclability is mainly attributable to the encapsulating carbon layers which are able to suppress the aggregation of the SnO2 nanoparticles and can preserve the electrochemical activity during the lithiation/delithiation process46. As stated in the literature, the main reason for the rapid fading of SnO2 electrodes is that a large volume expansion of the SnO2 material occurs during the battery cycling, leading to the pulverization of the electrode16. Here, in the case of SnO2/C as confirmed by HRTEM and N2 adsorption-desorption, the carbon layers homogeneously encapsulate the annealed SnO2 NCs separating them. The protecting layers are porous due to the pyrolysis of the ligands. Such an amorphous carbon layer could provide an elastic matrix which is helpful in tolerating the reversible volume change while avoiding the merging of adjacent SnO2 nanoparticles34. Furthermore, the porosity in SnO2/C offers lithium ions easier accessibility during the cycling process, which leads to increased accommodation behavior for lithium as well as improve the kinetics of the SnO2/C anode46. These results demonstrate that the carbon layer not only enhances the electrochemical activity of the SnO2 NCs, but also improve cycling performance significantly34.

(a) CV profiles of half-cell SnO2/Li during the first three cycles at a rate of 0.5 mV/s; (b) Voltage profiles of the N2-annealed SnO2/C in half cells cycled between 0.01 and 2.0 V at a current density of 0.5 C; (c) Cycling performance showing the charge/discharge capacities of the half cell at a current density of 0.5 C for 100 cycles; (d) Electrode cycled at various current densities.
In order to better understand the cycling stability of the SnO2/C electrodes, the lithium driven microstructural evolution of the SnO2 electrode was studied by ex-situ TEM and SEM imaging (Figure 4 and Figure S12 in the SI), which was performed on the SnO2 electrodes after electrochemical cycling by stopping the cells at 2.0 V. Examinations of the electrode after 20 cycles indicated that the uniform size of the NCs was preserved and that they were still contained within the layered carbon. The average observed diameter of the NCs was around 3.0 nm and the lattice distance measured can be ascribed to the tetragonal SnO2 and α-Sn (Figure 4b), which demonstrates there are phase transitions during the lithiation/delithiation process as expected33,34. The EDS spectrum and mapping profiles obtained in connection with the TEM observation are presented in Figure S13 (see SI). The results reveal that other substances such as F, P and adventitious Si are present in the anode materials which originate from the electrolyte of LIBs (Table S4, SI). It is notable that the complete structural integrity of the interconnected SnO2/C active electrode materials was preserved. It is usually thought that the SnO2 nanoparticles form interconnected aggregates due to the repeatable volume expansion and shrinkage during charge/discharge42. However, we do not find any significant aggregation under TEM observation. The large layered substance is likely acetylene black and there are still amorphous carbon layers on the NCs after cycling as evidenced by EDS mapping (Figure S13). Therefore it is reasonable to infer that the carbon encapsulation of the SnO2 NCs functions as a physical matrix or buffer layer and strengthens the stability of the electrodes, which highlights the merits of the SnO2/C electrodes in enhancing the cycling performance. Moreover, the interconnectivity of these discrete SnO2 NCs through the carbon layers facilitates electron transfer as an electrode material and presence of inherent vacancies inside both the amorphous carbon and acetylene black sheaths are another plus for alleviating the problems resulting from the large volume change of SnO2, which gives their superior cycling performance. The smaller Ohmic contact resistance Rs (0.94 Ω) and charge-transfer resistance Rct (24.4 Ω) as compared to those of SnO2 NCs annealed in O2 (Rs = 2.46 Ω, Rct = 56.9 Ω) supports this conclusion. It was found that the Rs and Rct increase notably after 20 runs of charge/discharge in case of SnO2 nanoparticles annealed from O2 atmosphere while the resistance in the SnO2/C electrode does not show much change. The lower Rs and Rct values of the assembled SnO2/C electrode can be attributed to the homogeneous conductive carbon sheaths which show good electronic conductivity. The Nyquist plots and the corresponding fitted data of the dummy cells consisting of two identical electrodes made with SnO2/C and SnO2 nanoparticles are presented in the SI (Figure S14, Table S5).

TEM (a) and HRTEM images (b) of SnO2/C nanocrystal anodes by an ex-situ analysis after 20 cycles.
Discussion
Based on the analyses, we propose an O2-induced hydrolysis mechanism to rationalize the synthesis of colloidal SnO2 NCs in the hot-bubbling process. As identified by Peng and co-workers47, in a mixed alcohol/n-octadecene (ODE)/OLA system, the formation of In2O3 nanocrystals takes place via the pathways of hydrolysis and esterification, in which the alcohol is the activating reagent. We thus propose that the formation of SnO2 NCs proceeds by the following reactions and that O2 is an activator during the high-temperature hydrolysis.



The freshly prepared stannic hydroxide was obtained by hydrolysis of SnCl4·5H2O and we term the product of the hydrolysis as SnOx(OH)2(2−x)48. Stannic hydroxide can be dissolved by oleic acid (HOA) in ODE at elevated temperatures (Eq. 5). The photographic images of the phase transitions are shown in Figure S15 (SI). As demonstrated, the turbid suspension of stannic hydroxide in ODE/OA and/or OLA becomes clear at about 220°C. This provides evidence for the formation of the Sn-oleate complex (Sn(OA)2x(OH)2(2−x)) in the hot surfactant solution similar to that of Kovalenko's Sn-oleylamide complex49. The bubbled air (containing O2) works as an activator to deprotonate the Sn(OA)2x(OH)2(2−x) which in turn accelerates the hydrolysis reactions and promotes the burst of nucleation at elevated temperature (Eq. 6). Equation 7 denotes the hydrolysis when other carboxylic acids were used as anions for the tin precursor. The ligands, namely OA and OLA, are suspected to coordinate to the SnO2 NCs surface and thus can affect the oriented attachment during the growth4. The presence of large crystals was corroborated by TEM observation at prolonged growth time (Figure S11a, SI). This phenomenon has also been observed by Zhou and co-workers when they synthesized micron sized plate-shaped SnO2 crystals34. Under conditions where no air is bubbled through the solution it is possible to receive the hydrolysis products, however, the products are large-sized and irregularly shaped if the reaction time is more than 3.0 hrs (Figure S11b, SI). No particles are produced if the reaction time is less than 2.0 hrs. This suggests that the bubbled O2 is a good protonating agent to induce the hydrolysis of Sn-oleate complexes. The morphology of colloidal NCs is also dependent on the nature of solvents used. For example, when ODE was replaced completely by OLA and tin(II) acetylacetonate (Sn(acac)2) was used as the Sn precursor, only laminated SnO2 nanoparticles were obtained and flower-shaped SnO2 particles of micron size were produced after calcination. Figure S11c in the SI provides an SEM image of this sample and the corresponding EDS profile (Figure S11d, SI). These results clearly demonstrate that the polarity of the solvent and the solubility of reactants affect the intermediates in solvothermal synthesis, thus yielding different morphologies of the final products19,50.
In summary, for the first time ultrasmall SnO2 nanocrystals were successfully prepared via a facile hot-bubbling route. The colloidal SnO2 NCs were synthesized by an O2-induced hydrolysis pathway in the hot surfactant solution. The high-temperature in the synthetic environment provides a high degree of crystallinity in the final products and the inert atmosphere annealing affords the formation of carbonaceous layers hybridized with nitrogen dosage which can prevent aggregation during the synthesis and annealing processes. The N2-annealed SnO2 NCs exhibit outstanding reversible capabilities and cyclability for Li-ion storage, which are attributed to the ultrasmall size of the as-synthesized NCs, large specific areas of the particles and their special characteristics of thin layered carbon shells and N-doped structures resulting from carbonization reactions. Using these SnO2/C composite nanocrystals as starting materials, the synthesis of other 0D nanomaterials using similar synthetic routes will certainly aid to extend their potential applications.
Methods
Hot-bubbling synthesis of SnO2 nanocrystals
The reagents including tin(IV) chloride pentahydrate (SnCl4·5H2O, AR. grade), oleic acid (OA), oleylamine (OLA, 80–90% C18 content), n-octadecene (ODE, 90%), LiPF6 (battery grade), ethylene carbon (EC, anhydrous, 99%), dimethyl carbonate (DMC, anhydrous, ≥99%), N-methyl-2-pyrrolidone (NMP, anhydrous, 99.5%), toluene (≥99.5%), isopropanol (AR.) and methanol (anhydrous) were purchased from Sigma-Aldrich. All of the chemicals were used as received without further purification. In a typical experiment, the tin precursor stannic hydroxide (SnOx(OH)2(2−x)) was prepared by hydrolysis of SnCl4·5H2O (3.50 g, 0.01 mol) in the presence of NaOH (0.16 g, 0.04 mol) in 200 mL aqueous solution. The greyish hydrolyzed SnOx(OH)2(2−x) particles were collected by filtration and purified by rinsing with cold water. The freshly prepared SnOx(OH)2(2−x) (0.180 g, 1.0 mmol), OA (2.6 mL, 4.0 mmol), ligand (OLA, 1.0 mL) and solvent (ODE, 10 mL) were loaded into a three-neck flask connected to a Schlenk line. Any volatile material was removed by vacuum distillation at 100°C at 0.1 bar. Subsequently the green turbid solution was switched to N2 flow and the mixture heated from 100 to 220°C until a colorless solution was obtained, where the Sn-OA complex was prepared. The solution was then bubbled with air at elevated temperature (280–320°C) during which the clear solution turned brownish red and finally dark with increasing time. The resulting nanocrystals were harvested by precipitation employing toluene as the solvent and isopropanol/methanol (1.0, v/v) as nonsolvents. The products were purified several times to remove excess ligands by the same means (dissolving in toluene and nonsolvent precipitation). The final products (0.174 g) were dried at 60°C in vacuum before they were used further. During the hot-bubbling synthesis aliquots were taken from the flask and quenched in room temperature toluene and were used for optical characterization. For the synthesis of SnO2 NCs with other precursors such as Tin (II) 2-ethylhexanoate and di-n-octyltin oxide, the procedures are the same with the exception that there was no need to synthesize stannic hydroxide as a precursor.
Characterizations
SEM measurements were performed on a CrossBeam Workstation NVision40 field emission scanning electron microscope operated at 5 kV. TEM and HRTEM characterizations were conducted on a JEM-2100F field emission transmission electron microscope operated at 200 kV. EDX analysis was obtained with an EDAX system (Oxford 7788) linked with TEM instrument. Thermogravimetric (TG) analysis was carried out on a SDT Q600 instrument and measurements were done under an O2 or N2 flow of 60 mL/min with a temperature ramp of 3°C/min. XPS spectra were collected on an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Mg Kα radiation. XRD measurements were performed on a Rigaku D/max2500 diffractometer using the Cu Kα radiation.
Lithium-ion batteries (half cells) fabrication
Electrochemical experiments were performed using Swagelok-type cells. Similarly to the procedures of Yan et al.51, lithium-ion batteries were fabricated using a lithium foil as the negative electrode, a 1.0 M solution of LiPF6 in EC/DMC (1:1, v/v) as the electrolyte and a pellet made of the hybrid SnO2 NCs, acetylene black and NMP in a weight ratio of 7:2:1 as the positive electrode. Each electrode was 8 mm in diameter and separated by two pieces of Celgard 2400 membranes. The battery cell was assembled in an argon-filled glovebox with the H2O and O2 concentrations below 1.0 ppm. The electrochemical properties were studied with a multichannel battery-testing system (Arbin BT 2000). Galvanostatic charge-discharge cycling was performed at a constant current density of 0.5 C (0.392 A·g−1) over a voltage window of 0.01–2.0 V. The working electrode was measured at room temperature during the whole process. Electrochemical measurements were performed by using a potentiostat (IM6, Zahner elektrik) connected to a personal computer and controlled by using the Thales software package. The electrochemical impedance spectroscopy (EIS) tests were carried out in a frequency range from 50 mHz to 100 kHz to evaluate the electrochemical behaviors of the dummy cells made with two identical the electrodes derived from SnO2 or SnO2/C NCs. The parameters of the equivalent circuit were fitted by using Zview software.
References
Idota, Y., Kubota, T., Matsufuji, A., Maekawa, Y. & Miyasaka, T. Tin-based Amorphous Oxide: A High-Capacity Lithium-ion-storage Material. Science 276, 1395–1397 (1997).
Zhou, Z. Y., Tian, N., Li, J. T., Broadwell, I. & Sun, S. G. Nanomaterials of High Surface Energy with Exceptional Properties in Catalysis and Energy Storage. Chem. Soc. Rev. 40, 4167–4185 (2011).
Lu, M. L. et al. A Facile Integration of Zero- (I–III–VI Quantum Dots) and One-(Single SnO2 Nanowire) Dimensional Nanomaterials: Fabrication of A Nanocomposite Photodetector with Ultrahigh Gain and Wide Spectral Response. Nano Lett. 13, 1920–1927 (2013).
Xu, X., Zhuang, J. & Wang, X. SnO2 Quantum Dots and Quantum Wires: Controllable Synthesis, Self-assembled 2D Architectures and Gas-Sensing Properties. J. Am. Chem. Soc. 130, 12527–12535 (2008).
Huang, J. Y. et al. In Situ Observation of the Electrochemical Lithiation of a Single SnO2 Nanowire Electrode. Science 330, 1515–1520 (2010).
Herrera, M., Maestre, D., Cremades, A. & Piqueras, J. Growth and Characterization of Mn Doped SnO2 Nanowires, Nanobelts and Microplates. J. Phys. Chem. C 117, 8997–9003 (2013).
Demir-Cakan, R., Hu, Y. S., Antonietti, M., Maier, J. & Titirici, M. M. Facile One-Pot Synthesis of Mesoporous SnO2 Microspheres via Nanoparticles Assembly and Lithium Storage Properties. Chem. Mater. 20, 1227–1229 (2008).
Zhuang, Z. Y., Huang, F., Lin, Z. & Zhang, H. Z. Aggregation-Induced Fast Crystal Growth of SnO2 Nanocrystals. J. Am. Chem. Soc. 134, 16228–16234 (2012).
Wang, Z., Luan, D., Boey, F. Y. C. & Lou, X. W. Fast Formation of SnO2 Nanoboxes with Enhanced Lithium Storage Capability. J. Am. Chem. Soc. 133, 4738–4741 (2012).
Xu, C., Sun, J. & Gao, L. Direct Growth of Monodisperse SnO2 Nanorods on Graphene as High Capacity Anode Materials for Lithium Ion Batteries. J. Mater. Chem. 22, 975–979 (2012).
Haag, J. M., Pattanaik, G. & Durstock, M. F. Nanostructured 3D Electrode Architectures for High-Rate Li-Ion Batteries. Adv. Mater. 25, 3238–3243 (2013).
Deng, J. et al. Sandwich-Stacked SnO2/Cu Hybrid Nanosheets as Multichannel Anodes for Lithium Ion Batteries. Acs Nano 7, 6948–6954 (2013).
Ding, S., Chen, J. S. & Lou, X. W. One-Dimensional Hierarchical Structures Composed of Novel Metal Oxide Nanosheets on a Carbon Nanotube Backbone and Their Lithium-Storage Properties. Adv. Funct. Mater. 21, 4120–4125 (2011).
Kim, H. & Cho, J. Hard Templating Synthesis of Mesoporous and Nanowire SnO2 Lithium Battery Anode Materials. J. Mater. Chem. 18, 771–775 (2008).
Ding, S. et al. Formation of SnO2 Hollow Nanospheres inside Mesoporous Silica Nanoreactors. J. Am. Chem. Soc. 133, 21–23 (2010).
Lou, X. W., Wang, Y., Yuan, C., Lee, J. Y. & Archer, L. A. Template-free Synthesis of SnO2 Hollow Nanostructures with High Lithium Storage Capacity. Adv. Mater. 18, 2325–2329 (2006).
Lee, K. T., Jung, Y. S. & Oh, S. M. Synthesis of Tin-Encapsulated Spherical Hollow Carbon for Anode Material in Lithium Secondary Batteries. J. Am. Chem. Soc. 125, 5652–5653 (2003).
Liu, J., Li, W. & Manthiram, A. Dense Core-Shell Structured Sno2/C Composites as High Performance Anodes for Lithium Ion Batteries. Chem. Commun. 46, 1437–1439 (2010).
Cui, G. et al. An One-Step Approach towards Carbon-Encapsulated Hollow Tin Nanoparticles and Their Application in Lithium Batteries. Small 3, 2066–2069 (2007).
Na, J. S., Gong, B., Scarel, G. & Parsons, G. N. Surface Polarity Shielding and Hierarchical ZnO Nano-Architectures Produced Using Sequential Hydrothermal Crystal Synthesis and Thin Film Atomic Layer Deposition. Acs Nano 3, 3191–3199 (2009).
Lizandara-Pueyo, C. et al. Shape Anisotropy Influencing Functional Properties: Trigonal Prismatic ZnO Nanoparticles as an Example. Adv. Funct. Mater. 21, 295–304 (2011).
Weller, H., Fojtik, A. & Henglein, A. Photochemistry of semiconductor colloids: properties of extremely small particles of cadmium phosphide (Cd3P2) and zinc phosphide (Zn3P2). Chem. Phys. Lett. 117, 485–488 (1985).
Miao, S. D. et al. Synthesis of Monodisperse Cadmium Phosphide Nanoparticles Using ex-Situ Produced Phosphine. Acs Nano 6, 7059–7065 (2012).
Li, L., Protière, M. & Reiss, P. Economic synthesis of high quality InP nanocrystals using calcium phosphide as the phosphorus precursor. Chem. Mater. 20, 2621–2623 (2008).
Fojtik, A., Weller, H. & Henglein, A. Photochemistry of Semiconductor Colloids. Size Quantification Effects in Q-Cadmium Arsenide. Chem. Phys. Lett. 120, 552–554 (1985).
Wang, H. et al. Hydrothermal Synthesis of Hierarchical SnO2 Microspheres for Gas Sensing and Lithium-ion Batteries Applications: Fluoride-Mediated Formation of Solid and Hollow Structures. J. Mater. Chem. 22, 2140–2148 (2012).
Liu, R. et al. J. Sodium Chloride Template Synthesis of Cubic Tin Dioxide Hollow Particles for Lithium Ion Battery Applications. Acs Appl. Mater. Interfaces 4, 1537–1542 (2012).
Azam, A., Ahmed, A. S., Habib, S. S. & Naqvi, A. H. Effect of Mn doping on the structural and optical properties of SnO2 nanoparticles. J. Alloys Compd. 523, 83–87 (2012).
Yin, Y. D. et al. Formation of hollow nanocrystals through the nanoscale Kirkendall Effect. Science 304, 711–714 (2004).
Yang, H. X., Qian, J. F., Chen, Z. X., Ai, X. P. & Cao, Y. L. Multilayered nanocrystalline SnO2 hollow microspheres synthesized by chemically induced self-assembly in the hydrothermal environment. J. Phys. Chem. C 111, 14067–14071 (2007).
Lou, X. W., Li, C. M. & Archer, L. A. Designed Synthesis of Coaxial SnO2@carbon Hollow Nanospheres for Highly Reversible Lithium Storage. Adv. Mater. 21, 2536–2539 (2009).
Kong, J. et al. One-dimensional Carbon-SnO2 and SnO2 Nanostructures via Single-Spinneret Electrospinning: Tunable Morphology and the Underlying Mechanism. J. Mater. Chem. 21, 15928–15934 (2011).
Lou, X. W., Li, C. M. & Archer, L. A. Designed Synthesis of Coaxial SnO2@carbon Hollow Nanospheres for Highly Reversible Lithium Storage. Adv. Mater. 21, 2536–2539 (2009).
Zhou, X., Wan, L. J. & Guo, Y. G. Binding SnO2 Nanocrystals in Nitrogen-Doped Graphene Sheets as Anode Materials for Lithium-Ion Batteries. Adv. Mater. 25, 2152–2157 (2013).
Bhattacharyya, S., Hong, J. & Turban, G. Determination of the Structure of Amorphous Nitrogenated Carbon Films by Combined Raman and X-Ray Photoemission Spectroscopy. J Appl. Phys. 83, 3917–3919 (1998).
Gross, T., Lippitz, A., Unger, W. & Guttler, B. Uncertainty in measurement of overlayer thickness of thermally oxidized silicon using X-ray photoelectron spectroscopy. Surf. Interface. Anal. 29, 891–894 (2000).
Lou, X. W., Chen, J. S., Chen, P. & Archer, L. A. One-Pot Synthesis of Carbon-Coated SnO2 Nanocolloids with Improved Reversible Lithium Storage Properties. Chem. Mater. 21, 2868–2874 (2009).
Wang, C. M. et al. In Situ Transmission Electron Microscopy Observation of Microstructure and Phase Evolution in a SnO2 Nanowire during Lithium Intercalation. Nano Lett. 11, 1874–1880 (2011).
Liu, N., Huo, K., McDowell, M. T., Zhao, J. & Cui, Y. Rice Husks as A Sustainable Source of Nanostructured Silicon for High Performance Li-Ion Battery Anodes. Sci. Rep. 3, 1–7 (2013).
Kim, J. C. et al. Superior Long-Term Cycling Stability of SnO2 Nanoparticle/Multiwalled Carbon Nanotube Heterostructured Electrodes for Li-Ion Rechargeable Batteries. Nanotechnology 23, 465402–465402 (2013).
Han, F., Li, W. C., Li, M. R. & Lu, A. H. Fabrication of superior-performance SnO2@C composites for lithium-ion anodes using tubular mesoporous carbon with thin carbon walls and high pore volume. J. Mater. Chem. 22, 9645–9651 (2012).
Li, J., Zhao, Y., Wang, N. & Guan, L. A high performance carrier for SnO2 nanoparticles used in lithium ion battery. Chem. Commun. 47, 5238–5240 (2011).
Sun, X., Liu, J. & Li, Y. Oxides@C Core–Shell Nanostructures: One-Pot Synthesis, Rational Conversion and Li Storage Property. Chem. Mater. 18, 3486–3494 (2006).
Yu, Y., Chen, C. H. & Shi, Y. A Tin-Based Amorphous Oxide Composite with a Porous, Spherical, Multideck-Cage Morphology as a Highly Reversible Anode Material for Lithium-Ion Batteries. Adv. Mater. 19, 993–997 (2007).
Wen, Z., Wang, Q., Zhang, Q. & Li, J. In Situ Growth of Mesoporous SnO2 on Multiwalled Carbon Nanotubes: A Novel Composite with Porous-Tube Structure as Anode for Lithium Batteries. Adv. Funct. Mater. 17, 2772–2778 (2007).
Wu, Z. S. et al. Three-Dimensional Graphene-Based Macro- and Mesoporous Frameworks for High-Performance Electrochemical Capacitive Energy Storage. J. Am. Chem. Soc. 134, 19532–19535 (2012).
Narayanaswamy, A., Xu, H., Pradhan, N., Kim, M. & Peng, X. Formation of Nearly Monodisperse In2O3 Nanodots and Oriented-attached Nanoflowers: Hydrolysis and Alcoholysis vs. Pyrolysis. J. Am. Chem. Soc. 128, 10310–10319 (2006).
Mal'chenko, S. N., Ivanovskaya, M. I., Orlik, D. R., Laykhov, A. S. & Grigorenko, V. I. Growth of SnO2 Crystals In Stabilized Sols of Hydrated Tin Dioxide. Inorg. Mater. 28, 81–84 (1992).
Kravchyk, K. et al. Monodisperse and Inorganically Capped Sn and Sn/SnO2 Nanocrystals for High-Performance Li-Ion Battery Anodes. J. Am. Chem. Soc. 135, 4199–4202 (2013).
Si, R., Zhang, Y. W., Zhou, H. P., Sun, L. D. & Yan, C. H. Controlled-Synthesis, Self-Assembly Behavior and Surface-Dependent Optical Properties of High-Quality Rare-Earth Oxide Nanocrystals. Chem. Mater. 19, 18–27 (2006).
Deng, J. et al. Naturally Rolled-Up C/Si/C Trilayer Nanomembranes as Stable Anodes for Lithium-Ion Batteries with Remarkable Cycling Performance. Angew. Chem. Int. Ed. 52, 2326–2330 (2013).
Acknowledgements
This project was financed by the National Natural Science Foundation of China (21103039), Anhui Province Natural Funds for Distinguished Young Scientists, Research Fund for the Doctoral Program of Higher Education of China (20110111120008), Beijing National Laboratory for Molecular Sciences (BNLMS) and Deutsche Forschungsgemeinschaft Grant (DFG): H1113/3-5. C.Y. acknowledges the support from the “Thousand Talents Program” and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
S.M., C.Y. and J.Y. conceived and coordinated the research. L.D., M.R.J., S.L. and S.H. contributed to the synthesis, structural and electrochemical characterization of the nanocrystals S.G.H., contributed to the theoretical calculation. L.H. and S.G.H. contributed to the fuel cell tests. The manuscript was primarily written by S.M. and revised by C.Y. All authors (including A.E. and O.G.S.) contributed to discussions and manuscript review.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supporting Information-R2
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Ding, L., He, S., Miao, S. et al. Ultrasmall SnO2 Nanocrystals: Hot-bubbling Synthesis, Encapsulation in Carbon Layers and Applications in High Capacity Li-Ion Storage. Sci Rep 4, 4647 (2014). https://doi.org/10.1038/srep04647
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04647
This article is cited by
-
Porous SnO2 nanoparticles based ion chromatographic determination of non-fluorescent antibiotic (chloramphenicol) in complex samples
Scientific Reports (2018)
-
High rate capability and cyclic stability of hierarchically porous Tin oxide (IV)–carbon nanofibers as anode in lithium ion batteries
Applied Nanoscience (2017)
-
High Efficiency Dye-sensitized Solar Cells Constructed with Composites of TiO2 and the Hot-bubbling Synthesized Ultra-Small SnO2 Nanocrystals
Scientific Reports (2016)
-
Octahedral Tin Dioxide Nanocrystals Anchored on Vertically Aligned Carbon Aerogels as High Capacity Anode Materials for Lithium-Ion Batteries
Scientific Reports (2016)
-
Confined SnO2 quantum-dot clusters in graphene sheets as high-performance anodes for lithium-ion batteries
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
