
Abstract
A new deep acceptor state is identified by density functional theory calculations and physically activated by an Au ion implantation technique to overcome the high energy barriers. And an acceptor-compensated charge transport mechanism that controls the chemical sensing performance of Au-implanted SnO2 nanowires is established. Subsequently, an equation of electrical resistance is set up as a function of the thermal vibrations, structural defects (Au implantation), surface chemistry (1 ppm NO2) and solute concentration. We show that the electrical resistivity is affected predominantly not by the thermal vibrations, structural defects, or solid solution, but the surface chemistry, which is the source of the improved chemical sensing. The response and recovery time of chemical sensing is respectively interpreted from the transport behaviors of major and minor semiconductor carriers. This acceptor-compensated charge transport mechanism provides novel insights not only for sensor development but also for research in charge and chemical dynamics of nano-semiconductors.
Similar content being viewed by others


Introduction
Fabrication of p-type bulk SnO2 is proven difficult due to the high formation energy of tin vacancies which are the only possible acceptor state in pure SnO21. Recent developments in controlled synthesis and fabrication of metal-oxide-based nanomaterials such as nanowires2, nanofibers3 and nanorods4 have, however, opened up new possibility to activate deep acceptor levels in nanocrystal SnO2, which influence the surface charge transport process and may further enhance surface chemical reactions that are critical to gas sensing5.
Ion implantation involves the incorporation of energetic ions in the nanowires, where the depth of penetration of the ions can be controlled easily. Although ion implantation is widely reported for improved sensing performances6,7, there has been no investigation about acceptor modified charge transport and the corresponding chemical reaction in metal-implanted nanowires.
In this work, an ion implantation technique is applied to activate a deep acceptor level in the electronic structure of SnO2. On the basis of experiment and modeling approaches, a charge transport mechanism and a predominant factor controlling the chemical sensing are established. The electron-carrier concentrations in the Au-implanted SnO2 nanowires are estimated and a relationship between the carrier concentrations and electron scattering (due to structural defects and surface chemistry) is obtained. Furthermore, we speculate that the experimental sensor responses and recovery times are influenced by the electron–phonon coupling for the embedded electron gas from the Au atoms. This model shows that the surface chemistry is the predominant factor affecting the electrical resistivity and is thus the source of the improved chemical sensing.
Results
DFT calculations
To locate the acceptor levels in Au-implanted SnO2, its electronic properties were calculated by DFT methods. SnO2 has a rutile crystal structure in the space group P42/mnm (No. 136). In this work, we have used a 2 × 2 × 3 unit cell of SnO2 with one Sn atom substituted with Au giving a stoichiometry of AuSn23O48 as shown in Fig. 1a.

DFT analysis.
(a) Crystal structure of Au-SnO2. (b) Density of states (DOS) of pristine SnO2 and Au-implanted SnO2. (c) Calculated formation energy for substitution Au impurity in SnO2 under O-rich conditions. The dotted line indicates the case of interstitial Au in SnO2.
The formation energy of an impurity Au in a charge state q occupying Sn site is calculated by8

where E[Au − SnO2] and E[SnO2] are the total energies of doped and perfect SnO2, respectively, μAu and μSn are the chemical potentials of Au and Sn, respectively. The last term describes the dependence on the Fermi level (EF) measured from the valence band edge. EVBM is a position of the valence band maximum (VBM) in the perfect system and ΔV is a correction term aligning the VBM in the doped and perfect crystal supercells. Chemical potential  where
where  is calculated for the bulk phases of Au and Sn, while Δμi may vary corresponding to certain growth conditions (e.g. Sn-rich/O-poor or Sn-poor/O-rich). Since Perdew-Burke-Ernzerhof (PBE) calculations typically underestimate band gaps, we use an extrapolation correction in order to compare transition levels and formation energies with experimental band gaps.
is calculated for the bulk phases of Au and Sn, while Δμi may vary corresponding to certain growth conditions (e.g. Sn-rich/O-poor or Sn-poor/O-rich). Since Perdew-Burke-Ernzerhof (PBE) calculations typically underestimate band gaps, we use an extrapolation correction in order to compare transition levels and formation energies with experimental band gaps.
The formation energy of neutral Au impurity under Sn-poor/O-rich conditions (which should be more suitable for doping at cation site in SnO2) is found to be about 1 eV. The calculated value is quite large due to the larger atomic size in Au than in Sn. The positive sign of Au formation energy suggests that spontaneous incorporation of Au in SnO2 is difficult to achieve under normal growth conditions. Therefore, high-energy implantation is required to activate the deep acceptors in Au-implanted SnO2 samples.
Density of states (DOS) analysis shows that pure defect-free SnO2 is a semiconductor with a band gap of 1.26 eV, being in reasonable agreement with existing theoretical studies on the same level of theory9,10,11. Meanwhile, Au substitution causes a shift of the Fermi level in the direction of the valence band indicating a charge transfer from SnO2 to Au atom. When Au substitutes Sn atoms, it produces extra electronic states inside the SnO2 band gap as shown in Fig. 1b. The impurity band contains a large contribution of Au 5d-orbitals and is spin-polarized. In addition, we observe hybridization between Au 5d and O 2p levels in the Fermi level region.
Figure 1(c) presents the formation energies of Au dopants in SnO2 as a function of the Fermi level position. The range of the Fermi energy is given by the calculated valence-band maximum (VBM, 0 eV) and conduction-band minimum (CBM, 1.26 eV). For each dopant, only the charge states that are most energetically favorable at a given Fermi energy are shown. We find that Au impurities prefer the substitutional site. The interstitial site is not favorable at any conditions due to the large atomic size of Au. The 0/−1 ionization level for Au impurity is 1.02 eV (Fig. 1c), indicating that Au is a deep acceptor. Therefore, Au doping in SnO2 may serve to “kill” the electrons in the system and shift the Fermi level towards the valence band side. This is schematically described in Fig. 2. These findings explain a decrease in concentration of electron carriers in Au-implanted SnO2 (to ~105 from ~106 cm−3) which will be described in later sessions.

Schematics of Au implantation.
Schematics describing the penetration of Au into the SnO2 nanowire, acting as an electron-killer layer.
Microstructure of Au-implanted SnO2 nanowires
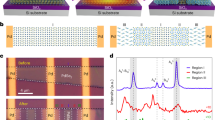
Figure 3 shows the microstructure of the networked SnO2 nanowires used for the sensor platform. Fig. 3a schematically shows the procedure used to grow the networked SnO2 nanowires, which is described in detail in Methods part. The plan-view FE-SEM image in Fig. 3b clearly demonstrates the entanglement of nanowires grown selectively on the adjacent electrode pads, leading to an immense number of nanowires involved in the gas-sensing process and thus, resulting in the fabrication of highly reproducible and reliable gas sensors. The inset figure shows the networking nature more clearly. Figure 3c shows a cross-section image showing the entanglement more clearly. Figure 3d shows a representative TEM image taken from an individual nanowire, revealing that it is single-crystalline. The inset figure is a selected-area electron diffraction pattern, again demonstrating its high crystalline quality. The shape and geometrical dimensions of the networked SnO2 nanowires for superior sensing performances are discussed in detail in our previous report12.

Microstructure of Au-implanted SnO2 nanowires(a) Schematic illustration of selectively-grown, networked SnO2 nanowires on a patterned electrode. (b) Plane and (c) Cross-section field-emission scanning electron micrographs of networked SnO2 nanowires. (d) Transmission electron micrograph of single SnO2 nanowire. The inset shows a selected-area electron-diffraction pattern.
Au ions were implanted into these networked SnO2 nanowires with different dose levels (1013 or 1014/cm2). First, we investigated whether the implantation process influenced the surface morphology of the nanowires. Supplementary Fig. S1 displays typical FE-SEM images of Au-implanted networked SnO2 nanowires. The insets are the corresponding high-magnification images showing the surface morphology more obviously. For comparison, an image showing the microstructure of pristine SnO2 nanowires (without Au implantation) is included in Supplementary Fig. S1a. As shown in Supplementary Fig. S1b, the surface of each SnO2 nanowire implanted with a dose of 1013/cm2 Au+ has a smooth surface similar to that of the pristine SnO2 nanowires shown in Supplementary Fig. S1a. SnO2 nanowires implanted with a dose of 1014/cm2 Au+ (Supplementary Fig. S1c) also show no distinctive difference in surface morphology from the pristine ones.
TEM observations were conducted for further investigation of the effect of Au implantation on the microstructure; the results are shown in Supplementary Fig. S2 (the insets are the corresponding low-magnification TEM images). As shown in Supplementary Fig. S2, there is no significant difference between the implanted samples and the pristine one. In addition, all the samples show clear lattice fringes, indicating that the subsequent thermal annealing completely eliminated any crystalline disorder in the nanowires that might have been incorporated during Au implantation. From these TEM results together with the FE-SEM results, we conclude that the Au implantation caused no detectable structural change in the pristine SnO2 nanowires.
XPS measurements were made to confirm the existence of Au-related species in the nanowires. Supplementary Fig. S3a shows the XPS spectrum taken from the networked SnO2 nanowires implanted with 1014/cm2 Au+. No peaks originating from Au-related species are observed. The spectrum from the pristine SnO2 nanowire sample showed the same feature (Supplementary Fig. S4). According to the Monte Carlo simulation code SRIM, when 1014/cm2 Au+ is implanted at an accelerating voltage of 30 kV into the SnO2 lattice, the atomic concentration of Au in the implanted region is about 0.36 at% and the penetration (implanted) depth is about 10 nm, which is deeper than the detection depth of XPS, meaning that most of the implanted Au ions are out of range for XPS measurements. Therefore, it is not so easy to detect the implanted Au in SnO2 by XPS. Hence, we performed EDS analysis to confirm the implantation of Au ions in the networked SnO2 nanowires using the EDS analyzer installed in the FE-SEM equipment. The EDS spectrum for SnO2 nanowires implanted with a dose of 1014/cm2 Au+ is shown in Supplementary Fig. S3b. The EDS analysis shows that the nanowires were composed of Sn, O and Au. The peak at around 2.1 keV, which corresponds to the energy level of gold, is observed clearly, indicating the presence of Au+ in the SnO2 nanowires. In order to further identify the presence of Au in the SnO2 nanowires, EDS line scans for elemental analysis were performed using the EDS analyzer installed in the TEM equipment. The EDS line spectra are shown in Figure 4, obviously revealing the presence of Au in the SnO2 nanowire.

Elemental analysis of Au-implanted SnO2 nanowires.
EDS line scans for (a) an Au-implanted SnO2 nanowire with a dose of 1014/cm2. (b, c, d) Line spectra of Sn, O and Au, respectively.
Sensing behavior of Au-implanted SnO2 nanowires
The sensing performances of the implanted nanowire sensors were investigated in terms of NO2 detection. Figures 5a, 5b and 5c shows resistance curves of the pristine SnO2 nanowires and the Au-implanted SnO2 nanowires with doses of 1013 and 1014/cm2, respectively, for various concentrations of NO2 at 300°C. For the pristine SnO2 nanowires, the resistance increases sharply to a high value upon exposure to NO2 gas and drops quickly to a low value upon removal of the NO2 gas, exhibiting a response of 18 for 1 ppm NO2. This resistance change is well known in n-type semiconductor gas sensors; NO2 molecules adsorbed on the surface of the SnO2 nanowires are likely to extract electrons from them, eventually leading to the surface depletion of each SnO2 nanowire. On the other hand, release of electrons occurs in conjunction with the desorption of NO2 molecules. This charge transfer accounts for the resistance change observed in the sensors.

Sensing performance of Au-implanted SnO2 nanowires.
Dynamic resistance curves of (a) pristine SnO2 nanowires and (b,c) Au-implanted SnO2 nanowires with doses of (b) 1013 and (c) 1014/cm2 for various NO2concentrations. (d) NO2 responses and (e) response and recovery times of pristine SnO2 nanowires and Au-implanted SnO2 nanowires with doses of 1013 and 1014/cm2.
All the NO2 sensing performances are summarized in Fig. 5d and 5e. It is evident that the Au implantation caused a significant change in the sensing performance. In the case of the Au-implanted SnO2 nanowires with a dose of 1013/cm2, the fabricated sensor showed a significant enhancement in sensing capability; the gas response, response time and recovery time changed from 18, 120 s and 241 s (pristine SnO2 nanowires) to 90, 60 s and 17 s, respectively, for 1 ppm NO2. Even at the extremely low NO2 concentration of 50 ppb, the Au-implanted SnO2 nanowires showed a clear response curve. In sharp contrast, the pristine SnO2 nanowires showed no meaningful response to NO2 for concentrations below 1 ppm. In the case of the Au-implanted SnO2 nanowires with a dose of 1014/cm2, the NO2 sensing capabilities were enhanced further; the gas response, response time and recovery time were 175, 48 s and 11 s, respectively, for 1 ppm NO2.
The responses of the Au-implanted SnO2 nanowires were significantly higher than that of the pristine SnO2 nanowires, suggesting that the implantation of Au ions in SnO2 nanowires is a very efficient way to enhance the NO2 sensing response. In addition, both the response and recovery times were shortened significantly upon Au implantation. These remarkable improvements are attributed to the amplification effects from both structural and surface chemical modifications, as shown below. The present results indicate that the synthesis of bimetallic catalysts would be a promising strategy for enhancing the sensing performances of nanowire gas sensors.
Electrical transport properties in Au-implanted SnO2 nanowires
In general, there are four components that contribute to the measured electrical resistivity:

in which ρ(i) (i = T, d, c) are the thermal, defect and surface chemical catalysis contributions, respectively. The last term is the solid solution contribution (by Nordheim's rule)13, C is a constant and X is the solute concentration (Au in this study). It is noteworthy that both the defect and surface chemical catalysis contribution terms are independent of the temperature. Besides the solid solution contribution, Au layers may also contribute to ρ(c) even though they are about 10 nm below the surface of the nanowires. However, as described later, we do not have to single out such a contribution in this work, since our model (without this contribution) reproduces well our experiments. The ρ(T) value is assigned as 2.63 ohm/cm (see details below), which is the value for pristine SnO2 nanowires. Therefore, ρ(d) takes pristine SnO2 as its reference state.
In order to study the electron transport process and calculate carrier concentrations, we must deduct the solid solution effect from the measured electrical resistivity data. Our measured resistivity of pristine SnO2 (n-type) can be reproduced reasonably from the semiconductor theory; specific resistivity = 1/(qμen), in which q, μe and n are the elementary charge, mobility and concentration of electrons, respectively. By taking the values of q = 1.6 × 10−19 C, μe = 72 cm2/(V·s)14 and n = 3.3 × 1016 cm−315, we obtain a specific resistivity of 2.63 ohm/cm. Using our measured resistivity, the equivalent length of nanowires is calculated to be 38023 cm, which will be applied to convert the measured resistance data to the specific resistivity, as shown below. By using equation (2) and assuming ρ2(d,c) = A × ρ3(d,c), where ρi(d,c) = ρi(d) + ρi(c) and A is a constant (A = 0.5), we can derive the values of C = 8219, ρ2(d,c) = 10.19 and ρ3(d,c) = 20.38. Furthermore, the electron carrier concentrations in SnO2, ni, are 3.3 × 106, 5.2 × 105 and 2.9 × 105 cm−3 for pristine SnO2 and SnO2 with 1013 and 1014/cm2 dosages, respectively. If we select A = 0.33, the above carrier concentrations are 3.3 × 106, 5.7 × 105 and 1.9 × 105 cm−3, respectively. Therefore, a different estimation of the constant A may not change significantly the magnitudes of the carrier concentrations. In this study A = 0.5 is applied, which will enable discussions on the basis of the carrier transport properties. For the partitioning of the measured overall resistivity into the four terms in equation (2), the following calculation procedures are used:
-
1
ρ(ohm/cm) at NO2 = 0 is 2.63 ( = 100000/38023), 15.78 ( = 600000/38023) and 52.60 ( = 2000000/38023), respectively, for the three dosages.
-
2
CX(1 − X) is 0 ( = 8219 × 0 × (1 − 0)), 2.96 ( = 8219 × 0.00036 × (1 − 0.00036)) and 29.55 ( = 8219 × 0.0036 × (1 − 0.0036)), respectively, for the three dosages.
-
3
ρ − CX(1 − X) at NO2 = 0 is 2.63, 12.82 ( = 15.78 − 2.96) and 23.05 ( = 52.60 − 29.55), respectively, for the three dosages.
-
4
ρ(T) = 2.63, which is the resistivity of nanowires without exposure to chemical and ionic irradiation.
-
5
At NO2 = 0, ρ(c) = 0, then ρ(d) = ρ − ρ(T) − CX(1 − X) is calculated using the data from steps 2 and 3 above. Through regression of the three sets of data, the following equation is derived: log ρ(d) = 7.533 − 1.142 × log(n), where n is the carrier concentration.
-
6
Repeating the above steps from 1 to 4 for measurements at NO2 = 1 ppm, the following equation is derived: logρ(c) = 13.890 − 1.872 × log (n)
Therefore, both ρ(d) and ρ(c) can be predicted from the carrier concentrations by using the following equations;


Equations (3) and (4) provide the relationship between the carrier concentration and resistivity caused by scattering from defects and the surface chemistry, respectively. Then, the overall resistivity of the SnO2 nanowires is described as

Figure 6 shows the relative contributions of each term (in log form). The surface chemistry term is the clear winner over the whole range of carrier concentrations and is the predominant control mechanism of the resistivity. The second place is occupied by the solid solution, defect and thermal scattering contributions in the ranges of 105 to 4 × 105, 4 × 105 to 1.7 × 106 and >1.7 × 106 cm−3, respectively. The revealed interplay between the carrier concentration, solid solution, defects and thermal and surface chemistry, will aid in further study on the coupling of electronic and ionic conduction in semiconductors and theoretical investigation of electron–phonon coupling.

Electrical resistivity contributions.
Changes in relative resistivity contributions (logρ(i) (i = T, d, c, or solid solution) with carrier concentrations.
Discussion
This research may also provide insights into the sensing dynamics. As shown in Fig. 5, the sensor response time decreases from ≈120 (pristine) to ≈60 s (1013/cm2 dose) for 1 ppm NO2, which may be speculated from the relative strengths of the thermal and solid solution factors shown in Fig. 6, which crossover at the 1013/cm2 dose. The response time records the speed of the physical event that leads to the increase of resistivity upon exposure to NO2 gas, which is believed to be the reduced oxygen vacancies at increasing oxygen partial pressures. SnO2 is an n-type semiconductor with tin vacancy as the only possible acceptor but of high formation energy1, therefore, electron (from oxygen vacancy) and hole (from tin vacancy) are respectively the major and minor carrier. Our earlier DFT calculation identified new holes (deep acceptors by Au-implantation) which do not yet contribute to the electrical conductivity but compensate the electron charge of oxygen vacancies. Therefore the measured response time may indicate the speed of the hole compensation process which is about 120 s associated with tin vacancies in pristine, 60 s and 48 s with Au-implanted acceptor respectively under 1013/cm2 and 1014/cm2 dosages. The reduction in response time of Au-implanted samples results from the activation of “shallower” holes by Au implantation than these by tin vacancies. The response time reduced a little further to 48 s in the case of 1014/cm2 dose where the new acceptor state may move closer to the valence band under the higher Au implantation dosage. In addition, the extra electrons (by Au implantation) at the region about 10 nm below the SnO2 surface via either interstitial or work function mechanisms have to couple with the surrounding lattice represented by the thermal scattering factor (2.63 ohm/cm). It seems that the crossover shown in Fig. 6 indicates the effective coupling of electrons and phonons, which also contributes to a short response time.
Similarly the recovery time records the speed of the physical event that removes the electron loss upon switching off the NO2 gas. The recovery time is dependent on the mobility of electrons (major carrier) which is higher than that of holes (minor carriers). Therefore, it is not surprised that the recovery time is about 4 times shorter than the corresponding response time under Au implantation (17 to 60 s and 11 to 48 s), considering the fact that the electron mobility is about 5 times higher than the hole mobility in SnO216,17. This trend does not hold for the pristine SnO2 somehow, which may involve more complicated mechanism that results in a strong dependence between the NO2 gas concentration and recovery time (Fig. 5e), unlike the other two Au-implanted cases. In general, the gas response and recovery times are closely associated with the exact surface catalysis involving electron and hole transport, which can be established by DFT calculations18,19,20. In the future work, detailed study of the surface catalysis under the influence of deep acceptors and the electron gas (produced by Au implantation) will be made through DFT calculations. In addition, the effect of hindering the adsorption of both atmospheric oxygen and NO2 by limiting the available free charge carriers on sensing performances needs to be investigated to further clarify the acceptor role of Au in SnO2.
Recently, it was revealed that the role of Au as an additive in SnO2 is mainly chemical and can be described in terms of enhanced reactivity to oxygen21, being in contrast to the effect of Pd and Pt promoters which were found to be distributed at an atomic level on the surface and in the bulk of the supporting sensing material and therefore have a tremendous effect on its bulk and surface electronic properties22. According to the results presented in the reference21, Au is present in the form of small metallic particles at the surface of the host metal oxide, leading to the “spill-over effect”, meaning that the Au particles enrich the surface of the active metal oxide with oxygen species. However, the Au-implanted SnO2 nanowires in this work showed no evidence of forming metallic particles either at the surface or in the interior of SnO2 nanowires. This is confirmed by the TEM observations. Figure 1d and Figure S5 show the detailed microstructures of an Au-implanted SnO2 nanowire with a dose of 1014/cm2, confirming no evidence of Au metallic particles or clusters. Usually in the implantation process, when the amount of an implanted element is very small or the post heat-treatment is not enough, it is more likely that they exist in the form of point defects such as interstial or substitutional impurities in the host material23,24. On the basis of the TEM observations, the general characteristic of the implantation process23,24 and the first principle calculations for the formation energies of Au dopants in SnO2 revealing that Au prefers the substitutional site (seeFigure 1), it is reasonable to claim that Au can play the role of a deep acceptor particularly in the Au-implanted SnO2 nanowires.
Another point that should be noted is the effect of the back to back Schottky barriers formed at the nanowires contacts. According to a literature survey25,26, the Schottky barriers can be one of the key sources of resistance change because the barrier height will be modulated by adsorption and desorption of gas molecules, consequently intensifying the resistance change. However, in our networked SnO2 nanowires, the contact is minimal. That is, one or two contacts are produced while making a linkage from one electrode to the other one12. In this case, the dominant source of resistance change is likely to come from the radial modulation of the electron-depleted layer in the surface region of nanowires, which was elucidated in our previous reports27,28. A systematic study based on the impedance spectroscopy may reveal more clearly the contributions from the Schottky barrier and the radial modulation of the electron-depleted layer, which needs to be performed in the future.
In summary, the NO2 sensing performances of SnO2 nanowires were improved greatly by Au implantation. The response of pristine SnO2 nanowires for 1 ppm NO2 was improved about ten folds upon Au implantation with a dose of 1014/cm2, while a clear response curve was shown even at 50 ppb NO2. In addition, the response and recovery times were shortened significantly by the Au implantation. The surface chemistry was found to be the predominant control mechanism of NO2-gas sensors based on the Au-implanted SnO2 nanowires. An equation of electrical resistance has been established as a function of thermal vibration, structural defects (Au implantation), surface chemistry (1 ppm NO2) and solute concentration. The electron carrier concentrations in the Au-implanted SnO2 nanowires were calculated and used to predict the strength of the electron scattering of structural defects and surface adsorbents. The sensor performance, in terms of response and recovery times, was discussed in relation to the acceptor compensation and the electron–phonon coupling mechanism. We further investigated the feasibility of using sensor response and recovery time to determine fundamental physics governing the electron/hole transport in semiconductors. This study provides novel insights into the fundamental carrier dynamics in semiconductors as well as aids the development of next-generation chemical sensors.
Methods
DFT calculation
Our spin-polarized calculations were performed using Quantum Espresso package. The exchange-correlation potential was approximated by generalized gradient approximation (GGA) using Perdew-Burke-Ernzerhof (PBE) functional. A Monkhorst-Pack mesh with (3 × 3 × 3) k-point sampling was used for geometry optimizations, while a denser k-point grid of (8 × 8 × 8) was employed in the case of density of states (DOS) calculations. The kinetic-energy cutoffs for valence electron wave functions and charge density were set as 37 Ry and 370 Ry, respectively. The optimized structures were obtained by relaxing all atomic positions using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) quasi-Newton algorithm until the interatomic forces are less than 0.01 eV Å−1.
Fabrication of nanowire sensors
For the sensor platform, SnO2 nanowires were grown selectively through the vapor–liquid–solid (VLS) method on an SiO2-grown Si(100) substrate, on which patterned interdigital electrodes (PIEs) had been formed by using a conventional photolithographic process. The electrode was a layer consisting of Au(3 nm)/Pt (200 nm)/Ti (50 nm), deposited in sequence through a sputtering method. The Si substrate was introduced into a horizontal quartz tube furnace in which an alumina crucible containing Sn powders (Aldrich, 99.9%) was placed. The furnace was then evacuated with a pump down to a pressure of 8 × 10−2 torr and heated to 900°C for 5 min. During the VLS growth of the SnO2 nanowires, N2 and O2 were flowed through the tube at rates of 300 and 10 standard cubic centimeters per minute, respectively. The experimental conditions used to synthesize the SnO2 nanowires and the sensor design are described in detail in our previous reports12,28.
Ion implantation
The Au implantation in SnO2 nanowires was performed with a conventional ion implanter (Nissin Electric Co., Ltd., Japan). Au+ ions with an energy of 30 keV were implanted into the SnO2 nanowires under a pressure of 1 × 10−7 torr with an ion current density of 10 nA/cm2, without any intentional heating. After the Au implantation, the samples were annealed at 500°C for 1 h in air to repair the structural damage that might be caused by the implantation process.
Materials characterization
The microstructure of the pristine and Au-implanted SnO2 nanowires was examined through field-emission scanning electron microscopy (FE-SEM, Hitachi, S-4200) and transmission electron microscopy (TEM, Philips CM-200). The bulk and surface chemistries were characterized through energy-dispersive X-ray spectroscopy (EDS, Oxford INCA energy) installed in the FE-SEM equipment and X-ray photoelectron spectroscopy (XPS, Thermo K-Alpha), respectively. For further elemental analysis, EDS line scans were performed using the EDS analyzer (EDS, Oxford INCA energy) installed in the TEM equipment.
Sensing measurement
The sensing performances of the sensors fabricated from the Au-implanted SnO2 nanowires were investigated for a representative oxidizing gas (NO2) with a custom-made gas-sensing system and compared with the performance of pristine SnO2 nanowires. The fabricated sensors were placed in a horizontal tube furnace and the operating temperature was fixed to 300°C because the NO2 sensing performances were found to be best at that temperature in preliminary tests. The NO2 gas has a concentration of 100 ppm in dry air and the gas concentration was controlled by changing the ratio of this mixture gas to dry air by using accurate mass flow controllers. The total flow rate was set to 500 sccm to avoid any possible variation in sensing properties. According to the specifications provided by the gas manufacturer (Daeduk Gas Co., Korea), the content of water vapor in both the dry-air-balanced NO2 gas and the dry air was negligible. The response (R) for NO2 was estimated with the following formula: R = Rg/Ra, where Ra and Rg are the resistances in the absence and presence of NO2, respectively. The response time and recovery time were defined as the time taken for a 90% variation of the resistances upon exposure to the target gas and air, respectively.
References
Godinho, K. G., Walsh, A. & Watson, G. W. Energetic and electronic structure analysis of intrinsic defects in SnO2 . J. Phys. Chem. C 113, 439–448 (2009).
Sysoev, V. V., Goschnick, J., Schneider, T. & Strelcov, E. A gradient microarray electronic nose based on percolating SnO2 nanowire sensing elements. Nano Lett. 7, 3182–3188 (2007).
Park, J. Y., Asokan, K., Choi, S.-W. & Kim, S. S. Growth kinetics of nanograins in SnO2 fibers and size dependent sensing properties. Sens. Actuators B: Chem. 152, 254–260 (2011).
Choi, S.-W., Katoch, A., Sun, G.-J., Wu, P. & Kim, S. S. NO2-sensing performance of SnO2 microrods by functionalization of Ag nanoparticles. J. Mater. Chem. C 1, 2834–2841 (2013).
Wang, B., Zhu, L. F., Yang, Y. H., Xu, N. S. & Yang, G. W. Fabrication of a SnO2 nanowire gas sensor and sensor performance for hydrogen. J. Phys. Chem. C 112, 6643–6647 (2008).
Gierak, J. et al. Focused gold ions beam for localized epitaxy of semiconductor nanowires. Microelectron. Eng. 87, 1386–1390 (2010).
Liao, L. et al. The sensitivity of gas sensor based on single ZnO nanowire modulated by helium ion radiation. Appl. Phys. Lett. 91, 173110 (2007).
Malyi, O. I., Wu, P., Kulish, V. V., Bai, K. W. & Chen, Z. Formation and migration of oxygen and zirconium vacancies in cubic zirconia and zirconium oxysulfide. Solid State Ionics 212, 117–122 (2012).
Kiliç, Ç. & Zunger, A. Origins of coexistence of conductivity and transparency in SnO2 . Phys. Rev. Lett. 88, 095501 (2002).
Godinho, K. G., Walsh, A. & Watson, W. Energetic and electronic structure analysis of intrinsic defects in SnO2 . J. Phys. Chem. C 113, 493–448 (2009).
Xia, W.-Z., Wang, L.-L., Xu, L., Li, X.-F. & Deng, H.-Q. First-principles study of magnetic properties in Ag-doped SnO2 . Phys. Status Solidi B 248, 1961–1966 (2011).
Park, J. Y., Choi, S.-W. & Kim, S. S. Junction-tuned SnO2 nanowires and their sensing properties. J. Phys. Chem. C 115, 12774–127781 (2011).
Fink, D. G. & Christiansen, D. Electronics engineer's handbook. 2nd edn. [Whitaker, J. C. (ed.)] (New York, McGraw-Hill, 1982).
Tsokkou, D., Othonos, A. & Zervos, M. Carrier dynamics and conductivity of SnO2 nanowires investigated by time-resolved terahertz spectroscopy. Appl. Phys. Lett. 100, 133101(2012).
Zervos, M. et al. Structural properties of SnO2 nanowires and the effect of donor like defects on its charge distribution. Phys. Stat. Sol. A 210, 226–229 (2013).
Mi, Y., Odaka, H. & Iwata, S. Electronic structure and optical properties of ZnO, SnO2 and In2O3 . Jpn. J. Appl. Phs. 38, 3453–3458 (1999).
Sanon, G., Rup, R. & Mansingh, A. Band gap narrowing and band structure in degenerated tin oxide (SnO2) films. Phys. Rev. B 44, 5672–5680 (1991).
Lim, F. C. H., Zhang, J., Jin, H. M., Sullivan, M. & Wu, P. A density functional theory study of CO oxidation on Pd-Ni alloy with sandwich structure. Appl. Cat. A 451, 79–85 (2013).
Zhang, J., Cao, X.-M., Hu, P. J. & Wu, P. Density functional theory studies of ethanol decomposition on Rh (211). J. Phys. Chem. C 115, 22429–22437 (2011).
Zhang, J., Jin, H. M., Sullivan, M., Lim, F. C. H. & Wu, P. Study of Pd–Au bimetallic catalysts for CO oxidation reaction by DFT calculations. Phys. Chem. Chem. Phys. 11, 1441–1446 (2009).
Hübner, M., Koziej, D., Grunwaldt, J.-D., Weimar, U. & Barsan, N. An Au clusters related spill-over sensitization mechanism in SnO2-based gas sensors identified by operando HERFD-XAS, work function changes, DC resistance and catalytic conversion studies. Phys. Chem. Chem. Phys. 14, 13249–13254 (2012).
Hübner, M., Barsan, N. & Weimar, U. Influences of Al, Pd and Pt additives on the conduction mechanism as well as the surface and bulk properties of SnO2 based polycrystalline thick film gas sensors. Sens. Actuators B: Chem. 171–172, 172–180 (2012).
Shuai, M., Liao, L., Lu, H. B., Zhang, L., LI, J. C. & Fu, D. J. Room-temperature ferromagnetism in Cu+ implanted ZnO nanowires. J. Phys. D: Appl. Phys. 41, 135010 (2008).
Müller, S., Zhou, M., Li, Q. & Ronning, C. Intra-shell luminescence of transition-metal-implanted zinc oxide nanowires. Nanotechnology 20, 135704 (2009).
Chen, Y.-J. et al. α-MoO3/TiO2 core/shell nanorods: Controlled-synthesized and low-temperature gas sensing properties. Sens. Actuators B: Chem. 155, 270–277 (2011).
Sun, P. et al. Synthesis of novel SnO2/ZnSnO3 core-shell microspheres and their gas sensing properties. Sens. Actuators B: Chem. 155, 606–611 (2011).
Choi, S.-W., Katoch, A., Sun, G.-J., Wu, P. & Kim, S. S. NO2-sensing performance of SnO2 microrods by functionalization of Ag nanoparticles. J. Mater. Chem. C 1, 2834–2841 (2013).
Choi, S.-W., Jung, S.-H. & Kim, S. S. Functionalization of selectively grown networked SnO2 nanowires with Pd nanodots by γ-ray radiolysis Nanotechnology. 22, 225501(2011).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No.2012R1A2A2A01013899). Dr. Ping Wu's research is partially sponsored by the SUTD-ZJU grant (SUTD-ZJU/RES/01/2012) on chemical thermodynamics and by a Singapore MOE Tier 2 grant (MOE 2012-T2-1-097) on carrier transport.
Author information
Authors and Affiliations
Contributions
S.S.K. designed the experiments, analyzed the data and prepared the manuscript. P.W. planned and performed the models and calculations, designed the theoretical work and prepared the manuscript. A.K., G.-J.S. and S.W.C. conducted the experiments. S.H. organized the implantation process. V.V.K. carried out the DFT calculations. All authors participated in discussion. A.K. and S.W.C. contributed equally as first authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported license. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Katoch, A., Sun, GJ., Choi, SW. et al. Acceptor-Compensated Charge Transport and Surface Chemical Reactions in Au-Implanted SnO2 Nanowires. Sci Rep 4, 4622 (2014). https://doi.org/10.1038/srep04622
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04622
This article is cited by
-
Effect of the Deposition Temperature on Ammonia Gas Sensing Based on SnO2/Porous Silicon
Plasmonics (2021)
-
Electrowetting-on-dielectric characteristics of ZnO nanorods
Scientific Reports (2020)
-
NO2 sensing studies of bio-green synthesized Au-doped SnO2
Journal of Materials Science: Materials in Electronics (2017)
-
Micro/Nano Gas Sensors: A New Strategy Towards In-Situ Wafer-Level Fabrication of High-Performance Gas Sensing Chips
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
