
Abstract
The high-resolution melting curve analysis (HRMA) might be a good alternative method for rapid detection of BRAF mutations. However, the accuracy of HRMA in detection of BRAF mutations has not been systematically evaluated. We performed a systematic review and meta-analysis involving 1324 samples from 14 separate studies. The overall sensitivity of HRMA was 0.99 (95% confidence interval (CI) = 0.75–0.82) and the overall specificity was very high at 0.99 (95% CI = 0.94–0.98). The values for the pooled positive likelihood ratio, negative likelihood ratio and diagnostic odds ratio were 68.01 (95% CI = 25.33–182.64), 0.06 (95% CI = 0.03–0.11), and1263.76 (95% CI = 393.91–4064.39), respectively. The summary receiver operating characteristic curve for the same data shows an area of 1.00 and a Q* value of 0.97. The high sensitivity and specificity, simplicity, low cost, less labor or time and rapid turnaround make HRMA a good alternative method for rapid detection of BRAF mutations in the clinical practice.
Similar content being viewed by others
Introduction
BRAF, a signal transduction protein which is downstream of KRAS in the mitogen-activated protein kinase (MAPK) pathway, is one of three members of the RAF kinase family: A-RAF, B-RAF and C-RAF1,2. It acts as direct effector of RAS and through the activation of MEK, promotes tumour growth and survival3. Since mutations in BRAF gene were originally identified in melanomas1, now they have been found in different kinds of cancers, predominantly in melanoma, metastatic colorectal cancer and papillary thyroid cancer4,5,6. In recent years, BRAF mutation has been found to correlate with an aggressive clinical outcome in patients with PTC and drug resistance of patients receiving anti-EGF-receptor (EGFR) therapies for colorectal cancer7. These findings not only make BRAF an attractive therapeutic target but also demonstrate the significant diagnostic and prognostic implications of BRAF mutation status in a wide variety of clinical settings7. Therefore, the detection of BRAF mutation is increasingly important and frequent in clinical practice for both diagnostics and screening of the patients who treatment with BRAF inhibitor.
Up to date, a number of methods for detection of BRAF mutations in different kinds of cancer have been described, including DNA sequencing, TaqMan array, single-strand conformation analysis, allele-specific PCR, pyrosequencing8,9,10,11,12. Although DNA sequencing, including direct DNA sequencing and pyrosequencing, is considered as the “gold standard” for known/unknown mutation scanning, but it is limited by high cost and low sensitivity for the clinical screening of BRAF mutation13. Other methods mentioned above are time consuming and require manipulation of amplified PCR products, which is a common source of sample contamination13. The most important is that BRAF mutation testing requires a much higher analytical sensitivity when dealing with cytological specimens where the cells in question may be a minor population among the vast majority of background non-neoplastic cells7. However, the methods above could not overcome this shortcoming. Furthermore, some of the methods need the expensive equipment or fluorescence-labeled probes may not be economical for all the patients with BRAF mutation.
In recent years, high-resolution melting curve analysis (HRMA) has been proved to be applicable for detection of various clinically relevant human mutations13. It is a cost-efficient, closed-tube system and very sensitive scanning method that allows rapid detection of DNA sequence variations without cumbersome post–polymerase chain reaction (PCR) methods, which is not achievable by other methods including direct sequencing14.
Therefore, HRMA might be a good alternative method for rapid detection of BRAF mutations in the clinical practice. However, the accuracy of HRMA in the detection of BRAF mutations has not been systematically evaluated. Thus, we performed this meta-analysis to evaluate the overall accuracy of HRMA for rapid detection of BRAF mutations.
Results
Selection characteristics
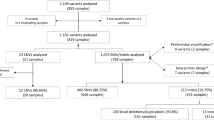
A total of 143 abstracts and titles were obtained by searching multiple databases and sources. 53 records were excluded because of duplicates. And then the remaining 90 records were screened by the titles and abstracts. After 63 studies were excluded, 27 full-text papers were deemed to be relevant and were examined in detail. 13 of full-text articles were excluded with the reasons described in Figure 1. Finally, 14 studies met the inclusion criteria and were included in our meta-analysis.

A flow chart highlighting study selection.
Study characteristics and quality of reporting
The main characteristics of the studies included were summarized in Table 1. A total 14 studies13,14,15,16,17,18,19,20,21,22,23,24,25,26 with 1324 samples were included in our study and the average sample number was 94.5 (range 13 to 201). The earliest study was in March 200515, while the latest was in April 201325,26. Thirteen studies were from western world country, while only one study is from China,which is in Asia20. Type of disease included colorectal cancers (CRCs), melanoma, thyroid nodules, endometrial cancers, gastrointestinal stromal tumors and lung cancers and the CRCs accounts for more than 57.1%. Specimen were mainly formalin-fixed paraffin-embedded (FFPE) (n = 8), the remaining were paraffin-embedded (PE) (n = 3) and fresh frozen tissue (FF) (n = 3). Instrument types included HR-1 (n = 2), LightCycler480 (n = 8), RotorGene6000 (n = 3) and Rotor-Gene Q (n = 1). The dye included LCGreen(n = 1), Syto 9(n = 5), LightCycler LC480 High Resolution Melting Master(n = 6),EvaGreen(n = 1),and one study didn't mention about it17. The final volumes (μL) of HRM were 10 μL (n = 8), 20 μL (n = 4), 25 μL (n = 1), while one study didn't mention about it19. The quality of individual studies was relatively high, as all studies met 8 or more of the QUADAS criteria. All the studies were Cross-sectiona and used the DNA sequencing as the reference method.
Diagnostic accuracy
The Spearman correlation coefficient is −0.130 (p = 0.657), which indicated absence of threshold effect. Therefore, we pooled the sensitivity, specificity, PLR, NLR and DOR by the data extracted from the included studies. Fig. 2 showed the results of the pooling sensitivity and specificity. The sensitivity ranged from 0.93 to 1.00 and the pooled sensitivity was 0.99 (95% confidence interval (CI) = 0.75–0.82) while the specificity from 0.88 to 1.00 and the pooled specificity was 0.99 (95% CI = 0.94–0.98). Fig. 3 showed the results of the pooling PLR and NLR. PLR was 68.01 (95% CI = 25.33–182.64), NLR was 0.06 (95% CI = 0.03–0.11). Fig. 4 showed that DOR was1263.76 (95% CI = 393.91–4064.39). The chi-square and I2 tests for heterogeneity in the summary results suggested significant heterogeneity for specificity (P = 0.00, I2 = 68.8%) and PLR (P = 0.00, I2 = 63.5%). The SROC curve, which is intended to represent the relationship between sensitivity and specificity across all included studies, is shown in Fig. 4. The SROC curve from our data showed that the Q value was 0.97, while the area under the curve (AUC) was close to 1.00 (0.995).

Forest plots estimates of sensitivity (a) and specificity (b) for high-resolution melting curve analysis with 95% confidence intervals (CI).

Forest plots estimates of positive likelihood ratio (PLR) (a) and negative likelihood ratio (NLR) (b) for high-resolution melting curve analysis with 95% confidence intervals (CI).

Forest plots estimates of diagnostic odds ratio (DOR) (a) and summary receiver operating characteristic (SROC) curves (b) for high-resolution melting curve analysis.
Multiple-regression analysis and publication bias
To investigate the source of the heterogeneity for specificity and PLR, Multivariate meta-regression analysis with the covariates including quality, specimen, instrument, dye, final volume of HRM and disease was performed. The quality for every study was scored using QUADAS criteria, where a 1 or a 0 was given if all criteria were fulfilled or not achieved, respectively27. The result of the Multivariate meta-regression analysis showed that there was no statistical significance in RDOR values between studies (Table 2). The subgroup was not performed because the source of the heterogeneity was not found. To our knowledge, biases are inherent within any meta-analysis during the process of locating, selecting and combining studies. In our study, the Deek's Funnel Plot Asymmetry Test showed that the biases were not very significant (P = 0.23). (Fig. 5)

Deek's Funnel Plot Asymmetry Test for the assessment of potential publication bias.
Discussion
Mutations in BRAF have been researched for many years in different kinds of the cancers28,29,30. Now, BRAF mutation has been found to correlate with an aggressive clinical outcome in patients with PTC and drug resistance of patients receiving anti-EGF-receptor (EGFR) therapies for colorectal cancer7. There is obviously an urgently need for fast, reliable and inexpensive methods for screening of BRAF mutation in the clinical practices. HRMA might be a good alternative method for rapid detection of BRAF mutations. However, the accuracy of HRMA in the detection of BRAF mutations has not been systematically evaluated. We performed a meta-analysis including 14 studies with 1324 samples to evaluate the overall accuracy of HRMA for rapid detection of BRAF mutations.
Our meta-analysis showed that the pooled values of the sensitivity and specificity of HRMA were 0.99 (95% CI = 0.75–0.82) and 0.99 (95% CI = 0.94–0.98), respectively. And the SROC showed that Q value was 0.97 and the AUC was 1.00. The results obtained here indicate a very high level of overall accuracy of HRMA. It suggests that the HRMA was a more appropriate method for screening of the BRAF mutation compared with the DNA sequencing. Although DNA sequencing is accepted as the gold standard, but mostly it is limited by higher cost and lower sensitivity in the clinical screening of BRAF mutations. The DOR is a single indicator of test accuracy that combines the data from sensitivity and specificity into a single number and the ratio of the odds of a positive test result for a patient with disease or without disease27. The value of a DOR ranges from 0 to infinity, with higher values indicating better discriminatory test performance27. In our present meta-analysis, we found that the pooled DOR was 1263.76 (95% CI = 393.91–4064.39). It further indicates that the HRMA is ideal methods with a high level of overall accuracy. HRMA also had a very high PLR and a very low NLR for the detection of BRAF mutation in our study, which indicates an excellent ability to both confirm and exclude the presence of BRAF mutation.
Since HRMA appeared as a rapid method for genotyping known variants or scanning for unknown variants, it has been as a screening technique for wider application31. HRMA is a simple, PCR-based method for detecting DNA sequence variation by measuring changes in the melting of a DNA duplex32. Duplex melting is generally monitored using intercalating dyes, which bind to double-stranded but not single-stranded DNA. Therefore, the PCR for the sample, carried out in the presence of a saturating DNA binding dye, is needed before HRMA. After this, the product is heated while the level of fluorescence is measured. As the temperature rises and the duplex passes through its melting transition, dye is released and fluorescence intensity is reduced32. The Tm of duplex is always different among the homozygous sequence, the homozygous sequence with variant and the heterozygous sequence and the fluorescence is distinct from each other during the product is subjected to increasing temperatures, resulting different shapes of the melting curve. The mutations are identified by a change in the melt curve shape compared with the reference profile. Therefore, the sensitivity of HRMA is very high. A previous study has demonstrated that HRM analysis were able to detect as little as 5%–6% tumor cell line DNA in a background of wild-type DNA and another study has a lower detection limit at 0.5%33,34. All categories of substitution are detectable, as are insertions and deletions small enough to be amplified by PCR32.
Therefore, HRMA has been an excellent new tool for BRAF mutation detection. Now, HRMA has been also developed for the detection of DNA sequence variants from different Samples from many sources, such as dried blood spots, buccal cells and frozen tumour specimens, Archived samples in the form of methanol-fixed tissue, formalinfixed paraffin-embedded tissue and cytology slides33. the advantage of simplicity, low cost, ease of use and a high sensitivity/specificity have made HRMA an attractive tool for genotyping and application in diagnostic labs35.
Though the accuracy of HRMA is very high, some factors had been reported that might affect the HRMA accuracy, such as sample source and preparation, amplicon length, GC content, dye, equipments and so on33,36,37. Takano et al. showed that a higher sensitivity could be achieved when fresh tumor samples were used for HRMA, because the DNA might have degenerated during sampling or the preservation of the archived samples36,38. Wittwer et al. found that although HRMA of fragments could be up to 600 bp and more, but the technology is more sensitive for smaller fragments39. When the fragments was below 400 bp, sensitivity and specificity were 100%, while sensitivity decreased to 96.1% and 99.4% for specificity when between 400 and 1000 bp36. And they also found it critical for high sensitivity that the melt profile contains not more than one to two melt domains39. Taylor et al described in their study that Lower GC content might have an association with false negative results32. Besides, dye and equipments are also very important in the accuracy of HRMA. Different dyes detect heteroduplexes to varying degrees and the most common real-time dye, SYBR Green I, does not detect them at all36. And instruments vary in their ability to resolve shape and absolute temperature differences, explained by their signal-to-noise ratios, acquisition rates, data density and software36. Furthermore, Tindall et al proposed that the shape of an HRMA curve in itself is usually not sufficient to type a specific variant, to achieve this either a melt probe should be added or the fragment should be sequenced40. However, we performed this meta-analysis including 14 studies with 1324 samples proved the high accuracy of HRMA for detection of BRAF mutation compared with the DNA sequencing.
An exploration of the reasons for heterogeneity rather than the computation of a single summary measure is an important goal of meta-analysis27. Although the accuracy of the HRMA showed a high level in our study, we found that there was heterogeneity in the pooled result of specificity and PLR. To investigate the source of the heterogeneity, we performed the multivariate meta-regression analysis with the covariates including quality, specimen, instrument, dye, final volume of HRM and disease. The results of the meta-regression analysis indicated there was no significant characteristic influencing diagnostic accuracy. Previous studies reported that the instrument type, sample source and dye may affect the accuracy37,39. The inconsistent result between our study and the previous studies may be explained by the sample size. Though we performed the multivariate meta-regression analysis, we still could not find the source of the heterogeneity. Therefore, the subgroup would not be performed according to the results of multivariate meta-regression analysis. Besides, the dependence of scanning accuracy on PCR product length has been studied on the HR-1 and LightScanners instruments, revealing more errors as the length increases above 400 bps39. Therefore, we may infer that another reason responsible for the heterogeneity is the length of the DNA product, but we could not get sufficient information in our included studies.
Our meta-analysis had several strengths. Firstly, we used a comprehensive search strategy with well defined inclusion criteria in such a meta-analysis to date. Secondly, two reviewers performed the study selection and data extraction dependently and discrepancies were resolved by consensus including a third author. Thirdly, we assessed the quality of included studies by the standard criteria and the score of included in our study was relatively higher. Lastly, we used a careful approach to selecting a fixed or random effects model for pooling studies by taking into account the presence or absence of significant heterogeneity. However, our systematic review also had several limitations. Firstly, we could not find the source of the heterogeneity and the subgroup was not performed according to the result of multivariate meta-regression analysis. Secondly, though our meta-analysis included fourteen available studies, but the number is still relatively small, which may affect the pooling accuracy of the HRMA. Lastly, we didn't collect enough information about other factors which may disrupts the accuracy of the HRMA, such as length of DNA products or the GC content.
Despite these limitations, this study proved the higher sensitivity and specificity of HRMA. The advantage high sensitivity and specificity, simplicity, low cost, less labor and time and rapid turnaround make HRMA a good alternative method for rapid detection of BRAF mutations in the clinical practice.
Methods
Search strategy
To identify relevant publications, PubMed (http://www.ncbi.nlm.nih.gov/pubmed), ISI Science Citation Index (http://apps.isiknowledge.com) and EMBASE (http://www.embase.com/home) databases were searched using the following search terms: “BRAF”, “B-RAF”, “HRMA”, “HRM”, “HRMCA”, “HRMC”, “high resolution melting analysis”, “high resolution melting”, “high resolution melting curve analysis”, “high resolution melting curve”. No limitations were placed on the language of publications and the type of studies. We also manually searched the reference lists of the articles identified in the searches for additional eligible studies.
Selection criteria
Studies eligible for inclusion in this meta-analysis met the following criteria: (1) HRMA was applied to the study of BRAF mutations in humans; (2) DNA Sequencing was used as a reference standard; (3) provided enough information to describe true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN); (4) it must be published as a full text in English. While the exclusion critseria were as follows: (1) studies were performed using only HRMA or comparing HRMA with non-sequencing techniques; (2) studies used samples with artificially created sequences or the cell lines; (3) The TP is 0; (4) meeting abstracts, case reports, letters and reviews were excluded due to limited data; (5) Publications identified as duplicates were excluded.
Data extraction and quality assessment
Information was carefully extracted from all eligible studies. The following data were collected from each study: first author's name, year of publication, study design, country where the study was conducted, type of disease, sources of sample, kits of DNA extraction, instruments, dye and final volume of HRMA, reference method, number of sample and number of samples with the indicated test result (TP, FP, FN, TN). Data extraction was done independently by two of the authors and discrepancies were resolved by consensus including a third author. We assessed the quality of the individual studies using criteria based on the Quality Assessment of Studies of Diagnostic Accuracy41.
Data synthesis and meta-analysis
We used the standard methods recommended for meta-analysis of diagnostic test evaluations for the measures of test accuracy for each study by Meta-Disc (version 1.4) and Stata (version 11.0) software42. The numbers of TP, FP, FN and TN were taken directly from the included studies and 0.5 was added to all cells to handle studies with empty cells.
First of all, we conducted analysis of diagnostic threshold to find weather there was a threshold effect in included studies. Threshold effect arises from different cut-offs used in different studies to define a positive test result, which was which was assessed by Spearman correlation coefficient.
Threshold effect was defined as a positive correlation (P < 0.05). If threshold effect was present, the accuracy data should be pooled by summary receiver operating characteristic (SROC) curve and calculating the area under the curve (AUC). If not, we performed the next pooling.
Study heterogeneity was assessed using the chi-squared test of heterogeneity (Q Cochran's Q statistic) and the Higgin's I2 measure. Taking into account the low statistical power of these tests of heterogeneity, significant heterogeneity was defined as a Q test P value of <0.10, or an I2 measure >30%. The sensitivity, specificity, positive likelihood ratio (PLR), negative-likelihood ratio (NLR) and diagnostic odds ratio (DOR) from the different studies were combined using fixed effects or random effects models with 95% confidence intervals (CIs). The choice of the fixed or random effects model was made on the absence or presence of significant heterogeneity based on the depended on the Q test, respectively.
Source of the heterogeneity were investigated by multivariate meta-regression analysis with the covariates including QUADAS scores, specimen, instrument, dye, final volume of HRM and disease if there was statistically significant heterogeneity across the included studies. The subgroup analysis was followed multivariate meta-regression analysis if factors of heterogeneity could be found. Because publication bias is of concern for meta-analysis of diagnostic studies, we tested the potential presence of this bias using the Deek's Funnel Plot Asymmetry Test.
References
Pao, W. & Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 12, 175–180 (2011).
Yaeger, R. & Saltz, L. BRAF mutations in colorectal cancer: clinical relevance and role in targeted therapy. J Natl Compr Canc Netw. 10, 1456–1458 (2012).
Sclafani, F., Gullo, G., Sheahan, K. & Crown, J. BRAF mutations in melanoma and colorectal cancer: A single oncogenic mutation with different tumour phenotypes and clinical implications. Crit Rev Oncol Hematol. 87, 55–68 (2013).
Tol, J., Nagtegaal, I. D. & Punt, C. J. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 361, 98–99 (2009).
Kumar, R. et al. BRAF mutations in metastatic melanoma: a possible association with clinical outcome. Clin Cancer Res. 9, 3362–3368 (2003).
Cohen, Y. et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 95, 625–627 (2003).
Ziai, J. & Hui, P. BRAF mutation testing in clinical practice. Expert Rev Mol Diagn. 12, 127–138 (2012).
Walther, Z., Ronski, K., Wan, M. & Sklar, J. A TaqMan array system for mutation profiling of solid tumors using routine surgical pathology and cytology specimens. J Mol Diagn. 14, 704 (2012).
Tan, Y. H. et al. Detection of BRAF V600E mutation by pyrosequencing. Pathology. 40, 295–298 (2008).
Adeniran, A. J. et al. Reflex BRAF testing in thyroid fine-needle aspiration biopsy with equivocal and positive interpretation: a prospective study. Thyroid. 21, 717–723 (2011).
Wooster, R., Futreal, A. P. & Stratton, M. R. Sequencing Analysis of BRAF Mutations in Human Cancers. Methods Enzymol. 407, 218–224 (2006).
Szankasi, P. et al. A Quantitative Allele-Specific PCR Test for the BRAF V600E Mutation Using a Single Heterozygous Control Plasmid for Quantitation: A Model for qPCR Testing without Standard Curves. J Mol Diagn. 15, 248–254 (2013).
Pichler, M. et al. Evaluation of High-Resolution Melting Analysis as a Diagnostic Tool to Detect the BRAF V600E Mutation in Colorectal Tumors. J Mol Diagn. 11, 140–147 (2009).
Simi, L. et al. High-resolution melting analysis for rapid detection of KRAS, BRAF and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 130, 247–253 (2008).
Willmore-Payne, C., Holden, J. A., Tripp, S. & Layfield, L. J. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 36, 486–493 (2005).
Seth, R. et al. Concomitant mutations and splice variants in KRAS and BRAF demonstrate complex perturbation of the Ras/Raf signalling pathway in advanced colorectal cancer. Gut. 58, 1234–1241 (2009).
Bennani, B. et al. Mutation analysis of BRAF exon 15 and KRAS codons 12 and 13 in Moroccan patients with colorectal cancer. Int J Biol Markers. 25, 179–184 (2010).
Borras, E. et al. Clinical pharmacogenomic testing of KRAS, BRAF and EGFR mutations by high resolution melting analysis and ultra-deep pyrosequencing. BMC cancer. 11, (2011).
Bozzao, C. et al. Survey of KRAS, BRAF and PIK3CA mutational status in 209 consecutive Italian colorectal cancer patients. Int J Biol Markers. 27, e366–374 (2012).
Hsieh, L.-L. et al. Characteristics and prevalence of KRAS, BRAF and PIK3CA mutations in colorectal cancer by high-resolution melting analysis in Taiwanese population. Clin Chim Acta. 413, 1605–1611 (2012).
Krol, L. C. et al. Concordance in KRAS and BRAF mutations in endoscopic biopsy samples and resection specimens of colorectal adenocarcinoma. Eur J Cancer. 48, 1108–1115 (2012).
Mancini, I. et al. A High-Resolution Melting Protocol for Rapid and Accurate Differential Diagnosis of Thyroid Nodules. J Mol Diagn. 14, 501–509 (2012).
Ney, J. T., Froehner, S., Roesler, A., Buettner, R. & Merkelbach-Bruse, S. High-Resolution Melting Analysis as a Sensitive Prescreening Diagnostic Tool to Detect KRAS, BRAF, PIK3CA and AKT1 Mutations in Formalin-Fixed, Paraffin-Embedded Tissues. Arch Pathol Lab Med. 136, 983–992 (2012).
Yang, L. et al. Identification of biomarkers to distinguish clear cell sarcoma from malignant melanoma. Hum Pathol. 43, 1463–1470 (2012).
Guedes, J. G. et al. High resolution melting analysis of KRAS, BRAF and PIK3CA in KRAS exon 2 wild-type metastatic colorectal cancer. BMC cancer. 13, (2013).
Richter, A. et al. A multisite blinded study for the detection of BRAF mutations in formalin-fixed, paraffin-embedded malignant melanoma. Sci. Rep. 3, 1659; 10.1038/srep01659 (2013).
Xu, H.-B., Jiang, R.-H., Sha, W., Li, L. & Xiao, H.-P. PCR-Single-Strand Conformational Polymorphism Method for Rapid Detection of Rifampin-Resistant Mycobacterium tuberculosis: Systematic Review and Meta-Analysis. J Clin Microbiol. 48, 3635–3640 (2010).
Boursault, L. et al. Tumor Homogeneity between Primary and Metastatic Sites for BRAF Status in Metastatic Melanoma Determined by Immunohistochemical and Molecular Testing. PLoS One. 8, e70826 (2013).
van Eijk, R. et al. Rapid KRAS, EGFR, BRAF and PIK3CA mutation analysis of fine needle aspirates from non-small-cell lung cancer using allele-specific qPCR. PLoS One. 6, e17791 (2011).
Libra, M. et al. Analysis of BRAF Mutation in Primary and Metastatic Melanoma. Cell Cycle. 4, 1382–1384 (2005).
Wittwer, C., Reed, G., Gundry, C., Vandersteen, J. & Pryor, R. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 49, 853–860 (2003).
Taylor, C. F. Mutation scanning using high-resolution melting. Biochem Soc Trans. 37, 433–437 (2009).
Krypuy, M., Newnham, G. M., Thomas, D. M., Conron, M. & Dobrovic, A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC cancer. 6, 295 (2006).
Bastien, R. et al. High-throughput amplicon scanning of the TP53 gene in breast cancer using high-resolution fluorescent melting curve analyses and automatic mutation calling. Hum Mutat. 29, 757–764 (2008).
Vossen, R. H. A. M., Aten, E., Roos, A. & den Dunnen, J. T. High-Resolution Melting Analysis (HRMA)—More than just sequence variant screening. Hum Mutat. 30, 860–866 (2009).
Montgomery, J. L., Sanford, L. N. & Wittwer, C. T. High-resolution DNA melting analysis in clinical research and diagnostics. Expert Rev Mol Diagn. 10, 219–240 (2010).
Li, B. S. et al. Is high resolution melting analysis (HRMA) accurate for detection of human disease-associated mutations? A meta analysis. PLoS one. 6, e28078 (2011).
Takano, T. et al. Epidermal Growth Factor Receptor Mutation Detection Using High-Resolution Melting Analysis Predicts Outcomes in Patients with Advanced Non–Small Cell Lung Cancer Treated with Gefitinib. Clin Cancer Res. 13, 5385–5390 (2007).
Wittwer, C. T. High-resolution DNA melting analysis: advancements and limitations. Hum Mutat. 30, 857–859 (2009).
Tindall, E. A., Petersen, D. C., Woodbridge, P., Schipany, K. & Hayes, V. M. Assessing high-resolution melt curve analysis for accurate detection of gene variants in complex DNA fragments. Hum Mutat. 30, 876–883 (2009).
Whiting, P., Rutjes, A. W., Reitsma, J. B., Bossuyt, P. M. & Kleijnen, J. The development of QUADAS: a tool for the quality assessment of studies of diagnostic accuracy included in systematic reviews. BMC Med Res Methodol. 3, 25 (2003).
Deville, W. L. et al. Conducting systematic reviews of diagnostic studies: didactic guidelines. BMC Med Res Methodol. 2, 9 (2002).
Acknowledgements
This work was supported in part by grants from the National High Technology Research and Development Program of China (National 863 Program, No. 2011AA02A121 and 2013AA020204) the Preferential Foundation of Science and Technology Activities for Students Studying Abroad of Chongqing (No. YuLiuZhu201201), the Project Sponsored by the Scientific Research Foundation for Returned Overseas Chinese Scholars from the State Education Ministry.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: D.C., W.F., Q.H. Performed the experiments: Y.Y.W., Y.X.W. Analyzed the data: K.L., J.H. Contributed reagents/material/analysis tools: D.S., Q.L. Wrote the manuscript: D.C., L.Z. Reference collection and data management: Z.C., Z.Y.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chen, D., Wang, YY., Chuai, ZR. et al. High-Resolution Melting Analysis for accurate detection of BRAF mutations: a systematic review and meta-analysis. Sci Rep 4, 4168 (2014). https://doi.org/10.1038/srep04168
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04168
This article is cited by
-
Surveillance of Plasmodium falciparum pfcrt haplotypes in southwestern Uganda by high‐resolution melt analysis
Malaria Journal (2021)
-
Mutation spectrum of COL1A1/COL1A2 screening by high-resolution melting analysis of Chinese patients with osteogenesis imperfecta
Journal of Bone and Mineral Metabolism (2020)
-
BRAF V600E mutation detection in hairy cell leukemia-utility of archival DNA from bone marrow aspirate/imprint smear and amplification refractory mutation system
Molecular Biology Reports (2020)
-
Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine
Modern Pathology (2018)
-
Two kinds of common prenatal screening tests for Down’s syndrome: a systematic review and meta-analysis
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
