
Abstract
Microbial strains with high substrate efficiency and excellent environmental tolerance are urgently needed for the production of platform bio-chemicals. Bacillus coagulans has these merits; however, little genetic information is available about this species. Here, we determined the genome sequences of five B. coagulans strains and used a comparative genomic approach to reconstruct the central carbon metabolism of this species to explain their fermentation features. A novel xylose isomerase in the xylose utilization pathway was identified in these strains. Based on a genome-wide positive selection scan, the selection pressure on amino acid metabolism may have played a significant role in the thermal adaptation. We also researched the immune systems of B. coagulans strains, which provide them with acquired resistance to phages and mobile genetic elements. Our genomic analysis provides comprehensive insights into the genetic characteristics of B. coagulans and paves the way for improving and extending the uses of this species.
Similar content being viewed by others


Introduction
White biotechnology, the clean industrial technology supported by several predominant political movements, will comprise no less than 20% of the chemical industry sales in the United States in 20201. To attempt to meet the dramatic future demands for these materials, researchers have used microbial strains to produce platform bio-chemicals2. Microbial strains with the characteristics of robust high substrate efficiency, low by product formation and excellent environmental tolerance are not easy to isolate from nature3. However, Bacillus coagulans is one such microorganism that has a number of these characteristics4,5,6,7. It is a spore-forming gram-positive soil bacterium, which can be found all over the world8. This species was first isolated in 1915 by Hammer, who isolated this organism from spoiled canned milk9. Recently, B. coagulans has been reported to possess many valuable fermentation features, such as growth at 50°C–55°C and high carbon-efficiency7. In addition, it can ferment various biomass-derived sugars to yield various platform bio-chemicals, such as lactic acid. Moreover, the high fermentation temperature of B. coagulans strains enables non-sterilized batch and fed-batch fermentation for l-lactic acid production4. For example, Qin et al. used B. coagulans 2-6 to obtain a maximum l-lactic acid concentration of 182.0 g/liter with an optical purity of 99.4% at 50°C4. Milind et al.7,10 and Wang et al.6 reported that B. coagulans strains produce lactic acid and that ~98% of xylose could be converted to l-lactic acid. In addition to the production of lactic acid, B. coagulans has also shown to be a source of many other commercially valuable products, such as thermostable enzymes8 and coagulin, an antimicrobial peptide11. More recently, this species has also been regarded as a novel safe probiotic12. These studies suggest that B. coagulans strains can readily achieve generally regarded as safe (GRAS) status required for large-scale commercial use. Compared to other probiotic strains, such as those belonging to Lactobacillus species, B. coagulans strains are able to survive as spores in the extreme environments, such as high heat or acidity12.
B. coagulans is one of the earliest isolated microorganisms and it is thought to be an ideal industrial organism with remarkable advantages in manufacturing of various chemicals and enzymes9,13. However, little genetic information about this species is currently available and there are still many questions to answer, such as why B. coagulans strains can produce high concentrations of optically pure L-lactic acid and why they can ferment openly without sterilization. We have recently determined the nucleotide sequences of B. coagulans strains 2-6, XZL4, XZL9, H-1 and DSM114,15,16,17. Comparative genomic analysis of these strains should provide us with comprehensive insights into the metabolic characteristics of B. coagulans and its niche-specialized adaptation. Ultimately, we hope that this analysis will help answer the questions stated above and lead to new strategies for using and genetically improving these already useful strains.
Results
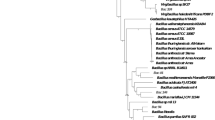
Genome sequence and phylogenetic analysis
Table 1 shows the genomic features of all B. coagulans strains examined in this study. B. coagulans strains share many characteristics with those from the B. subtilis groups, including B. subtilis, B. licheniformis and B. amyloliquefaciens. Their genomes have similar GC content (43% ~ 47%) and they grow well at a wide range of temperatures. However, the chromosome sizes of B. coagulans strains (~3 Mbp) are smaller than those of the B. subtilis group (~4 Mbp). The genomes of B. coagulans strains 36D1 and XZL9 are significantly larger than those in the other strains. In contrast, the size of B. coagulans 2-6 genome in GenBank is smaller than all other Bacillus strains previously reported16. Comparison of the six B. coagulans genomes showed a high degree of sequence similarity and gene synteny in genome core regions (Figure 1). For a comprehensive comparative analysis, we concatenated all conserved genes and constructed a phylogenetic tree (Figure 2). Unexpectedly, all B. coagulans strains, which are clustered together, are more closely related to B. cereus groups than to B. subtilis groups, which is consistent with the result of Mun et al.18. However, we did not find any gene related to the PlcR regulon, which is the main virulence system of B. cereus groups. Inside the B. coagulans strains, two strains (36D118 and XZL9) have nearly identical genome sizes (~3.5 Mbp), genomic context and gene orders and have diverged from the other B. coagulans strains quite recently; strain 2-6 diverged from XZL4, H-1 and DSM1.

Circular representation of the Bacillus coagulans 2-6 chromosome.
The nine circles (from outside to inside) show the following: (i) the predicted ORFs on the plus and minus strands based on the COG database (colors were assigned according to the colors of the COG functional classes, which are listed on the bottom); (ii–vi) homology of B. coagulans 2-6 CDSs identified using BLAST in the strains XZL4, XZL9, DSM1, H-1 and 36D1 (red-to-blue were assigned according to the similarity of homologs); (vii) the genomic islands predicted by IslandViewer; (viii) the value of the GC skew (G − C/G + C); and (ix) the percentage of GC content with a 10-kb window size.

Maximum likelihood tree of Bacillus strains.
Genes that are conserved in all strains were aligned and concatenated for tree construction. The B. coagulans strains are highlighted in green. And strains from B. cereus group are highlighted in pink. A scale bar for the genetic distance is shown at the bottom.
Central carbon metabolism
As industrial producers, B. coagulans strains have the ability to use a variety of carbohydrates. However, hexose and pentose are substrates that are involved in fermentation; therefore, these are the two sugars that we are most interested in. Based on our previous studies4,7, B. coagulans is a homofermentative Bacillus strain that has an efficient metabolic pathway for utilizing hexose. The primary product of glucose fermentation is L-lactic acid (ca. 97% of the fermentation products) and small amounts of acetate and succinate are also produced4. Our genomic analysis results indicate that the essential genes for the Embden-Meyerhof-Parnas (EMP) pathway are present, whereas those for the Entner-Doudoroff pathway are absent in B. coagulans. These suggest that most of the hexose goes through the EMP pathway, which is the most efficient pathway for converting hexose into lactic acid. Conversely, pentoses, such as xylose, usually go through two pathways, namely the phosphoketolase pathway (PKP) and pentose phosphate pathway (PPP) (Figure 3A). If xylose is metabolized through the PKP, the theoretical lactic acid yield is not expected to be higher than 60%. However, when xylose was provided as the carbon source for different B. coagulans strains, lactic acid represented 70% to 98% of the total fermentation products6,7. We found that in all strains except 36D1, only a fragment of the phosphoketolase gene was predicted. This gene, which encodes the enzyme that catalyzes the conversion of ribose-5-phosphate to 5-phospho-ribose-1-diphosphate, the first step of the PKP, is interrupted by transposases. Although strain 36D1 has a full-length predicted phosphoketolase gene, based on the result of 13C-NMR experiments, the PPP is still the main metabolic pathway for xylose utilization in the strain 36D17. It is not surprising that we found genes encoding the enzymes that catalyze the conversion of d-xylulose-5P to fructose-6P and glyceraldehyde-3P.

Comparative analysis of xylose metabolism in the B. coagulans strains.
(A) The metabolic pathway for xylose fermentation in lactic acid bacteria. (B) Schematic gene maps of the xylose-utilization genes found in the B. coagulans strains examined in this study. Genes, that were not filled, are pseudogenes. xylR: DNA-binding transcriptional activator; xylAI: xylose isomerase Type I; xylB: xylulokinase; xylT: Xylose H+-symporter; xylAIII: xylose isomerase Type III; P1: quinolinate synthetase; P2: iron-containing alcohol dehydrogenase; P3: peptidase; P4: hypothetical protein; P5: PfkB domain-containing protein; P6: LacI family transcriptional regulator; P7: beta-ketoacyl reductase; P8: hypothetical protein; P9: breakpoint of contigs; P10: hypothetical protein; P11: hypothetical protein; P12: 6-phospho-3-hexuloisomerase; (C) Maximum likelihood tree of xylA genes. The genes marked by filled circles ( ) are from B. coagulans strains and are homologs of xylA in B. subtilis. The genes marked with squares (
) are from B. coagulans strains and are homologs of xylA in B. subtilis. The genes marked with squares ( ) are from B. coagulans strains and are the novel xylose isomerases discovered in this study. (D) Maximum likelihood tree of xylB genes. The genes marked with filled circle (
) are from B. coagulans strains and are the novel xylose isomerases discovered in this study. (D) Maximum likelihood tree of xylB genes. The genes marked with filled circle ( ) are from B. coagulans strains. The accession numbers of these genes downloaded from the NCBI database are shown in the parentheses. A scale bar for the genetic distance is shown at the bottom.
) are from B. coagulans strains. The accession numbers of these genes downloaded from the NCBI database are shown in the parentheses. A scale bar for the genetic distance is shown at the bottom.
Our analysis showed that in addition to the highly efficient EMP and PPP pathways, all the studied B. coagulans strains contain l-lactate dehydrogenase and produce l-lactic acid with highly optical purity (more than 99%) under facultative anaerobic conditions4,19,20. Meanwhile all the strains also contain d-lactate dehydrogenase (d-LDH) genes, even though no d-lactate dehydrogenase activity was detected in these strains4. The d-LDH encoding gene in some strains (2-6, XZL9 and XZL4) is interrupted by a premature stop codon. We compared the remaining d-LDHs with those from L. bulgaricus21,22. Some residues of d-specific lactate dehydrogenase that are essential for substrate specificity and catalysis have changed (Tyr52Leu, Asn77Thr, Val78Ala and Trp135Val)23 (Figure 4). Furthermore, we could not find any genes encoding pyruvate decarboxylase, which is a part of the pyruvate dehydrogenase complex.

Multiple sequence alignment of d-lactate dehydrogenases.
d-Lactate dehydrogenases were aligned by using ClustalX. 2DLD and 1J4A are the accession numbers for d-Lactate dehydrogenase from Lactobacillus helveticus and Lactobacillus bulgaricus, respectively, in the PDB. Visualization of the multiple sequence alignment was performed by ESPript. Secondary structure elements were calculated based on the structure of 1J4A. The residues that are marked in green are the key active sites that may affect the function.
B. coagulans can also produce other bio-chemicals. For example, we found genes encoding proteins involved in the production of acetoin and butanediol, which are very important platform bio-chemicals24. According to our present experiments (Figure S1) and previous studies7,23, ethanol, acetoin and butanediol are the primary fermentation products under aerobic conditions.
Xylose metabolism
Previous studies showed that approximately half of B. coagulans species have the ability to metabolize d-xylose, which is the most important difference among the B. coagulans strains25. Genomic context analysis indicated that the xylose-utilization strains (36D1, XZL9 and XZL4) contain at least one copy of the xyl operon, which is similar to that in B. subtilis26. In the other B. coagulans strains, incomplete xyl operons, that lack the xylose H+-symporter (xylT), were found, likely causing their inability to utilize d-xylose. In strains 36D1 and XZL9, there are also some other genes related to the xylose metabolism, such as xylose regulators, a xylose ABC transporter (xylFGH) and a xyloside Na+(H+)-symporter (xynT). To obtain a full picture of xylose-utilization in B. coagulans, we compared the xyl operons in all B. coagulans strains (Figure 3B).
Xylose isomerase (a synonym for glucose isomerase) is required for the first step of the xylose utilization, conversion of d-xylose into d-xylulose. B. coagulans is a known source of xylose isomerase for industrial production27. This enzyme was characterized in detail in L. lactis28 and its orthologs are present in many bacteria. Using pan-genome analysis (Data S1), we identified two orthologous group of xylose isomerases, both of which belong to the xylose isomerase-like TIM barrel family (Pfam: PF01261). One of the orthologous group has homology to xylA from B. subtilis, with ~85% similarity. We performed a phylogenetic analysis and identified the other group as an alternative xylose isomerase that we termed xylA-III, which is not homologous to xylA from B. subtilis or to xylA-II from Clostridium acetobutylicum26 (Figure 3C). In the genomes of strains 36D1 and XZL9, xylA-III is not clustered with any other xylose-utilization genes. Although they were not predicted to be in a genomic island, we suppose that xylA-III is a part of a mobile genetic element that was obtained by horizontal gene transfer (HGT). In addition, we also found pseudo xylA-III genes in XZL4, H-1 and DSM1, which resulted from a premature stop codon. These genes were likely interrupted during integration into the chromosome. Xylulokinase is required for the phosphorylation of d-xylulose, yielding d-xylulose-5-phosphate, a key intermediate in the PPP. Unlike xylose isomerases, we found only one category of xylulokinase, which is very well conserved (>95% identity). The phylogenetic analysis showed that the second xylulokinase in strains 36D1 and XZL9 likely resulted from gene duplication (Figure 3D). In addition, we found a fragment of a xylA-III gene directly upstream of the second xylB in the strain 36D1, which may have been generated during integration.
Due to the lack of a xylose uptake system, B. subtilis is unable to grow on xylose as a sole carbon source26. In the B. coagulans strains, we identified three different types of xylose/xyloside transport systems. The xylose H+-symporter (xylT), which belongs to the Major Facilitator Superfamily (MFS) of transporters family, is the last gene of the xyl operon. This gene, which is crucial for xylose update, has been reported in B. megaterium29 and L. brevis30. Another gene, xynT, which also belongs to the MFS transporter family, imports xyloside across the membrane. However, we found no genes related to xyloside utilization. The ABC-type xylose transporter xylFGH was originally described in Escherichia coli31. The genes encoding this ABC transporter system are separated from other xylose-utilization gene and there is an AraC homolog next to the ABC transporter, which is associated with the control of xylose uptake.
Protein secretion systems
As a source for many thermostable industrial enzymes, the protein secretion systems are crucial for the B. coagulans strains. Two types of protein secretion, the Sec- and Tat-dependent secretion systems, were identified in the B. coagulans strains (Table S2). The protein secretion systems in B. coagulans strains are fully orthologous to those in B. subtilis32. Similar to B. subtilis, B. coagulans strains lack a secretion-specific targeting factor similar to the SecB protein of E. coli. However, in all B. coagulans strains, there are highly conserved signal recognition particle (SRP) pathways that play important roles in the translocation of pre-proteins. The SRP complex (Ffh), which acts as a cellular chaperone, binds to the signal peptide of an mRNA chain and is targeted to the membrane with the help of FtsY. The pre-protein translocation machinery of the Sec-dependent system consists of SecA, SecYEG and SecDF, which are present in all B. coagulans strains. SecYEG functions as a membrane channel for protein export with the aid of SecA. However, we found that the secE gene was missing in the genome of strain 2-6. In the Tat-dependent secretion system, pre-proteins with twin-arginine signal peptides fold in the cytoplasm and are translocated by the Tat complex (TatAC) in the membrane33. At the latest stage, SPases remove the signal peptide from pre-proteins. We identified two types of SPases (type I and II) in the genomes of all B. coagulans strains.
Natural competence
Natural competence is the ability of a cell to take up free DNA from the surrounding medium. To incorporate DNA from the medium, cells synthesize a specific DNA-binding and -uptake system to efficiently replace homologous regions of the chromosome, leading to a permanent change in cell phenotype. Currently, five different genes have been identified, which are essential for the DNA transport: comCEFG and nucA34. However, based on the result of orthologous analysis, we have identified four of these genes, comCEFG (Table S3). The comE operon encodes a polytopic transmembrane protein (ComEC), which is thought to form a pore that guides the DNA into the cell interior, where it may associate with the DNA-helicase-like protein encoded by comF. The comG-encoded protein takes up the DNA by using a pilin-like structure. ComC appears to be involved in the correct assembly of this structure35. In addition, we have also identified the transcriptional factor ComK in all strains, which regulates the expression of genes for DNA uptake and recombination in Bacilli. According to the research of Kovacs36, although the B. coagulans ComK recognized several elements similar to those of B. subtilis, activation of the transcription of genes coding for DNA uptake in B. coagulans might differ from that of B. subtilis.
Amino acid, cofactor and vitamin biosynthesis
Nutrient requirements are important in industrial microbial fermentations. We identified most of the amino acid biosynthetic pathways in the B. coagulans strains using the comparative pathway tool of PATRIC (Data S2). However, the synthetic pathways for l-histidine are incomplete in all sequenced strains. Histidine biosynthesis in B. subtilis is encoded by hisABCDEFGIJ37. The gene for histidinol-phosphatase (hisJ, EC: 3.1.3.15), which catalyzes the dephosphorylation of histidinol phosphate to histidinol, is absent from all the B. coagulans genomes. Moreover, there are no transport systems to import histidine through the membrane. In strain XZL4, we could not find the gene that encodes glutamate-ammonia ligase (EC: 6.3.1.2), which converts L-glutamate to L-glutamine. Pathways for the synthesis of several cofactors, such as biotin, vitamin B6 and lipoic acid, are absent from all B. coagulans strains. However, according to the knowledge of KEGG, we identified at least one biotin transport system (bioY), through which the cells could obtain biotin from the surrounding medium38. The biosynthesis pathways for other cofactors, such as pantothenate, CoA, riboflavin, FAD and FMN, are present in all strains.
Diversifying selection of genes associated with amino acid metabolism
Its thermophilic characteristic is a favorable fermentation feature of B. coagulans. According to Darwin, diversifying selection is the main driving force of evolution, in which the genes involved in environmental adaptation are usually under strong selection pressure39. Temperature, as a dominant selective pressure, could apply strong selection pressure on the genes that are important for thermal adaptation40. Therefore, calculating the positive selection pressure of each gene could help to identify the key genes that allow B. coagulans to survive at high temperatures39,41. Based on the genome-wide positive selection analysis, we found that there are a large number of genes that have significant evidence for positive selection in amino acid metabolism pathways (P-value < 0.01, Table 2. Detailed information is available at the following web site: http://202.120.45.186/~webserver/kaks/detail.php?jobId = s4ZLfBfSGJ). In addition, many of these genes are associated with stress resistance. For example, rocR encodes a 52-kDa polypeptide that belongs to the NtrC/NifA family of transcriptional activators. It has been reported that a B. subtilis strain, which contains a rocR null mutation, is unable to use arginine as the sole nitrogen source, suggesting that RocR is a positive regulator of arginine catabolism42. RocR is also thought to be essential for nitrogen metabolism in response to various stresses43. Histidinol dehydrogenase (HisD), which catalyzes the last step in histidine biosynthesis, was reported as a virulence factor in the intracellular pathogen Brucella suis44. In B. subtilis, in response to diverse growth-limiting stresses, hisD expression is controlled by σB, which governs a large set of general stress proteins45.
Restriction-modification and CRISPR-Cas systems
Fermentation failures due to bacteriophage attack result in substantial economic losses in fermentation industry46, especially during open fermen2tation. Restriction-modification (R-M) and CRISPR-Cas are two types of general defense systems that protect cells from foreign DNA. The systems are compatible and act together to increase the overall phage resistance of the cells47.
R-M systems are nearly universal and have been found in more than 90% of bacterial and archaeal genomes48. In general, within a cell, a methyltransferase protects host DNA by modifying a specific nucleic acid. The restriction endonuclease cleaves any foreign DNA that contains a specific recognition site, which is not protected by the modification47. By comparing the genome sequences to those in the REBASE database, we identified various R-M systems in the B. coagulans strains (Table 3), comprising approximately ~1% of the genome. The majority of the B. coagulans R-M systems are Type I (>50%). In these systems, cleavage occurs at variable distances from the recognition sequence.
The CRISPR-Cas systems, which are comprised of clustered regularly interspaced short palindromic repeats along with their associated (Cas) proteins, are hyper-variable genetic loci that are widely distributed in bacteria and archaea49. This defense mechanism requires immunity to against invading genetic elements. Because the phages that attack bacteria are abundant in soil habitats, many soil bacteria carry CRISPR sequences. CRISPR-Cas systems are commonly found in the B. coagulans strains. In the genome of strain 2-6, we identified two different confirmed CRISPR loci; in strain 36D1, we found four different confirmed CRISPR loci (Table 4). However, only one CRISPR locus in each genome was associated with cas genes (CRISPR_2-6_2 and CRISPR_36D1_2), both of which include more than 40 spacers (Figure 5). We carefully compared the direct repeat sequences (DR) of the different CRISPR loci and found that the DR of each CRISPR locus has no more than 3 SNPs, which are highly conserved. In the remaining strains, we could not identify any complete CRISPR-Cas systems due to incompleteness of the genomes. However, there are cas genes in all of the draft genomes except for strain XZL4 (Table S4), implying that a complete CRISPR-Cas system may exist in XZL9, H-1 and DSM1. Based on a BLAST search of the GenBank database, we found that some CRISPR spacers have homology to sequences from different sources, including phage sequences (CRISPR_2-6_2_S9, CRISPR_2-6_2_S43 and CRISPR_36d1_2_S10) and plasmids (CRISPR_36d1_2_S3 and CRISPR_36d1_2_S10) (Data S3). Moreover, these two strains (2-6 and 36D1) share some common spacers (>90% identity), whereas the other spacers have no homology in GenBank. However, some researches have recently suggested that yet unidentified spacers might mediate the interaction between CRISPR and the bacteriophage or the environment50. The results of IslandViewer indicate that the CRISPRs-Cas systems are located in genomic islands in both strains (2-6 and 36D1; Table S5) and they are flanked by transposases (Figure 5). The GC contents of the CRISPR-Cas systems are approximately 34.0% and 32.6%, whereas those of the entire genomes are 47.3% and 46.5%, in strains 2-6 and 36D1, respectively. In B. coagulans 2-6, six cas genes were identified downstream of small, host-encoded silencing RNAs, cas1-6. cas3 encodes a large protein with separate helicase and DNase activities, which is an important characteristic for CRISPR-Cas classification51. According to the classification of Makarova et al.51 and cas genes found in different strains, the CRISPR-Cas systems found in the strains 36D1, DSM1 and XZL9 may belong to the typical Type I CRISPR-Cas family, which contain the cas3 gene. However, those of strains 2-6 and H-1 belong to an unclassified CRISPR-Cas family without a cas2 gene.

Overview of the CRISPR-Cas systems in B. coagulans strains 2-6 and 36D1.
(A). Genetic map of the CRISPR-Cas systems detected in two B. coagulans strains (2-6 and 36D1). Cas genes were detected around the CRISPR loci. Different colors show the different CRISPR loci and cas genes: cas1: light purple; cas2: red; cas3: dark red; cas4: purple; cas5: gold; cas6: pink; cas7: green; cas8: orange; CRISPR: blue; IS3: black; IS4: gray; XRE transcriptional regulator: green. (B). Overview of the five CRISPR loci in the two B. coagulans strains. The repeats are shown as gray rectangles and the spacers are shown as white diamonds. Spacers with similar sequences (>90% identity) in the studied genomes are shown as the same color.
Discussion
As good platform chemical producers, B. coagulans strains have many of the necessary characteristics required to meet the needs of white biotechnology. High-throughput sequence technology and comparative genomic analysis have provided us with a full landscape of central carbon metabolism. In particular, highly efficient sugar metabolism pathways are the genetic foundation for high lactic acid yield. This may be because the genomes of B. coagulans strains have been shaped by the evolutional history. For example, to obtain a competitive advantage B. coagulans strains have designed a very efficient metabolism pathway (EMP and PPP) to produce high concentrations of lactic acid from various substrates as a means to inhibit the growth of other microorganisms. These pathways, which produce more by-products, have less selection pressure. In addition, the key enzymes in these pathways, such as phosphoketolase in the PKP, may easily lose their function. Besides producing lactic acid, B. coagulans strains also produce various other platform bio-chemicals, such as acetoin and butanediol. Considering their highly efficient sugar metabolism, if carbon flux is redirected towards the acetoin-butanediol pathway instead of the lactic acid pathway by knocking out l-lactate dehydrogenase, B. coagulans strains could become very good producers of these useful platform bio-chemicals from renewable resources20,23.
Genome-wide positive selection analysis led us to examine the relationship between amino acid metabolism and thermotolerance. In previous studies52,53, B. coagulans strains required additional nutrients, such as amino acids, to maintain their rapid growth during high-temperature fermentation. The research of Marshall and Beers showed that B. coagulans strains require different nutritional supplements at different temperatures54. Two cases could lead to such results: (i) the cell requires more nutrients to make up for proteins that are denatured by heat; (ii) the enzyme has less activity at higher temperature. These hypotheses need to be validated in further studies. However, the genes, which are under strong positive selection pressure and play a key role in amino acid metabolism, may also be very important in thermal adaptation. The information above may provide some clues for the further studies aimed at reducing the requirements for expensive additional nutrients.
The CRISPR-Cas and R-M systems have shown to work together to protect bacteria against invaders such as phage and plasmids47. These defense systems are a double-edged sword. On one hand, they keep foreign DNA from being incorporated into the cells. On the other hand, these systems also limit the genetic engineering of these strains for the production of other useful chemicals. Many researches have tried to develop highly efficient general genetic engineering systems. For example, Rhee et al.55 developed an electroporation method to transfer plasmid DNA into B. coagulans strains. In addtion, they also constructed a B. coagulans/E. coli shuttle vector that contains the rep region from a native plasmid of B. coagulans strain P4-102B. Wang et al.23 built a temperature sensitive plasmid to delete the native ldh and alsS (encoding acetolactate synthase) genes of strain P4-102B. The engineered bacteria can be used to produce either l- or d-lactic acid, respectively, at high titers and yields from nonfood carbohydrates. Kovacs et al.13 and van Kranenburg et al.19 also developed a targeted gene disruption system using pSH71 replicon. Moreover, in the research of Kovacs13, they have successfully applied the widely used Cre-lox system for genomic modifications and removal of selectable genes. However, highly efficient genetic tools of B. coagulans are still not currently available, which limits their potential as a next-generation production platform for building block chemicals or biofuels from renewable resources13. As mentioned above, the CRISPR-Cas systems can keep foreign genetic material from B. coagulans strains. A full understanding of the diverse spacers in the CRISPR-Cas immune system could provide us with useful suggestions for modifying the current genetic tools to expand their host range13. R-M systems act to protect the strains against invading DNA48. Exogenous DNA with foreign methylation patterns are recognized and rapidly degraded47. Zhang et al.48 described a new pipeline that could potentially use as a universal genetic engineering tool, to overcome the problem of multiple RM systems. Another very important feature of a highly efficient genetic tool is a good plasmid origin. However, with the limited knowledge of B. coagulans, we could not find any high efficient plasmid origin. As more sequence data are obtained, new information and materials may be available to improve genetic engineering tools to meet the requirements of commercial applications.
In summary, we examined the genomes of six B. coagulans strains in an attempt to explain its favorable fermentation features. Its rapid and efficient carbon metabolism may contribute to the efficient production of platform bio-chemicals. The ability to ferment at high temperature and their encoded immune systems could protect the B. coagulans strains from phage infection and contamination. It is suggested that these specific features could be attributed to utility of these B. coagulans strains as excellent industrial strains.
Methods
Genome sequencing
Four newly isolated Bacillus coagulans strains (2-6, H-1, XZL9 and XZL4) were identified by 16S rDNA sequencing, morphology and physiological analysis. The genomic DNA of these four strains and the type strain of B. coagulans DSM1 from DSMZ were extracted using the Wizard Genomic DNA Purification Kit (Promega, USA). Whole-genomes of these five B. coagulans strains were sequenced by Chinese National Human Genome Center at Shanghai, China. The pair-end reads were assembled de novo using the program Velvet with manually optimized settings. The Phred/Phrap/Consed package was used to finish genomes. To fill the gaps among the de novo assembled contigs in B. coagulans 2-6, we followed the method of the reference-guided mapping56. The genomes of 2-6, XZL4, XZL9, DSM1 and H-1 were submitted to the web service RAST for automatic annotation followed by manual checking. The annotation of these genomes can be publicly obtained at the RAST website with a guest account. The genome sequence of B. coagulans 36D1 was downloaded from GenBank. We use the PATRIC, which is the NIAID/PathoSystems Resource Integration Center and RAST for comparative genomic and metabolic pathways analysis. The IslandViewer was used to detect genomic islands (GIs) in the genomes. GIs that were predicted at least by one method (IslandPick, SIGI-HMM or IslandPath-DIMOB), were accepted. The CRISPR/Cas systems were identified with CRISPR Finder. The Restriction-Modification systems were predicted based on the data of REBASE57. The genomic context was visualized performed by using Circos.
Phylogenetic analysis
Orthologous relationships between protein-coding sequences in the genomes were determined by using OrthoMCL, with the following criteria: identity > 50% and e-value < 1e-5. Single-copy orthologs common in all genomes were used to construct genome-scale phylogenetic tree. Briefly, individual orthologs were aligned by using MUSCLE, back translated to DNA sequences by using ad hoc Perl scripts similar to the strategy of PAL2NAL58 and concatenated to obtain a “chromosomal” alignment. The best fitting model of sequence evolution was determined using jModelTest2 with 11 substitution schemes. Model selection was computed using the Akaike information criterion (AIC). The phylogenetic tree was constructed with PHYML under GTR + gamma + I model according to the result of jModelTest2.
Molecular evolutionary analysis
We performed a positive selection analysis with PSP59, which is a web tool designed for calculating the selection pressure across multiply closely related genomes (http://db-mml.sjtu.edu.cn/PSP/ or http://202.120.45.186/~webserver/kaks/). Using the branch-site strain-specific model, we analyzed the positive selection pressure across 26 Bacillus strains with B. coagulans as “foreground branches” (Table S1). To determine the level of significance for the LRTs, we calculated the P-value using a Χ2 distribution, with the number of degrees of freedom corresponding to the difference of parameters between the nested models. We used the conservative BEB approach to calculate the posterior probabilities of a specific codon site and to identify those with higher probabilities for being under diversifying selection.
Accession numbers
The genome sequences of B. coagulans 2-6, XZL4, XZL9, DSM1 and H-1 were deposited in NCBI database under the accession number CP002472, AFWM00000000, ANAP00000000, ALAS01000000 and ANAQ00000000, respectively.
References
Lorenz, P. & Zinke, H. White biotechnology: differences in US and EU approaches? Trends Biotechnol. 23, 570–574 (2005).
Gao, C., Ma, C. & Xu, P. Biotechnological routes based on lactic acid production from biomass. Biotechnol. Adv. 29, 930–939 (2011).
Teusink, B. & Smid, E. Modelling strategies for the industrial exploitation of lactic acid bacteria. Nat. Rev. Microbiol. 4, 46–56 (2006).
Qin, J. et al. Non-sterilized fermentative production of polymer-grade l-lactic acid by a newly isolated thermophilic strain Bacillus sp. 2-6. PLoS One 4, e4359 (2009).
Zhao, B. et al. Repeated open fermentative production of optically pure l-lactic acid using a thermophilic Bacillus sp. strain. Bioresour. Technol. 101, 6494–6498 (2010).
Wang, L. et al. Efficient production of l-lactic acid from corncob molasses, a waste by-product in xylitol production, by a newly isolated xylose utilizing Bacillus sp. strain. Bioresour. Technol. 101, 7908–7915 (2010).
Patel, M. A. et al. Isolation and characterization of acid-tolerant, thermophilic bacteria for effective fermentation of biomass-derived sugars to lactic acid. Appl. Environ. Microbiol. 72, 3228–3235 (2006).
Kanwar, S. S., Kaushal, R. K., Sultana, H. & Chimni, S. S. Purification of a moderate thermotolerant Bacillus coagulans BTS1 lipase and its properties in a hydro-gel system. Acta Microbiol. Immunol. Hung. 53, 77–87 (2006).
Nakamura, L., Blumenstock, I. & Claus, D. Taxonomic Study of Bacillus coagulans Hammer 1915 with a proposal for Bacillus smithii sp. nov. Int. J. Syst. Evol. Microbiol. 38, 63–73 (1988).
Patel, M. A., Ou, M. S., Ingram, L. O. & Shanmugam, K. T. Simultaneous saccharification and co-fermentation of crystalline cellulose and sugar cane bagasse hemicellulose hydrolysate to lactate by a thermotolerant acidophilic Bacillus sp. Biotechnol. Prog. 21, 1453–1460 (2005).
Le Marrec, C., Hyronimus, B., Bressollier, P., Verneuil, B. & Urdaci, M. C. Biochemical and genetic characterization of coagulin, a new antilisterial bacteriocin in the pediocin family of bacteriocins, produced by Bacillus coagulans I4 . Appl. Environ. Microbiol. 66, 5213–5220 (2000).
Endres, J. R. et al. Safety assessment of a proprietary preparation of a novel Probiotic, Bacillus coagulans, as a food ingredient. Food Chem. Toxicol. 47, 1231–1238 (2009).
Kovacs, A. T., van Hartskamp, M., Kuipers, O. P. & van Kranenburg, R. Genetic tool development for a new host for biotechnology, the thermotolerant bacterium Bacillus coagulans. Appl. Environ. Microbiol. 76, 4085–4088 (2010).
Xu, K. et al. Genome sequences of two morphologically distinct and thermophilic Bacillus coagulans strains, H-1 and XZL9. Genome Announc. 1, e00254–13 (2013).
Su, F., Tao, F., Tang, H. & Xu, P. Genome sequence of the thermophile Bacillus coagulans Hammer, the type strain of the species. J. Bacteriol. 194, 6294–6295 (2012).
Su, F. et al. Genome sequence of the thermophilic strain Bacillus coagulans 2-6, an efficient producer of high-optical-purity l-lactic acid. J. Bacteriol. 193, 4563–4564 (2011).
Su, F. et al. Genome sequence of the thermophilic strain Bacillus coagulans XZL4, an efficient pentose-utilizing producer of chemicals. J. Bacteriol. 193, 6398–6399 (2011).
Rhee, M. S. et al. Complete genome sequence of a thermotolerant sporogenic lactic acid bacterium, Bacillus coagulans strain 36D1. Stand. Genomic Sci. 5, 331–340 (2011).
Van Kranenburg, R. et al. Genetic modification of homolactic thermophilic Bacilli., WO Patent 2,007,085,443 (2007).
Su, Y., Rhee, M. S., Ingram, L. O. & Shanmugam, K. Physiological and fermentation properties of Bacillus coagulans and a mutant lacking fermentative lactate dehydrogenase activity. J. Ind. Microbiol. Biotechnol. 38, 441–450 (2011).
Holton, S. J., Anandhakrishnan, M., Geerlof, A. & Wilmanns, M. Structural characterization of a d-isomer specific 2-hydroxyacid dehydrogenase from Lactobacillus delbrueckii ssp. bulgaricus. J. Struct. Biol. 181, 179–184 (2013).
Razeto, A. et al. Domain closure, substrate specificity and catalysis of d-lactate dehydrogenase from Lactobacillus bulgaricus. J. Mol. Biol. 318, 109–119 (2002).
Wang, Q., Ingram, L. O. & Shanmugam, K. T. Evolution of d-lactate dehydrogenase activity from glycerol dehydrogenase and its utility for d-lactate production from lignocellulose. Proc. Natl. Acad. Sci. USA 108, 18920–18925 (2011).
Xiao, Z. & Xu, P. Acetoin metabolism in bacteria. Crit. Rev. Microbiol. 33, 127–140 (2007).
De Clerck, E. et al. Polyphasic characterization of Bacillus coagulans strains, illustrating heterogeneity within this species and emended description of the species. Syst. Appl. Microbiol. 27, 50–60 (2004).
Gu, Y. et al. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genomics 11, 255–269 (2010).
Bhosale, S. H., Rao, M. B. & Deshpande, V. V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 60, 280–300 (1996).
Park, J. H. & Batt, C. A. Restoration of a defective Lactococcus lactis xylose isomerase. Appl. Environ. Microbiol. 70, 4318–4325 (2004).
Schmiedel, D., Kintrup, M., Kuster, E. & Hillen, W. Regulation of expression, genetic organization and substrate specificity of xylose uptake in Bacillus megaterium. Mol. Microbiol. 23, 1053–1062 (1997).
Chaillou, S., Bor, Y. C., Batt, C. A., Postma, P. W. & Pouwels, P. H. Molecular cloning and functional expression in lactobacillus plantarum 80 of xylT, encoding the D-xylose-H+ symporter of Lactobacillus brevis. Appl. Environ. Microbiol. 64, 4720–4728 (1998).
Lin, E. Dissimilatory pathways for sugars, polyols and carboxylates. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC, 307–342 (1996).
Tjalsma, H. et al. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68, 207–233 (2004).
van Dijl, J. M. et al. Functional genomic analysis of the Bacillus subtilis Tat pathway for protein secretion. J. Biotechnol. 98, 243–254 (2002).
Hamoen, L. W., Venema, G. & Kuipers, O. P. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology 149, 9–17 (2003).
Dubnau, D. DNA uptake in bacteria. Ann. Rev. Microbiol. 53, 217–244 (1999).
Kovacs, A. T., Eckhardt, T. H., van Kranenburg, R. & Kuipers, O. P. Functional analysis of the ComK protein of Bacillus coagulans. PLoS One 8, e53471 (2013).
Caspi, R. et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 36, D623–631 (2008).
Hebbeln, P., Rodionov, D. A., Alfandega, A. & Eitinger, T. Biotin uptake in prokaryotes by solute transporters with an optional ATP-binding cassette-containing module. Proc. Natl. Acad. Sci. USA 104, 2909–2914 (2007).
Petersen, L., Bollback, J. P., Dimmic, M., Hubisz, M. & Nielsen, R. Genes under positive selection in Escherichia coli. Genome Res. 17, 1336–1343 (2007).
Valentine, D. L. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat. Rev. Microbiol. 5, 316–323 (2007).
Chen, S. L. et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl. Acad. Sci. USA 103, 5977–5982 (2006).
Calogero, S. et al. RocR, a novel regulatory protein controlling arginine utilization in Bacillus subtilis, belongs to the NtrC/NifA family of transcriptional activators. J. Bacteriol. 176, 1234–1241 (1994).
Voigt, B. et al. The response of Bacilluslicheniformis to heat and ethanol stress and the role of the SigB regulon. Proteomics 13, 2140–2161 (2013).
Joseph, P. et al. Targeting of the Brucella suis virulence factor histidinol dehydrogenase by histidinol analogues results in inhibition of intramacrophagic multiplication of the pathogen. Antimicrob. Agents Chemother. 51, 3752–3755 (2007).
Lan, E. I. & Liao, J. C. Metabolic engineering of cyanobacteria for 1-butanol production from carbon dioxide. Metab. Eng. 13, 353–363 (2011).
Klaenhammer, T. R. Plasmid-directed mechanisms for bacteriophage defense in lactic streptococci. FEMS Microbiol. Lett. 46, 313–325 (1987).
Dupuis, M. E., Villion, M., Magadan, A. H. & Moineau, S. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat. Commun. 4, 2087–2094 (2013).
Zhang, G. et al. A mimicking-of-DNA-methylation-patterns pipeline for overcoming the restriction barrier of bacteria. PLoS Genet. 8, e1002987 (2012).
Horvath, P. et al. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int. J. Food Microbiol. 131, 62–70 (2009).
Cady, K. C. & O'Toole, G. A. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol. 193, 3433–3445 (2011).
Makarova, K. S. et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477 (2011).
Cleverdon, R. C., Pelczar, M. J. & Doetsch, R. N. The vitamin requirements of stenothermophilic aerobic sporogenous Bacilli. J. Bacteriol. 58, 523–526 (1949).
Campbell, L. L. & Sniff, E. E. Folic acid requirement of Bacillus coagulans. J. Bacteriol. 78, 267–271 (1959).
Marshall, R. & Beers, R. J. Growth of Bacillus coagulans in chemically defined media. J. Bacteriol. 94, 517–521 (1967).
Rhee, M. S., Kim, J. W., Qian, Y., Ingram, L. O. & Shanmugam, K. T. Development of plasmid vector and electroporation condition for gene transfer in sporogenic lactic acid bacterium, Bacillus coagulans. Plasmid 58, 13–22 (2007).
Nishito, Y. et al. Whole genome assembly of a natto production strain Bacillus subtilis natto from very short read data. BMC Genomics 11, 243–255 (2010).
Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 38, D234–236 (2010).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–612 (2006).
Su, F. et al. PSP: rapid identification of orthologous coding genes under positive selection across multiple closely related prokaryotic genomes. BMC genomics 14, 924 (2013).
Acknowledgements
The authors acknowledge the National Basic Research Program of China (2013CB733901) from Ministry of Science and Technology of China and the grant from National Natural Science Foundation of China (31121064). This work was partially supported by the Chinese National Program for High Technology Research and Development (2011AA02A202).
Author information
Authors and Affiliations
Contributions
F.S. and P.X. conceived and designed the project. P.X. contributed reagents and materials. F.S. analyzed data. F.S. and P.X. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Dataset S1
Supplementary Information
Dataset S2
Supplementary Information
Dataset S3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Su, F., Xu, P. Genomic analysis of thermophilic Bacillus coagulans strains: efficient producers for platform bio-chemicals. Sci Rep 4, 3926 (2014). https://doi.org/10.1038/srep03926
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03926
This article is cited by
-
In vitro assessment of probiotic attributes for strains contained in commercial formulations
Scientific Reports (2022)
-
Prebiotic properties of Bacillus coagulans MA-13: production of galactoside hydrolyzing enzymes and characterization of the transglycosylation properties of a GH42 β-galactosidase
Microbial Cell Factories (2021)
-
A cold shock protein promotes high-temperature microbial growth through binding to diverse RNA species
Cell Discovery (2021)
-
Complete genome sequence of thermophilic Bacillus smithii type strain DSM 4216T
Standards in Genomic Sciences (2016)
-
Genomic analysis of a xylose operon and characterization of novel xylose isomerase and xylulokinase from Bacillus coagulans NL01
Biotechnology Letters (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
