
Abstract
While somatic DNA copy number variations (CNVs) have been identified in multiple tissues from normal people, they have not been well studied in brain tissues from individuals with psychiatric disorders. With ultrahigh depth sequencing data, we developed an integrated pipeline for calling somatic deletions using data from multiple tissues of the same individual or a single tissue type taken from multiple individuals. Using the pipelines, we identified 106 somatic deletions in DNA from prefrontal cortex (PFC) and/or cerebellum of two normal controls subjects and/or three individuals with schizophrenia. We then validated somatic deletions in 18 genic and in 1 intergenic region. Somatic deletions in BOD1 and CBX3 were reconfirmed using DNA isolated from non-pyramidal neurons and from cells in white matter using laser capture microdissection (LCM). Our results suggest that somatic deletions may affect metabolic processes and brain development in a region specific manner.
Similar content being viewed by others


Introduction
Except for some immune cells, it is generally believed that the DNA sequence and structure is the same in all normal cells within an individual. The adult human body goes through numerous rounds of cell division and DNA replication to reach approximately 1014 cells. Therefore, it may be expected that a substantial number of somatic mutations occur in tissues according to the mutation rate in the DNA replication system. Several recent studies provide evidence for this in healthy people e.g. somatic DNA copy number variations (CNV) occur in multiple tissues1,2, age-associated CNVs occur in blood cells3 and somatic retrotransposition occurs in the brain4. Any somatic variations, theoretically, can be involved in developmental processes and in generating complexity and diversity of cellular function. Such variation has been suggested as one of the mechanisms that may underlie the functional diversity of brain cells among normal people4,5.
A causal relation between somatic genome variation and complex diseases such as neuropsychiatric disorders have long been of interest5. Previous studies have revealed low level mosaic aneuploidy of chromosome 1, 18 and X in the brain of individuals with schizophrenia and a somatic mutation in AKT3 has been identified in a brain with Hemimegalencephaly (HMG)6,7. Moreover, somatic CNVs have been identified in monozygote twins, both concordant and discordant for Parkinson disease and indicates that somatic variations may occur in the same zygote8.
Numerous neuropathological abnormalities have been described in various brain regions of individuals with schizophrenia9,10 and include a reduction in the density of a subset of GABAergic neurons11 and of perineuronal oligodendrocytes12 in the PFC of individuals with schizophrenia as compared to unaffected controls. Furthermore, these abnormalities have been associated with biological processes related to nervous system development and apoptosis13. It is possible that these cell specific abnormalities are due to region specific somatic variations that occur in DNA of specific brain cells in individuals with schizophrenia. However, somatic variations in brain cells have not been well studied due to the technical limitations. Identifying somatic CNVs that occur in only a subset of cells from a complex tissue with mixed cell types is very challenging. In this study we first determined if we could identify somatic deletions by examining whole genome sequencing (WGS) data from two brain regions, prefrontal cortex (PFC) and cerebellum, from one individual with schizophrenia using blood as a reference tissue. By laser capture microdissection we determined which cell type in the brain harbored the somatic deletions. In the second phase of the study we identified and replicated somatic deletions in the PFC from two unaffected controls and two additional individuals with schizophrenia. To reliably call somatic deletions we sequenced the whole genomes at ultrahigh depth and then applied stringent filters for the variant using several different algorithms, including read depth based analysis14, paired end mapping15 and breakpoint mapping16. All these methods have been used successfully for CNV calling in WGS data.
Results
Identifying germline CNVs using sequencing data from three tissues of an individual with schizophrenia
In the discovery phase, we sequenced the whole genome from two brain areas and blood of a female patient with schizophrenia at ultrahigh depth (Case A9; Supplementary Table S1). The depth of coverage of WGS reads were 74×, 85× and 67× for PFC, cerebellum and blood respectively. The read depth of blood DNA was lower because the data was used as a reference for filtering out germline deletions within PFC or cerebellum. Germline CNVs were called using read depth analysis14 and paired end mapping15 (Supplementary Fig. S1). We identified 343 germline duplications, including 6 novel duplications that do not overlap with more than 50% of the genome locus of previously reported CNV regions in the Database of Genomic Variants (DGV, http://projects.tcag.ca/variation/)17. We also identified 405 germline deletions, including 14 novel deletions. We attempted to validate 4 germline deletions and the breakpoints of the deletions that disrupted the 4 known annotated genes; protein phosphatase 2, regulatory subunit B, gamma (PPP2R2C), anillin, actin binding protein (ANLN), MYC associate factor X (MAX) and type 1 insulin-like growth factor receptor (IGF1R) using PCR amplification and Sanger sequencing. The four germline deletions were verified in all three tissues. The read depth analysis showed a homozygote deletion in ANLN and heterozygote deletions in MAX, PPP2R2C and IGF1R (Supplementary Fig. S2). None of the germline CNVs from this schizophrenia case overlapped with previously identified CNV regions associated with schizophrenia18,19,20,21,22,23,24,25.
Exploratory calling of somatic CNVs using read depth based mapping
Tissue specific CNVs were previously detected by quantitatively comparing genomic DNA in various normal tissues1,2. Therefore, we called somatic CNV candidates specific to brain tissues, PFC and cerebellum, in the schizophrenia case A9, using a read depth based mapping method. Eleven somatic duplication candidates specific to PFC and 10 specific to cerebellum were called. Sixty-three somatic deletion candidates specific to cerebellum were also called. We attempted to validate a total of 6 brain specific CNVs using quantitative (q) PCR (Supplementary Fig. S3). Five candidates were unable to be validated. The amount of DNA detected for the two somatic duplications specific to PFC were changed in the opposite direction to that expected for a duplication (Supplementary Fig. S3a). While the amount of DNA detected for three of the cerebellum specific somatic CNV candidates was changed in the appropriate direction there was no quantitative difference in the amount of DNA between the PFC and the cerebellum (Supplementary Fig. S3b, c) and thus they could not be validated as cerebellum specific. One cerebellum specific somatic deletion candidate in the C3P1 gene was validated using qPCR. However, we were unable to map the breakpoint and confirm it as a cerebellum specific deletion. Thus, the validation results suggest that the somatic CNV calling process based on read depth mapping alone called many false positives and required that we develop a more rigorous integrated somatic deletion calling pipeline. Moreover, the subtle changes in the amount of DNA which contain somatic CNV candidate regions indicates that a majority of somatic CNVs may occur only in a small fraction of cells within the brain regions.
Discovery of somatic deletions specific to brain tissues using an integrated somatic deletion calling pipeline
Genomic variations can be called more reliably by using an integrated pipeline of multiple variant calling algorithms than a method using a single algorithm in WGS data26,27. Thus, we developed an integrated somatic DNA deletion calling pipeline for multiple tissue sequencing data from the same individual (Fig. 1). While this works well for calling somatic deletions, we were unable to call somatic duplications because the current algorithms cannot reliably distinguish somatic duplications which occur in only a fraction of the cells in a tissue. We called 7 somatic deletions specific to PFC, 3 specific to cerebellum and 10 common to both PFC and cerebellum in case A9 using the pipeline (Supplementary Table S2). We also called 12 somatic deletions in blood DNA (Supplementary Table S3). We then validated 1 PFC specific deletion and 3 somatic deletions with different breakpoints in the PFC and cerebellum (Table 1, Supplementary Table S4). The 500 bp somatic deletion which disrupts the protein kinase interferon-inducible double stranded RNA dependent activator (PRKRA) gene and MIR548N occurred only in DNA from PFC and not in DNA from cerebellum or blood of this case A9 (Table 1). We found different sized somatic deletions in the coding regions of two genes; biorientation of chromosomes in cell division 1 (BOD1) and chromobox homolog 3 (CBX3) that occurred in DNA from PFC and cerebellum (Table 1). Unlike the germline deletions, the read depth analysis indicated that these deletions appear to occur in only a fraction of cells in the brain (Supplementary Fig. S4) as may be expected. We used whole genome amplified DNA for our validation as limited amounts of DNA were available from the same batch of extractions. To determine if the whole genome amplification could cause a difference in the validation results or not, we conducted PCR amplification with breakpoint specific primers using unamplified chromosomal DNA as a template (Fig. 2, Supplementary Fig. S5). Amplifying the specific DNA fragment in PFC only, reconfirmed the PFC specific deletion as well as proved there is no difference between results when using amplified or unamplified chromosomal DNA for validation (Fig. 2, Supplementary Fig. S5).

Procedures for calling somatic deletions in whole genome sequencing data from multiple tissues from one individual or from a single tissue from multiple individuals.
* All deletion candidates and selected candidates (read count ≤6) used for downstream filtering in sequencing data from multiple tissues and single tissue respectively.

Validation of somatic deletions in brain DNA of an individual with schizophrenia.
(a), PFC specific deletion in PRKRA and annotated genes were visualized using the UCSC genome browser. (b), 844 bp DNA fragment was amplified by nested PCR using amplified DNA from PFC as template. (c), The 1309 bp DNA fragment was amplified by first round PCR with nested primers using unamplified DNA from all three tissues as templates (top). The 299 bp somatic deletion specific DNA fragment was amplified with breakpoint specific primers using unamplified DNA from PFC only as template (bottom). (d). Validation of breakpoints in PFC DNA by Sanger sequencing of 845 bp DNA fragment amplified by nested PCR amplification. NC: no template control, PFC: prefrontal cortex, Cere: cerebellum. Gel images are cropped to highlight relevant bands and images of original full gels are presented in Supplementary Figure S5.
Validation of somatic deletions using DNA from cells isolated by laser capture microdissection
We then revalidated the somatic deletions in the BOD1 gene using an independent method (Fig. 3a). A 908 bp and a 1303 bp somatic deletion were validated in the BOD1 gene in the DNA from cerebellum and PFC respectively (Fig. 3b–d, Supplementary Fig. S6). PCR validation was then performed using DNA from cells isolated by laser capture microdissection (LCM) to reconfirm the PFC specific deletions and to determine what types of brain cells may harbor the somatic deletions. Ten cells from each type; pyramidal neuron, non-pyramidal neuron or white matter cells, were collected per cap from PFC sections (Fig. 3e). DNA was extracted from 10 caps per cell type. A 1577 bp wild-type DNA fragment from the BOD1 gene was amplified in DNA from 5 pyramidal neuron caps, from 1 non-pyramidal neuron cap and from 6 white matter cell caps by PCR with primers localized to the BOD1 somatic deletion region. The wild-type DNA fragment was not amplified in DNA from 11 caps out of 30 caps, indicating the overall locus dropout rate of the chromosomal region is approximately 60% during this process. Somatic deletions in BOD1 were reconfirmed in DNA from non-pyramidal cells and from white matter cells (Fig. 3f–h, Table 1, Supplementary Fig. S6). The 1303 bp somatic deletion found in BOD1 in DNA from white matter cells (Fig. 3h, Supplementary Fig. S6) has identical breakpoints to those found in our previously validated deletion using DNA from PFC (Fig. 3d). Moreover, we identified a novel 1451 bp somatic deletion in the same region in DNA from non-pyramidal cells (Fig. 3f and g, Supplementary Fig. S6). We did not validate the somatic deletion in pyramidal cells. We also revalidated a somatic deletion in CBX3 in DNA from white matter cells of the PFC (Table 1).

Revalidation of a somatic deletion in PFC of an individual with schizophrenia using cells isolated by laser capture microdissection.
(a), PFC specific deletions in BOD1 and annotated coding regions were visualized using the UCSC genome browser. (b), 275 bp and 685 bp DNA fragment were amplified by nested PCR using DNA from PFC and cerebellum as templates respectively. (c), Validation of breakpoints of somatic deletion in cerebellum DNA (685 bp fragment) by Sanger sequencing. (d). Validation of breakpoints of somatic deletion in PFC DNA (275 bp fragment) by Sanger sequencing (e). Microscopic images showing a pyramidal neuron, a non-pyramidal cell and a cell in white matter in PFC after firing laser. (f). 143 bp and 275 bp DNA fragment were amplified by nested PCR using DNA from non-pyramidal cells and cells in white matter as templates respectively. (g), Validation of breakpoints of somatic deletion in non-pyramidal cells (143 bp fragment) by Sanger sequencing. (h), Validation of breakpoints of somatic deletion in cells in white matter (275 bp fragment) by Sanger sequencing. NC: no template control, PFC: prefrontal cortex, Cere: cerebellum, BP: break point, Ins: insertion, non-Py; non-pyramidal cells, WM; cells in white matter. Gel images are cropped to highlight relevant bands (images of entire original gels are presented in Supplementary Figure S6).
Further identification of somatic deletions in brain from additional schizophrenia cases and unaffected controls
To determine if our somatic deletion findings in PFC were specific to individuals with schizophrenia or common to PFC in general we completed whole genome sequencing of PFC DNA from two additional schizophrenia cases and two unaffected controls (Supplementary Table S1). We called 640 and 646 germline deletions and 909 and 804 germline duplications in PFC of the two individuals with schizophrenia, respectively (Supplementary Fig. S7). Similarly we called 688 and 673 germline deletions and 818 and 823 germline duplications in PFC of the two unaffected controls respectively (Supplementary Fig. S7). While the germline CNVs have a global effect on many biological processes (Supplementary Fig. S8), there was no overlap between the germline CNVs from these schizophrenia cases and the previously identified rare CNVs associated with schizophrenia18,19,20,21,22,23,24,25. We then modified the integrated somatic deletion calling pipeline that we used for multiple tissue sequencing data from the same individual, to call somatic DNA deletions in data from single tissue sequencing without reference data (Fig. 1). To examine the performance and detection power of the pipeline, we attempted to call somatic DNA deletions using only PFC sequencing data from the case A9. A total of 16 somatic deletions candidates were detected - including 10 candidates that were specific to PFC or common to PFC and cerebellum that were called when we used the pipeline for multiple tissue data (Supplementary Table S5). Furthermore, one newly called candidate in MRPL42 was successfully validated (Table 1). These results suggest that the somatic deletion calling pipeline for single tissue data is as robust as the pipeline for multiple tissue data.
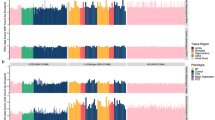
Using the pipeline for single tissue data, we then identified 29, 18, 15 and 18 somatic deletion candidates in the PFC of the two unaffected controls and the two schizophrenia cases respectively (Fig. 4a, Supplementary Table S6). Approximately 50% of the somatic deletions disrupted genes while the remaining deletions were localized in genic regions (Fig. 4a). There was no significant difference in the number of somatic deletions between the schizophrenia cases and unaffected controls. We successfully confirmed 8 somatic deletions; one intergenic deletion and 7 deletions that disrupted genes, including BCL2 associated transcription factor 1 (BCLAF1), thymine-DNA glycosylase (TDG) and succinate-CoA ligase, GDP forming, beta subunit (SUCLG2) (FDR = 0.1) (Table 1 and Fig. 5, Supplementary Fig. S9). Moreover, somatic deletions in two genes, CBX3 and PRKRA, which were validated in the initial schizophrenia case A9, were also confirmed in an unaffected individual (CBX3) and in an additional schizophrenia case (PRKRA). This suggests that chromosomal regions in these genes may be hot spots for somatic deletions in brain DNA.

Total number of somatic deletions in PFC of two unaffected controls and two schizophrenia cases and the biological processes associated with somatic deletions in the schizophrenia and unaffected controls.
(a), Number of somatic deletions in genic and intergenic chromosomal regions in PFC. (B), Biological processes related to genes disrupted by somatic DNA deletion candidates in the PFC. Classification of the Gene Ontology biological processes was done by using Panther software42.

Validation of somatic deletions in PFC of two individuals with schizophrenia and two unaffected controls.
PFC specific somatic deletions in BCLAF1, CBX3, PRKRA, SUCLG2 were confirmed by PCR validation. Two independent somatic deletions in PFC and cerebellum were validated in TDG and TYRO3. *The deletions with the same break points in TDG and intergenic region were validated in PFC and cerebellum. However, the deletions were considered somatic deletions because the read depth analysis indicated there was no clear decline in depth of coverage and deleted fragments were not amplified in our first PCR. Neg: no template control, PFC: prefrontal cortex, Cere: cerebellum. Gel images are cropped to highlight relevant bands (images of entire original gels are presented in Supplementary Figure S9).
Simulation to validate methodology
To determine the false positive rate and the false negative rate of our integrated deletion calling pipelines, we generated simulated whole genome sequencing data of chromosome 1 from a single tissue that included 100 germline deletions and 100 somatic deletions. The size range of both types of deletions was from 500 bp to 10 kb. The simulated occurrence of somatic deletions was set to 10% of a total cell population of the tissue. Using our integrated pipelines, we detected 96 (96%) of the germline deletions and 78 (78%) of the somatic deletions (Supplementary Fig. S10). There were no false positives in either calling method.
The distribution of the number of supporting read pairs for both the germline and somatic deletions were clearly separated at the threshold (supporting number of read pairs n = 6, see methods) that we set in the pipeline (Supplementary Fig. S11). A germline deletion was called by 2 read pairs (Supplementary Fig. S11). The read depth of the genome of the germline deletion declined approximately 50%, indicating heterogyzote deletion (Supplementary Fig. S12). Conversely, somatic deletions which were called by 2 read pairs did not show a clear decline in read depth (Supplementary Fig. S12). Moreover, the distribution of the number of supporting reads for germline and somatic deletions in Pindel calls were well separated at the threshold that we set (Supplementary Fig. S13). Most germline deletions were called by more than 9 supporting reads but none of the somatic deletions were called by that criterion (Supplementary Fig. S13). Our simulation results indicate that the integrated pipelines can robustly detect germline deletions as well as somatic deletions using whole genome sequencing data from a single tissue.
Functional annotation of genes disrupted by the germline CNVs and the somatic deletions in PFC
To explore the possible effect that somatic deletions may have on brain function we performed a functional annotation analysis of all the genes that we found disrupted by somatic deletions in the two unaffected controls and the two schizophrenia cases. Metabolic process, cell communication, developmental process, immune response and cell cycle were the functions primarily affected by the somatic deletions in the PFC (Fig. 4b). This indicates that somatic deletions may affect brain functions, such as metabolism and immune response, in a region specific manner and may also contribute to the functional diversity of specific subtypes of brain cells in an individual.
We further analyzed the biological processes that were significantly associated with somatic deletions in schizophrenia and controls independantly. While there was no biological process significantly over-represented in the genes disrupted by somatic deletions in the PFC of unaffected controls, a total of 7 biological processes were significantly over-represented by the genes in the two schizophrenia cases (FDR < 0.05, Supplementary Table S7). However, the genes related to the processes were linked on chromosome 11 and were disrupted by one large deletion, which indicates possible bias in the result. A larger sample size will be necessary for future studies to reliably identify biological processes associated with somatic deletions in schizophrenia.
Discussion
Somatic mutations may contribute to neuronal diversity in the normal population and may also pose a risk factor for neuropsychiatric diseases28,29. Previous studies have detected somatic CNVs in multiple human tissues, including brain, by comparing the quantitative amount of DNA between two tissues from the same individual1,2. The recent implementation of massively parallel sequencing techniques with chip-based enrichment4, stem cell techniques30 and whole genome amplification of single cells31 have provided further evidence for somatic variation in human tissues. Previous studies of individuals with HMG identified somatic mutations in 8–40% of sequenced alleles within the affected brain regions6,7. Because the somatic mutation alleles are present in only 8–40% of the sequenced alleles, even in the diseased brain regions of individuals with HMG, the somatic variations are very likely to occur in only a small fraction of brain cells in people with schizophrenia and unaffected controls1,2. A recent somatic CNV study also shows that large somatic CNVs occurred in 13 to 41% of neurons in post-mortem frontal cortex neurons32. In this study, we focused on somatic DNA deletions which are also likely to occur in a small fraction of brain cells (less than 25%). We developed an integrated pipeline for calling somatic deletions using ultrahigh depth sequencing data from multiple tissues from a single individual or from a single tissue type from multiple individuals. The advantage of our pipeline is that somatic deletions are efficiently called using WGS data of tissue by increasing read depth and without introducing additional confounds such as inducing stem cells, using chip-based enrichment or single cell isolation. Moreover, our somatic deletion calling pipeline for single tissue sequencing data can detect somatic DNA deletions without any reference sequencing data. In our validation experiment, we obtained robust results using the somatic calling pipeline (FDR = 0.1, Supplementary Table S4). Moreover, our simulation results showed that the integrated pipelines called somatic deletions at high sensitivity (78%) without any false positives. This indicates that the integrated somatic calling method can be used to detect somatic deletions using WGS data from various tissues without any reference sequencing data derived from the same individual.
Identifying somatic CNVs that occur in only a subset of cells from a complex tissue with mixed cell types is technically challenging. Therefore, robust validation experiments are essential for discovery of somatic CNVs. Non-random DNA sample degradation can lead to false positive CNVs in quantitative PCR33. This may be particularly problematic in the quantitative comparison of target DNA and reference DNA from human post-mortem tissue that is often stored in the freezer for extended periods of time. Thus, we validated a total of 19 somatic deletion candidates by direct sequencing of the breakpoints. Furthermore, since deletion breakpoints are not generated during the in vitro DNA amplification process, the method can be applied to amplified chromosomal DNA.
The PFC develops from the prosencephalon, while the cerebellum is derived from the metencephalon34. The PFC specific deletion in PRKRA (A9, C16), CBX3 (C21) and SUCLG2 (C17) and the different sized deletions in BOD1 (A9), CBX3 (A9), BCLAF1 (C13), TDG (C21) and TYRO (C21) in PFC and cerebellum suggest that these brain region specific somatic deletions may occur independently during or after the developmental stage when the three primary brain vesicles subdivide. Among 10 somatic deletions common to both PFC and cerebellum in case A9 identified in the discovery phase, 9 somatic deletions showed different breakpoints between the two brain regions (Supplementary Table S2). One somatic deletion common to both brain regions in the BOD1 gene was originally called as a cerebellum specific somatic deletion but additional somatic deletions in PFC were confirmed during the validation experiment (Fig. 3a). Two somatic deletions with the same break points in PFC and cerebellum were also validated, which indicates that some minor somatic deletions may occur in a very early developmental stage. The validated somatic deletions may be generated by nonhomologous end joining (NHEJ)36,35 which suggests that somatic deletions in brain cells may be formed by the same mechanism as germline deletions. Thirteen somatic deletions out of a total of 19 somatic deletions which were validated in this study reciprocally overlap with more than 50% of the genome locus of deletions previously reported in the Database of Genomic Variants. This raised the possibility that some somatic deletions likely occur in hotspot regions where germline deletions also occurred in the general population. However, based on our findings in both the first discovery phase as well as the second phase, there is a low probability that somatic deletions and germline deletions in the general population will share the exact same breakpoints. Our second phase showed that even when comparing the breakpoints of two tissues from the same individual, they often did not share identical CNVs. The somatic deletions that we identified here are unlikely to be caused by the confounding effects of variables such as medications or substance abuse because similar numbers of deletions were found in both the unaffected controls and the schizophrenia cases.
Somatic deletions in BOD1 and CBX3 occurred in non-pyramidal cells and/or cells in white matter but did not occur in pyramidal neurons of the PFC of the schizophrenia case (A9). These results are generally consistent with previous studies regarding somatic variation in the PFC4,31 that found numerous widespread somatic LINE-1 retrotransposons in the DNA from frontal tissues4, but such retrotransposons could not be detected in the DNA from isolated pyramidal neurons in the same brain region31. Thus, the interneurons and glial cells, in both gray and white matter, may be more vulnerable to somatic deletions than pyramidal neurons in the PFC of the schizophrenia cases. Deficits of GABAergic interneurons and oligodendrocytes have been widely reported in previous neuropathology studies in PFC of schizophrenia11,12,38,37. In addition, there is an increase in the density of interstitial white matter neurons (IWMN), which are aberrantly located immature neurons, in the PFC of schizophrenia cases40,39. Our results suggest that somatic variations in the DNA of specific brain cells such as GABAergic interneurons, oligodendrocytes or IWMN could be a novel mechanism to explain some of the pathological abnormalities found in the PFC of schizophrenia cases.
In this study, we identified 106 somatic deletions in DNA from two brain regions, the prefrontal cortex and cerebellum, of two normal controls subjects and three individuals with schizophrenia using an integrated calling pipeline. We then extensively validated somatic deletions in 18 genic and in 1 intergenic region. Our results suggest that somatic deletions may contribute to cellular diversity in both normal and schizophrenia affected brains and may consequently affect metabolic processes and brain development in a region specific manner. The three individuals with schizophrenia, whom we sequenced here, did not carry any germline CNVs previously identified as significantly associated with the disease18,19,20,21,22,23,24,25. Therefore, our results may provide an alternative hypothesis for the pathophysiology of the schizophrenia cases which cannot currently be explained by rare structural variants.
Methods
Brain DNA samples
For the discovery phase, a female case was selected from the Stanley Medical Research Institute (SMRI) Array Collection (AC). The case was diagnosed with schizophrenia, had psychotic symptoms and died from suicide. DNA was extracted from prefrontal cortex (PFC), cerebellum and blood from this case. For the second phase, two individuals with schizophrenia and two unaffected controls were selected from the SMRI Neuropathology Consortium (SNC). DNA was extracted from the PFC of these cases. Demographic and clinical information of each sample are listed in Supplementary Table S1. A detailed description of the selection process, clinical information, diagnoses of patients and processing of tissues has been described previously41. Genomic DNA was extracted from PFC, cerebellum and blood with the Wizard Genomic DNA Purification Kit (Promega) and was further cleaned with the QIAamp DNA kit (Qiagen). The purity and concentration of chromosomal DNA were determined by Nano Drop (NanoDrop Technologies). The DNA concentrations were re-quantified with Quanti-iT Pico Green dsDNA assay (Invitrogen).
Whole genome sequencing and paired-end read alignment
Genomic DNA was sequenced using a combination of Illumina GAIIx and HiSeq2000 instruments following the manufacturer's standard protocols. The detailed whole genome sequencing and paired-end read alignment are described in the Supplementary Methods.
Calling germline copy number variations and somatic deletions
Germline CNVs were called using read depth analysis14 and paired end mapping15 as outlined in Supplementary Fig. S1. We called a germline deletion if a deletion was detected using BreakDancer15 (paired end mapping) and CNVnator14 (read depth analysis). On the other hand, somatic deletions in brain DNA were called using an integrated method that included paired end mapping, split reads and read depth analysis. We initially called somatic deletion candidates if a deletion was detected in Breakdancer15 and then we filtered out possible false positive candidates using Pindel16 and CNVnator14. The Blat and size filter methods were also included in the somatic deletion calling pipeline to reduce false positive findings as outlined in Fig. 1. Aberrant deletion candidates were removed by Blat and size filtering (<400 bp). This method was applied to call somatic deletions in sequencing data from multiple tissues from one individual and a single tissue from multiple individuals (Fig. 1). The mean insert sizes, the standard deviation of the insert sizes and the minimal size of detectable deletions in individual libraries were calculated using Breakdancer15 (Supplementary Table S8). The detailed germline CNV and somatic deletion calling methods are described in the Supplementary Methods.
Validating somatic CNVs by quantitative PCR using SYBR green dye
Primer sets were designed to selectively amplify our CNV candidate regions: FLG2, ZNF438, NKX2-2, C3P1, LOC348120 and SLC4A2. Real-time PCR was carried out on 3 DNA samples each originating from the same individual but differing in the area of its extraction: Blood, Cerebellum and Prefrontal Cortex. RNAase P (RPP14) gene was used for internal control locus. The calculated ΔΔCt values for the blood DNA were used as a reference in determining any copy number variability in the candidate regions of either the cerebellum or PFC. 5 ng template DNA was used for qPCR with SYBR Select Master Mix (ABI). Each sample was run 4 times in 20 μL qPCR reactions (SYBR Select 2×, 12 pmol, 5 ng DNA) and loaded onto a 384 well plate. Fluorescence detection and qPCR were carried out in an ABI Prism 7900HT Sequence Detection System (ABI) and Ct values calculated with the machines corresponding software (SDS v2.2).
Deletion calling validation with simulated data
In order to validate our deletion calling pipelines, we simulated deletions in diploid genomes using human chromosome 1 (hg18) as a template. We randomly generated 100 germline and 100 somatic deletions with a size range of 500-bp to 10-kb, excluding the gap regions, for the answer set. All generated deletions were assumed as heterozygous deletions. Two genomes were constructed using the generated deletions: the first carried the germline deletions only and the second carried both the germline and somatic deletions. The overall processes to simulate genomes were implemented by Python.
Since our simulation was designed to determine our ability to call somatic deletions accurately which occur in only a fraction of the cells in tissue, we set the relative abundance of the genome carrying both germline and somatic deletions to 10% with that of the germline only deletions by using the metagenomic mode of GenSim41. We then generated sequencing data of the mixed sample. GemSim42 was used to generate paired-end reads of the mixed sample to match the conditions of the sequencing data obtained during our experiment. Read length was set to 101-bp and fragment size was set to 500-bp with a standard deviation of 20-bp. The average depth of coverage was set to 70×, as was the average depth of the experimental data. The Generated reads were used as input in our method pipeline.
Validating breakpoints of germline and somatic deletions by PCR and Sanger sequencing
Deletion breakpoints were confirmed by PCR amplification and Sanger sequencing. PCR primers are listed in Supplementary Table S9 and the detailed methods are described in the Supplementary Methods.
Laser capture microdissection
Sections of PFC were cut at 8 μm thick onto Arcturus HistoGene Slides at −20°C for LCM on a Leica CM 1950 Cryostat after being embedded in M1 Embedding Matrix (Thermo Scientific). Staining of the slides was done with the Arcturus HistoGene Frozen Section Staining Kit (Life Technologies) using the manufacturer protocol. Laser Capture Microdissection was performed on an Arcturus PixCell IIe with CapSure HS LCM caps. Capturing was done at 20× optics using a 15 μm spot size. The target parameter was set to 0.200 V with a power of 35 mW and a duration of 0.7 ms. Ten cells of a specific type were captured per cap followed by lysis directly on the cap. Whole genome amplification was performed using a user-developed protocol of the Repli-g Mini Kit (Qiagen) with a 16 hour amplification time. DNA clean up was done using the QIAmp DNA Micro Kit (Qiagen) and quantified using Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies). Deletion validation PCR was done using 100 ng template material.
Functional annotation
Panther software was used for classification of the Gene Ontology biological processes of genes that were disrupted by somatic deletions in the PFC of the two schizophrenia cases and two unaffected controls43. DAVID was used to identify the biological processes that were significantly over-represented by the genes in the two schizophrenia cases and two unaffected controls respectively44. False discovery rates less than 0.05 were considered significant.
Equipment and settings
Laser Capture Microdissection was done with an Arcturus PixCell IIe. Target parameters were set to 0.200 V with a 0.7 ms duration at 35 mW power. Images were captured using the LCM's built in CCD camera (Hitachi K.P-D590-V1) and processed using Arcturus' LCM control software (version 2.0). DNA agarose gel pictures were taken using an 8-megapixel digital camera. Color images were then converted to greyscale using Adobe Photoshop software.
Ethical considerations
Ethical approval for the Stanley Brain Collection was obtained through the Uniformed Services University of the Health Sciences, Bethesda, MD who determined that IRB approval was not needed (during the collection period of 1998–2004) because the human subjects were deceased and all work was being done on de-identified specimens that were simply numbered. Consent to donate the specimens was obtained from next-of-kin and witnessed by two people who signed a form verifying the fact.
References
Piotrowski, A. et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat 29, 1118–1124 (2008).
O'Huallachain, M., Karczewski, K. J., Weissman, S. M., Urban, A. E. & Snyder, M. P. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci U S A 109, 18018–18023 (2012).
Forsberg, L. A. et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet 90, 217–228 (2012).
Baillie, J. K. et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537 (2011).
Iourov, I. Y., Vorsanova, S. G. & Yurov, Y. B. Somatic genome variations in health and disease. Curr Genomics 11, 387–396 (2010).
Poduri, A. et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48 (2012).
Lee, J. H. et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 44, 941–945 (2012).
Bruder, C. E. et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet 82, 763–771 (2008).
Knable, M. B., Barci, B. M., Bartko, J. J., Webster, M. J. & Torrey, E. F. Molecular abnormalities in the major psychiatric illnesses: Classification and Regression Tree (CRT) analysis of post-mortem prefrontal markers. Mol Psychiatry 7, 392–404 (2002).
Knable, M. B., Barci, B. M., Webster, M. J., Meador-Woodruff, J. & Torrey, E. F. Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry 9, 609–620 (2004).
Beasley, C. L., Zhang, Z. J., Patten, I. & Reynolds, G. P. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 52, 708–715 (2002).
Vostrikov, V. M., Uranova, N. A. & Orlovskaya, D. D. Deficit of perineuronal oligodendrocytes in the prefrontal cortex in schizophrenia and mood disorders. Schizophr Res 94, 273–280 (2007).
Kim, S. & Webster, M. J. Correlation analysis between genome-wide expression profiles and cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol Psychiatry 15, 326–336 (2010).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21, 974–984 (2011).
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6, 677–681 (2009).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nat Genet 36, 949–951 (2004).
ISC. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241 (2008).
Mulle, J. G. et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet 87, 229–236 (2010).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236 (2008).
Walsh, T. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008).
Kirov, G. et al. Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull 35, 851–854 (2009).
McCarthy, S. E. et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 41, 1223–1227 (2009).
Ingason, A. et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry 16, 17–25 (2011).
Vacic, V. et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503 (2011).
Mills, R. E. et al. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65 (2011).
Lam, H. Y. et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol 30, 226–229 (2012).
Erickson, R. P. Somatic gene mutation and human disease other than cancer: an update. Mutat Res 705, 96–106 (2010).
Poduri, A., Evrony, G. D., Cai, X. & Walsh, C. A. Somatic mutation, genomic variation and neurological disease. Science 341, 1237758 (2013).
Abyzov, A. et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature (2012).
Evrony, G. D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012).
McConnell, M. J. et al. Mosaic copy number variation in human neurons. Science 342, 632–637 (2013).
Cukier, H. N., Pericak-Vance, M. A., Gilbert, J. R. & Hedges, D. J. Sample degradation leads to false-positive copy number variation calls in multiplex real-time polymerase chain reaction assays. Anal Biochem 386, 288–290 (2009).
Rice, D. & Barone, S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 108 (Suppl 3), 511–533 (2000).
Rothkamm, K., Kruger, I., Thompson, L. H. & Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23, 5706–5715 (2003).
Perry, G. H. et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 82, 685–695 (2008).
Lewis, D. A. & Sweet, R. A. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest 119, 706–716 (2009).
Uranova, N. A., Vikhreva, O. V., Rachmanova, V. I. & Orlovskaya, D. D. Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: a postmortem morphometric study. Schizophr Res Treatment 2011, 325789 (2011).
Eastwood, S. L. & Harrison, P. J. Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry 8, 769, 821–731 (2003).
Yang, Y., Fung, S. J., Rothwell, A., Tianmei, S. & Weickert, C. S. Increased interstitial white matter neuron density in the dorsolateral prefrontal cortex of people with schizophrenia. Biol Psychiatry 69, 63–70 (2011).
Torrey, E. F., Webster, M., Knable, M., Johnston, N. & Yolken, R. H. The stanley foundation brain collection and neuropathology consortium. Schizophr Res 44, 151–155 (2000).
McElroy, K. E., Luciani, F. & Thomas, T. GemSIM: general, error-model based simulator of next-generation sequencing data. BMC genomics 13, 74, 10.1186/1471-2164-13-74 (2012).
Thomas, P. D. et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res 31, 334–341 (2003).
Dennis, G., Jr et al. DAVID: Database for Annotation, Visualization and Integrated Discovery. Genome Biol 4, P3 (2003).
Acknowledgements
This study was supported by Stanley Medical Research Institute. This work was also supported by the Bio-Synergy Research Project (NRF-2012M3A9C4048758) of the Ministry of Science, ICT and Future Planning through the National Research Foundation. We specially thank Drs. Robert Yolken and Sarven Sabunciyan for helpful comments on study design and interpretation of results. We also thank Jonathan Cohen for technical support.
Author information
Authors and Affiliations
Contributions
J.K., J.-I.K., J.-S.S., D.L. and S.K. designed this study. J.-Y.S. and S.K. performed the experiment and J.K., J.-S.S., M.J.W. and S.K. analyzed the data. J.K., M.J.W., D.L. and S.K. wrote the manuscript and all authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kim, J., Shin, JY., Kim, JI. et al. Somatic deletions implicated in functional diversity of brain cells of individuals with schizophrenia and unaffected controls. Sci Rep 4, 3807 (2014). https://doi.org/10.1038/srep03807
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03807
This article is cited by
-
Chromosome instability and aneuploidy in the mammalian brain
Chromosome Research (2023)
-
Somatic mosaicism in the diseased brain
Molecular Cytogenetics (2022)
-
Assessment of somatic single-nucleotide variation in brain tissue of cases with schizophrenia
Translational Psychiatry (2019)
-
Human Molecular Neurocytogenetics
Current Genetic Medicine Reports (2018)
-
Somatic mutations in disorders with disrupted brain connectivity
Experimental & Molecular Medicine (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
