
Abstract
Recently, transcription activator–like effector nucleases (TALENs) have emerged as a highly effective tool for genomic editing. A pair of TALENs binds to two DNA recognition sites separated by a spacer sequence and the dimerized FokI nucleases at the C terminal then cleave DNA in the spacer. Because of its modular design and capacity to precisely target almost any desired genomic locus, TALEN is a technology that can revolutionize the entire biomedical research field. Currently, for genomic editing in cultured cells, two plasmids encoding a pair of TALENs are co-transfected, followed by limited dilution to isolate cell colonies with the intended genomic manipulation. However, uncertain transfection efficiency becomes a bottleneck, especially in hard-to-transfect cells, reducing the overall efficiency of genome editing. We have developed a robust TALENs system in which each TALEN plasmid also encodes a fluorescence protein. Thus, cells transfected with both TALEN plasmids, a prerequisite for genomic editing, can be isolated by fluorescence-activated cell sorting. Our improved TALENs system can be applied to all cultured cells to achieve highly efficient genomic editing. Furthermore, an optimized procedure for genomic editing using TALENs is also presented. We expect our system to be widely adopted by the scientific community.
Similar content being viewed by others


Introduction
Loss of function (LOF) is a powerful approach in the study of gene function. To achieve this, numerous technologies have been developed, such as RNA interference to downregulate the RNA transcripts of target genes. Recently, site-specific nucleases, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), have been used to disrupt target gene function by creating DNA insertions or mutations1,2. Since ZFNs or TALENs can completely abolish the function of targeted genes, when used in LOF studies, the results are much cleaner and easier to interpret than with RNA interference. In the past 15 years, significant effort has been invested in ZFNs, in which the DNA-binding property of the zinc finger domain of a class of transcription factors is exploited to bind specifically to the intended genomic sequence3. However, due to the complexity of the zinc finger domain's DNA-binding code, significant effort and expertise in the design and optimization of ZFN activity and specificity are usually required, preventing the wide adoption of ZFNs to date.
Recently, TALENs have emerged as a breakthrough technology for genomic editing1, allowing precise gene mutation or deletion at almost any specific genomic locus. The TALE protein consists of a translocation domain in the N-terminal, a nuclear localization signal in the C-terminal and various tandems of 33–34 amino acid repeats in the middle for target DNA sequence detection and binding1. Each tandem repeat has two variable amino acid residues at positions 12 and 13, designated repeat variable diresidues (RVDs)4. The RVDs NI, NG, HD and NN preferentially recognize adenine (A), thymine (T), cytosine (C) and guanine (G), respectively (Fig. 1A)5,6. When fused with the nonspecific FokI endonuclease catalytic domain at the C-terminal, TALENs form dimers through binding to two target sequences separated by ~17 bases. Between the pair of binding sites, the FokI catalytic domains dimerize and function as molecular scissors by introducing double-strand breaks (DSBs). The resulting DSBs are usually repaired by the error-prone non-homologous end-joining pathway (NHEJ), frequently leading to small DNA deletion or insertion and consequent frameshift in the open reading frames (ORFs) of targeted genes and thus, functional gene knockout1.

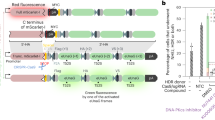
Our improved TALENs system.
(A) Schematic view of the TALEN structure with the DNA-binding modules highlighted. Each DNA-binding module consists of 34 amino acids, where the RVDs in the 12th and 13th amino acid positions of each repeat specify the DNA base being targeted according to the cipher NG = T, HD = C, NI = A and NN = G or A. (B) The TALENs system we have improved. A widely-used plasmid backbone, pcDNA3.1(−), was chosen for the TALEN vector. The red asterisks indicate where the BsaI sites were in the original pcDNA3.1(−) plasmid. They have been destroyed by mutagenesis. The original neomycin-resistant gene was replaced by a gene encoding EGFP or DsRed. The TALEN coding sequence and an adjacent upstream T7 promoter can be isolated by digesting the construct with SacI and PmeI (in red) as a template for in vitro transcription of TALEN mRNAs.
Due to their modular design, ease of use, high efficiency, low off-target rate and capacity to precisely target almost any genomic locus1, TALENs are superior to ZFNs for genomic editing. TALENs are also highly effective tool in introducing mutations into an endogenous genome, making the study of gene mutants in an endogenous physiological setting much easier7. Furthermore, when mRNAs encoding TALENs are directly injected into the cytoplasm of one-cell-stage embryos, homozygous gene mutation or knockout can be achieved with a relatively high efficiency8, saving months, even years of work compared to more traditional mouse genetics approaches.
When establishing a cell line with somatic gene knockout or knockin, two plasmids that encode a pair of TALENs need to be co-transfected into cells. The cells are then diluted to isolate clones with the intended genomic editing. Usually, a large number of cell clones need to be screened to select cells with successful gene disruption, especially in hard-to-transfect cells. In addition, because mammalian cells usually contain two copies of each chromosome, homozygous gene editing requires that both alleles be targeted. Due to genomic instability, aneuploidy is frequent in cancer cells9, making homozygous genomic editing even more challenging.
To improve TALEN targeting efficiency in these cells, a more robust TALENs system is clearly needed. Recently, a strategy was reported by co-transfecting a surrogate reporter, in which a piece of DNA that is homologous to the intended ZFNs or TALENs target sequence is cloned between a RFP and GFP gene, allowing enriching cells with nuclease-induced mutations by isolating dual-labelled cells with fluorescence-activated cell sorting (FACS)10. However, a major caveat of this approach is that distinct surrogate reporters are required for every gene targeting experiment, especially when multiple genes need to be targeted or multiple designs of TALENs targeting the same gene need to be tested.
To establish a simple and efficient TALENs system, we introduced genes that encode different fluorescent proteins into the TALEN-encoding plasmids. No additional work is required for this system other than essential TALENs cloning for genomic editing experiment. After co-transfection, TALENs-transfected cells can be enriched by isolating dual-labelled cells by FACS, greatly improving the efficiency of genomic editing. We also provide a practical guide on the use of this TALEN system for laboratories without prior experience with this powerful new technology.
Results
Constructing a pcDNA3.1-based EGFP/DsRed-encoding TALENs system
The TALEN toolkit developed by the Zhang lab provides a quick and convenient means for constructing custom TALENs for genomic editing11. In the original toolkit, four plasmids that encode NN, NI, HD and NG monomers were used to generate a library of all possible monomers by PCR for subsequent construction of 18-mer TALE DNA-binding domains. However, 18 different primers need to be synthesized with some as long as 79 nucleotides11, which is not economic for individual laboratories and errors could be introduced during DNA synthesis of long oligonucleotides. To simplify this procedure, we cloned the PCR products of all 40 monomers that may be used for assembling any 18-mer TALE DNA-binding domains into a pGEM-T vector (Supplementary Figure 1). This system allows us to amplify any monomers with two commonly used DNA sequencing primers, T7 and sp6.
Despite being a highly effective genomic editing tool, when TALENs are used in cultured cells cell clones still need to be screened to identify the ones with intended genome modifications, a potentially tedious process largely determined by the efficiency of TALEN plasmids transfection. We reasoned that, if untransfected cells and cells transfected with only one of the two TALEN plasmids can be excluded, cells transfected with both TALEN plasmids, a prerequisite for successful genome editing, will be highly enriched, thus greatly improving the efficiency of clone selection. To achieve this, we decided to introduce fluorescent protein markers into the TALEN plasmids to indicate their presence in cells.
The pcDNA3.1 plasmid is one of the most widely used mammalian gene expression vectors. The plasmid is well characterized and its physical map and DNA sequence are readily available from various sources, making additional modifications, if necessary, easy to carry out. We replaced the neomycin-resistant gene in pcDNA3.1(−) with an EGFP or DsRed gene, which allows us to estimate the transfection efficiency of the TALEN plasmids under a fluorescent microscope and isolate EGFP and DsRed dual-labelled cells by FACS. We then cloned the TALEN backbones11 into the EGFP- or DsRed-encoding pcDNA3.1(−) plasmid (Fig. 1B). Our modification also kept the T7 promoter upstream of the TALEN ORF intact. The T7 promoter will be used for the in vitro transcription of TALENs.
Fluorescence labelling improves the efficiency of selecting positive clones
A major bottleneck, lowering the efficiency of genomic editing in cultured cells, is the selection of clones that harbour a desired genomic modification. Usually, a large number of single cells need to be plated and screened following transfection of the TALEN plasmids, especially for cells that are difficult to transfect.
To validate our improved system, SKOV3 ovarian cancer cells were transfected with a pair of EGFP- or DsRed-encoding TALEN plasmids targeting human miR-210 (Fig. 2A). Forty-eight hours later, half of the transfected cells were sorted while the other half remained unsorted (Fig. 2B). After isolating DNA from these cells, Surveyor assay was performed to determine whether genomic editing had been effected at the miR-210 locus. A 3.8-fold increase of genomic editing efficiency at the miR-210 locus was detected in the sorted cells compared to that of in the unsorted ones (Fig. 2C).

Enrichment of SKOV3 cells transfected with both TALEN constructs.
(A) Schematic view of TALENs that target human miR-210. The highlighted region indicates 22 nucleotides of mature miR-210 sequence. (B) Top panel, transfected SKOV3 cells before FACS. Approximately 50% and 20% of cells were transfected with a TALEN-L construct encoding EGFP and a TALEN-R construct encoding DsRed, respectively. Bottom panel, SKOV3 cells plated right after FACS. All cells are labelled by both EGFP and DsRed. Size of the bar, 100 μm. (C) Surveyor assay of DNA extracted from SKOV3 cells before and after FACS. The numbers at the bottom of the gel indicate mutation percentages measured by band intensities. Genomic editing is enhanced 3.8-fold following cell sorting.
Strategy to efficiently isolate cells with genomic modification
Currently, the standard approach to verify whether cells harbour a desired genomic modification is to isolate genomic DNA from cell clones, amplify the targeted locus by PCR, clone the PCR product into plasmids and sequence randomly picked plasmids to assess whether the desired genomic modification is present.
Because of extensive genomic instability9, aneuploidy is common in cancer cells. To achieve homozygous genomic targeting in these cells, usually three or more copies of genes need to be targeted, demanding that more clones be sequenced. When tens or even hundreds of cell clones need to be verified, this process becomes extremely tedious and expensive.
To address this problem, we developed a PCR strategy to screen these clones first, before proceeding to cloning and sequencing. We designed a pair of PCR primers with the 3′ of one of the primers crossing the spacer region (Fig. 3A), assuming that successful genomic targeting by TALENs would introduce insertion or deletion in the spacer region, which would compromise primer annealing to the TALENs-edited DNA template during PCR reaction, completely abolish PCR amplification or reduce PCR efficiency and result in no or fewer PCR products.

Effective miR-210 targeting by our improved TALENs system.
(A) The principle of designing PCR primers that facilitate cell clone screening for potential genomic editing. The 3′ end of the forward or reverse primer should cross the spacer region. (B) PCR screening of 10 SKOV3 cell clones to identify ones with edited miR-210 sequence. Clones 1, 2, 4, 7 and 9, marked by asterisks, produced fewer PCR products than the other five clones. After sequencing, Clones 1, 4, 7 and 9 have successful miR-210 targeting with homozygous editing of miR-210 in Clones 1 and 4. (C) Chromatogram indicates successful homozygous editing of miR-210 sequence in Clone 1 by TALENs as designed in Fig. 2A. Top panel, wild type miR-210 sequence. The highlighted region indicates 22 nucleotides of mature miR-210; lower three panels, truncated miR-210 sequences detected in Clone 1, indicating SKOV3 is aneuploid of chromosome 11. (D) RT-qPCR indicates that miR-210 expression was completely abolished in Clone #1, but parental SKOV3 cells have robust miR-210 expression under 0.5% oxygen (H) and normoxia (N). Arrows in the top panel (amplification plot) indicate the amplification signals from cells with homozygous deletion of miR-210. Arrows in the bottom panel (dissociation curve) indicate that these amplification signals are actually background noises.
To avoid accumulation of PCR products from imperfect primer-template match due to subtle genomic editing, such as single nucleotide deletion or insertion, in the late rounds of PCR, only 30 cycles of PCR reaction were performed. When a high primer annealing temperature (66.5°C) was used, we identified 5 of the 10 clones screened as candidates (Clones 1, 2, 4, 7 and 9) as having modification at the miR-210 locus, based on the signal intensity of the PCR products in the agarose gel (Fig. 3B). The miR-210 locus in each of these five clones was then amplified and cloned into a pGEM-T vector.
Plasmids from 20 randomly selected bacterial clones were sequenced. Clones 1, 4, 7 and 9 contained miR-210 deletion, of which Clones 1 and 4 harboured homozygous miR-210 deletion while Clones 7 and 9 had heterozygous miR-210 deletion (Fig. 3C).
To further validate miR-210 disruption, we performed reverse transcriptase quantitative PCR (RT-qPCR) to examine miR-210 expression in these cells. Since miR-210 is a hypoxia-inducible microRNA12, expression of miR-210 was induced approximately 4-fold when parental SKOV3 cells were exposed to 0.5% oxygen for 24 hours. However, neither baseline expression nor hypoxia induction was detected for either Clone 1 or 4 (Fig. 3D), suggesting that expression of miR-210 was completely abolished in these two clones due to genomic editing at the miR-210 genomic locus.
A versatile system for producing mRNAs encoding TALENs for embryo microinjection
The potential of TALENs for in vivo genomic editing is highly promising13. Recently, injection of mRNAs encoding TALENs into the cytoplasm of one-cell-stage embryos has been reported to generate knockout rats and mice with high efficiency8,14,15,16, greatly shortening the time to generate knockout animals compared to traditional approaches. We have designed our TALENs system to accommodate this application.
Because an intact T7 promoter was retained in our TALEN plasmids (Fig. 1B), it allows us to perform in vitro transcription to generate TALEN mRNAs for embryo injection. Because RNA is prone to degradation, the quality of TALEN mRNAs is critical in generating animals with successful genomic modification. The presence of a 3XFLAG tag at the N-terminal of TALEN constructs allowed us to perform a quality control experiment by Western blotting to determine whether in vitro transcribed mRNAs can be translated into TALEN proteins.
As a proof of principle, we prepared a DNA template by digesting the pair of TALEN constructs targeting miR-210 with SacI and PmeI (Fig. 1B), followed by in vitro transcription, addition of 5′ G-cap and polyadenylation of the mRNAs, in vitro translation and Western blotting using an anti-FLAG antibody. Despite a relatively high background, a single band of the expected size was observed for both TALENs (Fig. 4), indicating that our TALENs system is well suited to generate mRNAs for mouse embryo microinjection.

In vitro translated TALEN proteins as an indicator for the quality of TALEN mRNAs.
The mRNAs encoding miR-210-targeting TALENs were transcribed in vitro. To check the quality of in vitro transcribed TALEN mRNAs, in vitro translation was performed and the translated proteins were separated on a 10% SDS-PAGE gel and detected by Western blotting using an anti-FLAG antibody. Proteins were successfully produced from both TALEN mRNAs, suggesting that they can be used for microinjection.
Discussion
In this report, we have detailed our improvement of the current TALENs system by the introduction of fluorescent markers into TALEN vectors to facilitate selection of those cells manifesting the intended genomic manipulation. We offer our delineation of the steps involved as a practical guideline for laboratories without prior experience with TALENs. As demonstrated, our system is highly effective in generating miR-210 knockouts in SKOV3 cells, with two clones harbouring homozygous and two clones harbouring heterozygous, miR-210 deletion out of 10 clones examined.
A surrogate reporter system as a functional readout of transfected ZFN or TALEN plasmids was reported recently10. The authors achieved over 10-fold enrichment of genomic targeting in sorted cells compared to unsorted cells measured by the T7 endonuclease I assay. As a comparison, our TALENs system achieved a 3.8-fold enrichment when measured by the Surveyor assay. The lower enrichment efficiency of our system may be explained by differential plasmid transfection efficiency prior to cell enrichment or our particular TALENs design targeting miR-210 is not as effective as the ZFNs used by Kim et al. in genomic editing. However, a distinct advantage of our system is that no additional cloning step is required, making it more versatile and less labour-intensive. When combined with the PCR strategy for clone selection, our system can achieve highly efficient genomic editing.
The system described is particularly useful with cells that cannot be transfected with high efficiency. Theoretically, if the transfection efficiency for a cell line is 90%, 81% of the cells (90% × 90%) are expected to be transfected with both TALEN constructs. Therefore, when our system is used a 19% improvement can be achieved. However, when the transfection efficiency of a cell line is 10%, only 1% of cells (10% × 10%) are expected to be transfected with both TALEN constructs and a large number of cell clones will need to be screened to select cells with the intended genomic editing. With our TALENs system, dual-transfected single cells can be isolated by FACS, with a 99% improvement of clone-selection efficiency. Highly efficient TALENs delivery can also be achieved by using viral vectors, such as lentivirus or adenovirus. However, lentiviral sequences usually integrate into the host genome, which may lead to unintended disruption of host gene function at the site of integration and generate ambiguous experimental results. And, although adenovirus does not integrate into host genome, it requires a complex procedure and at least 4 to 5 weeks of time to prepare adenoviral particles17. Thus, our TALENs system is a convenient and efficient tool for genomic editing.
During the selection of functional TALEN constructs, we found that sequencing the DNA-binding domains of TALENs alone as suggested11 sometimes is not sufficient. In addition to DNA sequencing, we also performed restriction digestion of 10 TALEN plasmids with AfeI and MluI. However, two clones (Clones #5 and #10) with correct DNA-binding domain sequences demonstrated abnormal DNA digestion patterns (Supplementary Fig. 3A), suggesting that unexpected rearrangements or assembly errors had occurred outside of the sequenced region during Golden Gate ligation. Thus, these clones should be excluded from TALEN experiment. Since a 3XFLAG tag is present at the N-terminal of the TALENs (Fig. 1B), we further examined two clones from each DsRed- or EGFP-encoding plasmid to determine whether they could be translated into TALEN proteins. Interestingly, although plasmid #2 was free of mutation in the DNA-binding domain and demonstrated a normal restriction digestion pattern, it did not express the encoded TALEN protein (Supplementary Fig. 3B). Thus, our data suggest that strict quality control assays for the TALEN constructs is essential to ensure successful genomic editing. These additional steps may seem somewhat redundant, but we believe that the extra scrutiny is warranted given the overall effort and time generally invested in experiments involving TALENs.
To date, the vast majority of published studies have focused on optimizing TALENs assembly and its versatile application in genomic editing18,19,20,21. In our study, we focused on optimizing the downstream application of TALENs and developed an effective PCR strategy for identifying single cell clones that possess desired genomic modifications. By designing one of the PCR primers across the spacer region between the DNA-binding sites of two TALENs, we screened cell clones for miR-210 deletion under a stringent PCR condition. With reduced cycle numbers of PCR reaction, our strategy worked well, out of five cell clones with no, or less abundant, PCR products, four had miR-210 modification. Our approach greatly improved the clone selection process by rendering it much less labour intensive.
Recently, the type II bacterial CRISPR/Cas system has emerged as another efficient gene-targeting technology22,23. The CRISPR (clustered regularly interspaced short palindromic repeat) and Cas (CRISPR-associated) proteins belong to an RNA-based adaptive immune system in bacteria and archaea for detecting and destroying invading viruses and plasmids24. Cas proteins, CRISPR RNAs (crRNAs) and trans-activating crRNA (tracrRNA) form ribonucleoprotein complexes, which target and degrade foreign nucleic acids, guided by crRNAs24. Provocatively, Cas9 and a crRNA-tracrRNA fusion transcript termed guide RNA (gRNA) are the only components necessary and sufficient for induction of targeted DNA cleavage in cultured human cells23, making it a much simpler system for genomic editing. However, despite an exciting and promising technology, recent reports suggest that CRISPR/Cas9 is prone to a high frequency of off-target cleavage25, limiting its application when highly specific genomic editing is required. Thus, a TALENs system, such as the one described in this report, will be the tool of preference for highly specific and accurate genomic editing.
Mouse or rat models can be established by injecting TALEN plasmids or mRNAs into the cytoplasm of one-cell-stage embryos, followed by implantation into pseudo-pregnant mothers8,15. However, injecting mRNAs has been shown to be more efficient than plasmid DNA in producing animals with intended genomic modifications15. RNAs are labile molecules, prone to rapid degradation by ubiquitous ribonucleases, but whether an animal experiment succeeded or not could only be determined after the pups were born and weaned. The complex experimental procedure required for generating mRNAs, including in vitro transcription, 5′ cap addition and polyadenylation, warrants a careful mRNA quality control prior to commitment to expensive microinjection and time-consuming animal breeding. As an indicator of mRNA quality, we examined the capacity of mRNAs encoding TALENs that target miR-210 as templates for protein translation. The successful translation of TALEN proteins suggests that the mRNAs are ready for microinjection. However, if TALEN proteins, at expected size, are not detectable, it may indicate that the mRNAs are of poor quality or that errors had occurred during in vitro transcription and, therefore, that the preparation of the mRNAs in question needs to be repeated. We believe that this critical quality control step is warranted for all experiments intended to establish animal models through embryo microinjection of TALEN mRNAs.
In summary, we have improved the current TALENs system for enriching cells with more efficient genomic editing and also presented a practical guide for TALENs experiments, which will be highly valuable for laboratories without prior experience but would like to adopt this exciting technology.
Methods
Cell lines and culture conditions
We purchased SKOV3 cells from ATCC (Manassas, VA). The cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal calf serum, 2 mM L-Glutamine and an antibiotic combination of 0.1 mg/ml streptomycin and 100 U/ml penicillin (all from Invitrogen, Carlsbad, CA). The cells were incubated at 37°C in a humidified air atmosphere containing 5% CO2. For transfection, 1 × 106 SKOV3 cells were plated onto a 6-well plate in DMEM without antibiotics approximately 24 hours prior to transfection. A mix of 2 μg left and 2 μg right TALEN plasmids were co-transfected into SKOV3 cells using the FuGENE HD Transfection Reagent (Promega, Madison, WI) following the manufacturer's protocol.
Molecular biology reagents
All restriction enzymes used in this study were purchased from New England Biolabs (Ipswich, MA). T4 ligase, buffer and the pGEM-T vector were purchased from Promega. PfuUltra high-fidelity DNA polymerase was purchased from Agilent (Santa Clara, CA). EconoTaq PLUS GREEN 2× Master Mix was purchased from Lucigen (Middleton, WI). Plasmids were isolated using Wizard Plus SV Minipreps DNA Purification System (Promega) according to the manufacturer's instructions. The pcDNA 3.1(−) vector was purchased from Invitrogen. The TALENs toolkit, including four monomer fragments (NI, HD, NG and NN) and the TALEN backbone plasmids, was kindly provided by Dr. Feng Zhang of the Broad Institute of MIT and Harvard.
Plasmid construction and validation
To prepare the monomer library, the primers were synthesized (Integrated DNA technologies, Coralville, IA) according to published report11. The monomers were PCR amplified using the PfuUltra high-fidelity DNA polymerase (Agilent) and gel purified. An “A” overhang was added at each end of the PCR products by incubating with EconoTaq (Lucigen) at 72°C for 10 minutes. The PCR fragments were then ligated with the pGEM-T vector for E. coli transformation. All resulting monomer constructs were verified by DNA sequencing.
To customize the pcDNA3.1(−) plasmid for Golden Gate ligation, an essential procedure for TALEN plasmid construction, we first destroyed the two BsaI sites in the plasmid by (1) mutating one BsaI site from GAGACC to GAGAGC in the ampicillin-resistant gene without changing the encoded amino acid; and (2) deleting the other BsaI site adjacent to the T7 promoter site by digesting the plasmid with BsaI and NotI, filling the sticky ends with PfuUltra high-fidelity DNA polymerase and ligating the blunt ends of the modified plasmid. The EGFP and DsRed genes were amplified from pEGFP-N1 (Clontech, Mountain View, CA) and pDsRed2-Mito (Clontech), respectively. The modified pcDNA3.1(−) vector was digested by SmaI and BstBI to remove the neomycin-resistant gene and ligated with SmaI- and BstBI-digested EGFP or DsRed PCR product. The four TALEN backbones were PCR amplified from corresponding pTALEN vectors11. The modified pcDNA3.1(−)-EGFP or pcDNA3.1(−)-DsRed vector and the TALEN backbone PCR products were then digested by EcoRV and HindIII followed by ligation. Four TALEN backbone plasmids were constructed with an EGFP label and four with a DsRed label. All plasmids were sequenced to verify that no mutation was introduced during cloning.
For constructing the miR-210-targeting TALEN plasmids, the monomers were PCR amplified from corresponding monomer plasmids using T7 and sp6 primers, gel purified and assembled into hexamers by Golden Gate ligation11. The hexamers were then further PCR amplified, gel purified and assembled into 18-mers and cloned into a DsRed- or EGFP-encoding TALEN backbone plasmid by Golden Gate ligation11. Positive colonies were sequenced, as recommended, to verify that no mutation was present in the DNA-binding domains of the TALENs. Restriction digestion with AfeI and MluI was performed to further verify no structural change was introduced during plasmid construction. The expected digestion products are 117, 165, 886, 2001 and 5200 base pairs for the DsRed-encoding vector and 117, 165, 886, 2001 and 5242 base pairs for the EGFP-encoding vector. But because the 117-bp and 165-bp fragments are part of the TALEN ORF that is verified by DNA sequencing, information from the three larger fragments was sufficient to determine whether the other parts of the TALEN constructs were compromised during our construct assembly. DsRed- or EGFP-encoding TALEN plasmids with expected restriction digestion pattern were then transfected into the SKOV3 cells followed by Western blotting using an anti-FLAG antibody to further validate that TALEN proteins can be successfully translated from these plasmids.
PCR primers and conditions
5′-TCGGACGCCCAAGTTGGA- 3′ (forward) and 5′-ACACCCCGTCCATAGGGCA- 3′ (reverse) were used to PCR amplify the miR-210 genomic sequence for Surveyor assay. 5′-TCGGACGCCCAAGTTGGA- 3′ (forward) and 5′-CCGCTGTCACACGCACAG- 3′ (reverse) were used for PCR screening potential cell clones with disrupted miR-210 sequence. RNA was harvested using TRIzol (Invitrogen) and processed according to the manufacturer's instructions. miR-210 expression was detected by real-time PCR using 2× RT2 SYBR Green qPCR Master mix (QIAGEN, Valencia, CA) on a 7900HT Fast Real-time PCR System (Invitrogen).
Surveyor assay
Genomic DNA in cells transfected with TALEN plasmids before or after FACS sorting was extracted using a Wizard Genomic DNA Purification Kit (Promega). The genomic region encompassing the mature human miR-210 sequence targeted by the TALENs was PCR amplified. Modification of the miR-210 sequence was detected by a SURVEYOR Mutation Detection Kit (Transgenomic, Omaha, NE) according to the manufacturer's instruction.
In vitro transcription and translation
The DNA fragments encoding the TALENs protein and an upstream T7 promoter were isolated by digesting the vectors with SacI and PmeI. The TALEN genes were transcribed in vitro, capped at 5′ and polyadenylated using an mMESSAGE mMACHINE T7 Ultra Kit (Invitrogen) according to the manufacturer's instructions. The mRNAs were translated in vitro using a Retic Lysate IVT Kit (Invitrogen) following the manufacturer's protocol.
Imaging and image processing
The dual-transfected SKOV3 cells were sorted by a BD LSRII flow cytometer (BD Biosciences, San Jose, CA) at the Flow Cytometry Facility in the Department of Immunology of the University of Pittsburgh School of Medicine. Cells were imaged by a Carl Zeiss Axiovert 40 CFL inverted microscope equipped with a Nikon DS-Qi1Mc digital video camera.
DNA band intensity in the Surveyor assay was estimated by the ImageJ software (http://rsb.info.nih.gov/ij/).
Western blotting
Cells were harvested in cold 1XPBS and centrifuged at 500 g for 2 minutes. The cell pellets were lysed in urea buffer (9 M Urea, 0.075 M Tris buffer, pH 7.6). Protein lysates were quantified with a BCA assay kit (Pierce, Rockford, IL) and separated on 10% SDS-PAGE. Antibodies used were as follows: anti-FLAG (F1804), Sigma-Aldrich; GAPDH (sc-47724), Santa Cruz Biotechnology.
References
Bogdanove, A. J. & Voytas, D. F. TAL Effectors: Customizable Proteins for DNA Targeting. Science 333, 1843–1846 (2011).
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S. & Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11, 636–646 (2010).
Miller, J. C. et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotech 25, 778–785 (2007).
Gaj, T., Gersbach, C. A. & Barbas, C. F. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends in Biotechnology 31, 397–405 (2013).
Boch, J. et al. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 326, 1509–1512 (2009).
Moscou, M. J. & Bogdanove, A. J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 326, 1501 (2009).
Wei, Q.-X., van der Hoeven, F., Hollstein, M. & Odell, A. F. Efficient introduction of specific TP53 mutations into mouse embryonic fibroblasts and embryonic stem cells. Nat. Protocols 7, 1145–1160 (2012).
Sung, Y. H. et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotech 31, 23–24 (2013).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Kim, H. et al. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Meth 8, 941–943 (2011).
Sanjana, N. E. et al. A transcription activator-like effector toolbox for genome engineering. Nat. Protocols 7, 171–192 (2012).
Huang, X. et al. Hypoxia-Inducible mir-210 Regulates Normoxic Gene Expression Involved in Tumor Initiation. Molecular Cell 35, 856–867 (2009).
Bedell, V. M. et al. In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114–118 (2012).
Wefers, B. et al. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proceedings of the National Academy of Sciences 110, 3782–3787 (2013).
Tesson, L. et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotech 29, 695–696 (2011).
Wang, H. et al. TALEN-mediated editing of the mouse Y chromosome. Nat Biotech 31, 530–532 (2013).
Luo, J. et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protocols 2, 1236–1247 (2007).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Research 39, e82 (2011).
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotech 30, 460–465 (2012).
Li, T. et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Research 39, 6315–6325 (2011).
Zhang, F. et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotech 29, 149–153 (2011).
Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-Guided Human Genome Engineering via Cas9. Science 339, 823–826 (2013).
Wiedenheft, B., Sternberg, S. H. & Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 (2012).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotech 31, 822–826 (2013).
Acknowledgements
We are grateful to Dr. Feng Zhang at the Broad Institute of MIT and Harvard for providing the TALENs toolkit to allow us to initiate this work. We would like to thank Mr. Bruce Campbell for manuscript editing. This work was supported by an American Cancer Society Research Scholar Grant and the Liz Tilberis Scholars Award from Ovarian Cancer Research Fund (XH).
Author information
Authors and Affiliations
Contributions
Y. F. and S.Z. contributed equally to this work. Y.F. and S.Z. designed and performed most of the experiments. Y.F., S.Z. and X.H. analyzed the data. X.H. conceived and initiated the research. X.H. wrote the manuscript with the help from all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemmentary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Feng, Y., Zhang, S. & Huang, X. A robust TALENs system for highly efficient mammalian genome editing. Sci Rep 4, 3632 (2014). https://doi.org/10.1038/srep03632
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03632
This article is cited by
-
The CRISPR–Cas9, genome editing approach: a promising tool for drafting defense strategy against begomoviruses including cotton leaf curl viruses
Journal of Plant Biochemistry and Biotechnology (2019)
-
Current status and perspectives of genome editing technology for microalgae
Biotechnology for Biofuels (2017)
-
A novel technique based on in vitro oocyte injection to improve CRISPR/Cas9 gene editing in zebrafish
Scientific Reports (2016)
-
Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system
Cellular and Molecular Life Sciences (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
