
Abstract
Although the plant lipidome show an enormous level of structural and functional diversity, our knowledge about its genetic control and its connection to whole-plant phenotypes is very limited. Here, we profiled 563 lipid species with UPLC-FT-MS in 289 field-grown inbred lines genotyped with 56,110 SNPs. Genome-wide association study identified 174 associations for 76 lipids explaining up to 31.4% of the genetic variance (P-value 8.4 × 10−18). Candidate genes were found for lipid synthesis, breakdown, transfer and protection against peroxidation. The detected SNP-lipid associations could be grouped into associations with 1) individual lipids, 2) lipids from one biochemical class and 3) lipids from several classes, suggesting a multilevel genetic control architecture. We further found a strong connection between the lipidome and agronomic traits in field-evaluated hybrid progeny. A cross-validated prediction model yielded correlations of up to 0.78 suggesting that the lipidome accurately predicts agronomic traits relevant in hybrid maize breeding.
Similar content being viewed by others
Introduction
Living organisms produce a plethora of chemically distinct lipid species, commonly referred to as the lipidome1. Lipids are fascinating for their chemical diversity which is especially enormous in plant domain due to the high plasticity of their biosynthetic machinery2. In addition, lipids are involved in various cellular processes including cell integrity, membrane formation and scaffolding for membrane proteins, energy storage and cell signaling3.Lipid composition has been demonstrated to change with environmental conditions, e.g. under various abiotic stresses and has actually been demonstrated to serve as an indicator for the respective stress4. Recent progress in mass spectrometry allows measuring simultaneously hundreds of chemically different lipid species in a single sample of plant tissue, which though impressive, covers only a part of the complexity of the lipidome5,6,7,8,9.
Although in Arabidopsis thaliana, most of the biochemical steps involved in lipid biosynthesis are known and the key genes have been identified, the regulation of the processes that results in the final lipid composition is only weakly understood. Quantitative genetic approaches have been followed more recently in Arabidopsis thaliana10 and maize11 to identify loci involved in storage oil formation; however this analysis was limited to one class of lipids, i.e. triacylglycerides. Recently, we have described results obtained for mapping primary metabolites in maize12 via genome-wide association (GWA) mapping. The GWA mapping approach relies on ancestral linkage disequilibrium (LD) in a diverse population and can yield a mapping resolution up to the single nucleotide level.
Using the same mapping population we here extend this study to identify numerous loci involved in lipid metabolism. Using UPLC-FT-MS, we measured 563 distinct leaf lipids in 289 diverse maize inbred lines genotyped with 56,110 SNPs. In addition we asked the question whether or not lipid profiles can be used for predicting general combining ability (GCA) of the parent lines reflecting the average performance of their hybrid progeny as previously successfully shown for primary metabolites13. GCA was estimated for four traits by crossing 285 lines from the Dent heterotic pool with two single-cross testers from the Flint heterotic pool and evaluating their testcross progeny in three locations over two years.
Results
Lipid profiling
Using ultra-performance liquid chromatography separation coupled to a high-resolution Fourier transform mass spectrometer (UPLC-FT-MS), 563 distinct leaf lipids were measured in the 289 maize inbred lines from the diversity panel of worldwide sources ( Supplementary Table S1 ). It is clear that these 563 species scratch only the surface of the lipidome, however, as demonstrated below, they already allowed to identify a number of loci/regions in the maize chromosome associated with their abundance in maize leaves. From the mass spectra of the detected compounds, 16 could be annotated to be diacylglycerols (DAG), 15 as digalactosyl-diacylglycerols (DGDG), 23 as monogalactosyl-diacylglycerols (MGDG), 24 as phosphatitylcholines (PC), 11 as phosphatidylethanolamines (PE), six as phosphatidylglycerols (PG), three as phosphatitylinositols (PI), nine as sulfoquinovosyldiacyl-glycerols (SQDG) and 48 as triacylglycerides (TAG). The remaining lipids could not be annotated at present, however, specifically for lipids showing a strong association with gene loci, efforts are presently undertaken to clarify their structure. Lipid concentrations showed an average repeatability (w2) of 0.66 ( Supplementary Fig. S1 ), with a total of 205 compounds (36.4%) having w2 > 0.8. The lipids showed a correlation pattern with intense substructuring ( Fig. 1a ). Notably, all annotated TAGs clustered together into one highly positively correlated group.

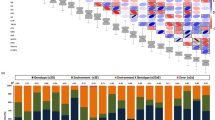
Correlation and colocalization pattern of lipids.
(a) Pairwise Pearson correlations (r) among all 563 measured lipids. Lipids are ordered using Ward clustering on pairwise dissimilarity, calculated as 1 – r. The cluster on the right is colored according to the chemical class of the lipids. (b–d) Colocalization of GWA mapping hits on chromosomes 3,5 and 8. Circles refer to –log10(P-value) of SNP-lipid associations which were significant with FDR ≤ 0.025. Dotted lines indicate genomic positions with association signals of multiple lipids. Candidate genes with known relationships to lipid metabolism or protection against peroxidation are shown at the bottom. PL, phospholipase; 12-ODR, 12-oxophytodienoate reductase; GST, gluthation-S-transferase; ERT, ethylene-responsive transcription factor; TLC, TRAM/LAG1/CLN8 (TLC) lipid-sensing domain containing protein; LTP, lipid transfer protein; INV, invertase; GDSL, GDSL esterase/lipase; VTE1, tocopherol cyclase; SERINC, serinc-domain containing sphingolipid biosynthesis protein; 3-HACD, 3-hydroxyacyl-CoA dehydratase. Details of all association hits can be found in Supplementary Table S2 and candidate genes in Table 1. Colocalizations on all other chromosomes are presented in Supplementary Fig. S2 .
Genome-wide association mapping
Using the Q10 + K mixed model and a FDR threshold of 2.5%, we detected 174 SNP-lipid associations for 76 lipids, explaining up to 31.6% of the genetic variance ( Supplementary Table S2 and Fig. 2 ). A total of 23 lipids showed at least one signal explaining more than 15% of the genetic variance. For the lipids with significant association signals, the average inflation factor λ was 1.14. From the annotated lipids, most significant hits were found for TAG and MGDG (Table S2). Associations were found on all chromosomes, with an intense co-localization of the same signal positions for groups of lipids ( Fig. 1b–d and Supplementary Fig. S2 ).

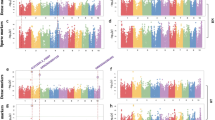
Five examples of GWA mapping results.
(a–e) Manhattan plots for lipids 5645, 5447, 8765, 6235 and 6109. P-values obtained from a Q10 + K mixed model analysis are shown on a –log10-scale and colored in red if significant with FDR ≤ 0.025. (f) Regional association plot for lipid 8765. The positions of the candiate genes GDSL esterlase/lipase and invertase (INV) are shown as triangles. Level of linkage disequilibrium (r2) with the top hit SNP is shown in red color.
The strongest SNP-lipid associations were found for three lipid species with unknown chemical structure, which were highly correlated with each other (0.96 < r < 0.99). All three showed their strongest associations (P-values ranging from 8.8 × 10−14 to 8.4 × 10−18) at position 66.8 Mb on chromosome 5, nearby (1.07 Mb) a lipolytic GDSL esterase/lipase and very close (4.5 kb) to an invertase gene ( Fig. 2c,f ).
Candidate genes involved in lipid synthesis, transfer, breakdown, signaling and protection against peroxidation were identified for many detected associations ( Table 1 ). Candidate genes were grouped into three hierarchy levels depending on the biochemical composition of the compound(s) for which the associations were detected ( Fig. 3 ). Level 1 candidate genes correspond to associations uniquely found for one lipid species. Level 2 candidate genes represent associations specific to several lipids from one chemical class. Finally, level 3 candidate genes correspond to associations found for lipids from multiple chemical classes.

Examples for the control levels 1–3 which have been assigned to the detected candidate genes.
A TLC lipid sensing protein was detected for lipids with the same fatty acid residual but different backbones and hence different chemical classes. A lipid transporter was associated with three strongly correlated TAGs with 56 to 60 C atomes. In contrast, a peroxidase was uniquely found for a single lipid species. Different chemical classes are shown as different colors. Red values on the edges correspond to pairwise correlations. The shown P-value reflects the lowest value for all associations with the candidate gene.
Level 1 candidate genes include a peroxidase on chromosome 2 which was uniquely associated with DGDG 34:4 and a phospholipase, which was found for an unknown lipid (ID 6151).
The biochemically closely related TAGs 56:4, 58:4 and 60:4 showed two associations leading to level 2 candidate genes. The first association was found at the beginning of chromosome 3 at 8.6 Mb, very close (268.3 kb) to a phospholipase and the second at the end of the chromosome at 228.4 Mb, nearby a lipid transporter.
Examples for level 3 candidate genes include amongst others a sphingosine kinase, found 294.5 kb away from an association at position 33.56 Mb on chromosome 4, for which one PG, one MGDG and three unknown lipids mapped together. One PG, one MGDG, one SQDG and nine unknown lipids mapped to a region from 41.4 to 42.4 Mb on chromosome 10, harboring two genes, the products of which have a high similarity (Protein-Protein BLAST E-values 4 × 10−64 and 5 × 10−86) to a fatty acid desaturase in Arabidopsis. Nine out of these 13 lipids also showed a common association on chromosome 1 nearby (538.1 kb) a lipid transfer protein. In addition, eight lipids from three chemical classes mapped to the same position at 115.5 Mb on chromosome 8, in close vicinity (4.9 kb) of a 3-hydroxyacyl-CoA dehydratase, a key component in the elongase complex for synthesizing very-long-chained fatty acids14. Finally, one DGDG, one MGDG and one DAG, all sharing the same fatty acid residual (36:4), mapped besides two unknown lipids to the same location at 173.1 Mb on chromosome 3, close (81.5 kb) to a protein containing a TLC (TRAM/LAG1/CLN8) lipid-sensing domain ( Fig. 3 ).
Other candidate genes with known relationship to lipid metabolism include ethylene-responsive transcription factors, tocopherol cyclase, gluthation-S-transferase, as well as 12-oxophytodienoate reductase, which participates in the conversion of linolenic acid into jasmonic acid, a fatty acid derivate acting as an important plant hormone.
Whole-lipidome prediction of general combining ability
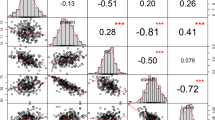
Correlations between individual lipids with GCA values ranged from –0.54 to 0.48 ( Fig. 4a–d ). To predict GCA with the lipidome, we applied a statistical prediction model that assumes that all lipids have small, normally distributed effects associated with the target trait.

Whole-lipidome prediction of GCA for dry matter yield, flowering time, fat and sugar contents.
(a–d) Histograms showing Pearson correlations between individual lipids and GCA values. Correlations significant at a Bonferroni-corrected 5% threshold (P-value < 8.9 × 10−5) are colored in red. (e–h) Observed vs. predicted GCA values using whole-lipidome prediction. Average values obtained from a 20 times repeated 5-fold cross-validation are presented. Genotypes with a correct classification of GCA values are colored in green.
Using a 20-times repeated 5-fold cross-validation scheme, the correlations between observed and predicted GCA values ranged from 0.47 for dry matter yield to 0.78 for flowering time, with quality traits being better predictable than dry matter yield ( Fig. 4e–h ). The predictive ability matched with the number of Bonferroni-corrected significant correlations with GCA (Fig. 4A–D, red portion), which was highest for flowering time (190) and considerably higher for sugar (130) and fat content (162) compared to dry matter yield (31).
Discussion
The composition of the detected, genetically co-regulated modules in the lipidome provided hints about the hierarchical position of the underlying controlling genes. Whereas level 1 suggests genes operating at the most elementary level for modifying or controlling individual lipids, level 2 candidate genes are assumed to control sets of biochemically closely related lipids of one chemical class. On top, associations found for large groups of structurally distinct lipids (level 3) suggest underlying causal genes regulating the lipidome at a higher hierarchical level.
Currently, incomplete annotations limit our ability to distinguish perfectly the level 3 from level 2 candidate genes. Nevertheless, the suggested hierarchy levels largely corresponded with the specificity of the associated candidate genes. For example, the lipid signaling molecule sphingosine-1-phosphate has recently received great attention because of its involvement in many physiological processes in yeast, mammals and plants15,16, suggesting a higher hierarchical control position conserved over species. It was therefore not surprising that the underlying gene sphingosine kinase was associated with lipids from multiple chemical classes. Likewise, the conserved TRAM/LAG1/CLN8 (TLC) lipid-sensing domain, reported to be involved in several lipid processes17, was found in a product of a level 3 candidate gene associated with three lipids with different backbones but the same fatty acid residual ( Fig. 3 ). Similarly, the level 3 ethylene-responsive transcription factor was described to control different lipid pathways in Arabidopsis18 and barley19. On the contrary, lipid transporters are known to be more substrate specific20, especially if covalently modified21. This makes the detected level 2 triacylglyceride specific transporter a promising candidate for further functional investigations.
Two candidate genes, GRMZM2G084264, GRMZM2G073258 ( Table 1 ), show high homology to the three members (SHN1, SHN2 and SHN3) of the Arabidopsis SHN transcription factors which are involved in the regulation of a variety of lipid metabolism processes including lipid transport, fatty acid elongation and the synthesis of wax, suberin and cutin18,22,23. The gene GRMZM2G176175 shows high homology (Protein-Protein BLAST E-values 8 × 10-33) to the gene WRI1 that belongs to the protein family of the AP2/EREBP transcription factors in Arabidopsis which have been demonstrated to be involved in fatty acid synthesis and triacylglycerol biosynthesis processes24,25,26.
On chromosome 4, a candidate gene showed a high similarity (Protein-Protein BLAST E-value 1 × 10−12) to CER2, a gene studied in detail in Arabidopsis, which is required for the elongation of fatty acids of exceptional length27. Our results also propose that protection against peroxidation needs to be particularly considered. In addition to several glutathion-S-transferases28, we also detected an invertase gene that reduces membrane lipid peroxidation during chilling temperatures if artificially expressed in the apoplast of potato29.
Lipid synthesis occurs via sequential addition of two-carbon units derived from acetyl CoA. which is nicely reflected in the mapping of biochemically closely related lipids from specific lipid classes such as TAGs 56:4, 58:4 and 60:4 to just one locus ( Fig. 1b ). The DAG moiety has been proposed as the major pathway for TAG synthesis in excised linseed (Linum usitatissimum) and soybean (Glycine max) cotyledons30. The DAG moieties derived from phospholipid hydrolysis are used in galactolipid biosynthesis31. We identified a phospholipase in close vicinity to the respective SNP which might carry the causal variant of this association ( Fig. 1b ). It is obvious that like in any association study, the loci identified remain candidate genes, which need further validation via e.g. reverse genetics approaches for final annotation.
The rich functional diversity of the lipidome and its association to a diversity of biological processes suggests that it might be used for the prediction of phenotypic performance. We tested this hypothesis by predicting agronomic relevant traits evaluated in hybrid progeny in multi-environment field trials. The high prediction accuracy confirmed by cross-validation showed that such an approach is very promising. In addition, the higher absolute correlation coefficients and cross-validated predictions for flowering time and sugar and fat content compared to dry matter yield indicated that the leaf lipidome is in general more closely connected to quality or developmental traits than to biomass.
In conclusion, our study demonstrates that lipidomics is not only a powerful tool for shedding light on the genetic control of the lipid metabolism in crop plants, but it also provides access to a novel layer of biochemical information exploitable for predicting agronomic traits relevant in hybrid maize breeding.
Methods
Genetic material
The genetic material consisted of (i) a previously described diversity panel of 285 Dent inbred lines12 with additional four genotypes as checks and (ii) the 570 testcrosses produced by crossing the 285 inbred lines to two single cross European Flint testers13.
Genotyping
Genotyping was performed using the Illumina SNP chip Maize-SNP50 (Illumina Inc.) containing 56,110 unique SNPs32. A quality preprocessing was done by applying the following criteria: (i) call rate above 0.95, (ii) unique allele assignment for the 22 replicated checks of genotype B73, (iii) minor allele frequency greater than 2.5% and (iv) no more than three heterozygous genotypes. A total of 37,282 SNPs met these criteria. Five genotypes with a residual heterozygosity above 5% were excluded. The chromosomal positions of the SNPs refer to the B73 reference genome (B73 RefGen_v1). Candidate genes were taken from the B73 filtered gene set (release 4a.53).
Lipid profiling
Inbred lines in field trials were subject to lipid profiling. Genotypes were divided into three maturity groups and randomized as three adjacent 20 × 5 α-lattice designs with two replications and planted in two-row plots. Leaf samples of ≈ 5 cm were cut from the middle part of the fully developed third leaf of 10 plants per plot, bulked and immediately frozen using dry ice. The five plots of every incomplete block were sampled within a period of 15 s to minimize within-block error due to metabolic changes over time. The 50 samples from 10 randomly chosen blocks of one field replication of one maturity group were subsequently processed together as one batch. With this blocking structure, we could account for systematic shifts among batches while keeping the field randomization intact. Samples on batches were lyophilized and stored for the subsequent analysis. Samples were processed using ultra-performance liquid chromatography coupled with Fourier transform mass spectrometry (UPLC-FT-MS, ref. 6) on a C8 reverse phase column coupled with an Exactive mass spectrometer (Thermo-Fisher, http://www.thermofisher.com) in positive ionization mode. Processing of chromatograms, peak detection and integration were performed using REFINER MS® 5.3 (GeneData, http://www.genedata.com). Processing of mass spectrometry data included the removal of the fragmentation information, isotopic peaks, as well as chemical noise. Obtained features (m/z at a certain retention time) were queried against an in-house lipid database for further annotation. Remaining unknown features were crossed checked with a different, on-line database using an in-house developed database search tool (Golm Biochemical Space, GOBIOSPACE) for putative annotation.
Multi environment trials of testcrosses
The 570 testcrosses were used to estimate GCA values in multi-environment field trials in three agroecologically diverse locations for two years (2008 and 2009) in Germany, as described previously13.Dry matter yield of whole-plant biomass (t/ha) and female flowering (d) were measured per field plot. Fat and sugar contents (%) were measured in 1 kg of harvested plant material from each plot using calibrated near-infrared spectroscopy (NIRS33).
Phenotypic analysis
Mass intensities were scaled by the median of each sample. Linear mixed model analysis was used to obtain least squares means of lipid levels and GCA values. The model for the lipids was G + M + M·R + M·R·A + M·R·A·B, with the following effects: genotype (G), trial of maturity group (M), field replication (R), batch (A) and block (B). Random effects are underlined. To achieve homoscedasticity of the model residuals for the lipids, the flexible Box-Cox power transformation was applied. For each lipid, the optimum transformation parameter was determined using the maximum likelihood method described previously34. Repeatabilities were calculated as w2 = σ2g/(σ2g + σ2e/2) where σ2g is the genotypic variance estimated by REML, by setting factor G as random and σ2e is the residual variance. Following established analysis, the testcross genotype was decomposed as G = GCA + T + SCA, where GCA is the main effect of the line, T is the main effect of the tester genotype and SCA (specific combining ability) is the line × tester interaction. Including an effect for the environment (E), defined as the year × location combination, the final model for estimating GCA was therefore GCA + T + SCA + GCA·E + T·E + SCA·E + E + E·M + E·M·R + E·M·R·B.
Genome-wide association mapping
The GWA mapping with plant populations is often hampered by population structure and cryptic relatedness, resulting in spurious associations35. However, powerful techniques are available to decouple genetic associations from confounding factors36. We used a mixed model approach which corrects simultaneously for the main directions of population substructure by regression on the first ten principal components (Q10), as well as pairwise relatedness by using an allele-sharing kinship matrix K as covariance matrix for the random genotype effects37. For an appropriate significance threshold for SNP–trait associations, we controlled the false discovery rate (FDR), as previously suggested for GWA mapping38. Genome-wide inflation factors (λ) were calculated as the regression coefficient in the QQ plot with a zero intercept. The GWA models were fitted using the maximum likelihood implementation in the function polygenic of GenABEL39. P-values were obtained with the 1 degree of freedom fast association score test–based analysis (FASTA), implemented in the function mmscore of GenABEL. P-values were transformed to q-values and regarded significant if smaller than 0.025 to control for a false discovery rate (FDR) of 2.5%. The proportion of genetic variance explained by a certain SNP was calculated as ρ = R2LR/w2 using the likelihood-ratio (LR) statistic R2LR = 1 – exp(–LR/n) with LR = 2 × log(LSNP/L0), where L0 is the maximum likelihood of the baseline Q10 + K model without considering the SNP, LSNP is the maximum likelihood of the full Q10 + K model including the SNP as cofactor and n is the number of genotypes40.
Whole-lipidome prediction of GCA
We applied ridge regression BLUP (RR-BLUP41), for prediction of GCA values using all lipids as predictor variables. The model makes two assumptions: (i) normally distributed predictor effects and (ii) normally distributed errors. Validation was performed using a 20 times repeated 5-fold cross validation. The data set was divided into five disjoint subsets of genotypes, where one subset was left out for validation. The other four subsets were used as the training population to estimate the model parameters for predicting the observations of the left-out genotypes in the validation population. In each of the five rounds, the Pearson correlation between observed and predicted GCA values was calculated. Twenty different randomizations for assigning the genotypes to five different subsets were used to yield 100 cross-validation runs. The predictive ability was then calculated as the Pearson correlation between the observed and predicted GCA values averaged over all cross-validation runs.
References
Mutch, D. M., Fauconnot, L., Grigorow, M. & Fay, L. B. Putting the ‘Ome’ in lipid metabolism. Biotechnol. Annu. Rev. 12, 67–84 (2006).
Broun, P., Shanklin, J., Whittle, E. & Somerville, C. Catalytic plasticity of fatty acid modification enzymes underlying chemical diversity of plant lipids. Science 282, 1315–1317 (1998).
Brown, H. A. & Murphy, R. C. Working toward an exegesis for lipids in biology. Nat. Chem. Biol. 9, 602–606 (2009).
Vu, H. S. et al. Direct infusion mass spectrometry of oxylipin-containing Arabidopsis membrane lipids reveals varied patterns in different stress responses. Plant Physiol. 158, 324–339 (2012).
Welti, R. & Wang, X. Lipid species profiling: a high-throughput approach to identify lipid compositional changes and determine the function of genes involved in lipid metabolism and signaling. Curr. Opin. Plant Biol. 7, 337–344 (2004).
Hummel, J. et al. Ultra Performance Liquid Chromatography and High Resolution Mass Spectrometry for the Analysis of Plant Lipids. Front. Plant Sci. 2, 1–17 (2011).
Horn, P. J. & Chapman, K. D. Lipidomics in tissues, cells and subcellular compartments. Plant J. 70, 69–80 (2012).
Wenk, M. R. Lipidomics: new tools and applications. Cell 143, 888-895 (2010).
Wenk, M. R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 4, 594–610 (2005).
O'Neill, C. M. et al. Towards the genetic architecture of seed lipid biosynthesis and accumulation in Arabidopsis thaliana. Heredity (Edinb) 108, 115–123 (2012).
Li, H. et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50 (2013).
Riedelsheimer, C. et al. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 109, 8872–8877 (2012).
Riedelsheimer, C. et al. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 44, 217–220 (2012).
Bach, L. et al. The very-long-chain hydroxy fatty acyl-CoA dehydratase PASTICCINO2 is essential and limiting for plant development. Proc. Natl. Acad. Sci. USA 105, 14727–14731 (2008).
Spiegel, S. & Milstien, S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Bio. 4, 397–407 (2003).
Fyrst, H. & Saba, J. D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 6, 489–497 (2010).
Winter, E. & Ponting, C. P. TRAM, LAG1 and CLN8: members of a novel family of lipid-sensing domains? Trends Biochem. Sci. 27, 381–383 (2002).
Aharoni, A., Dixit, S. & Jetter, R. The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties and confers drought tolerance when overexpressed in Arabidopsis. Plant Cell 16, 2463–2480 (2004).
Taketa, S. et al. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. USA 105, 4062–4067 (2008).
Kader, J. C. Lipid-transfer proteins in plants. Annu. Rev. Plant Physiol. Plant. Mol. Biol. 47, 627–654 (1996).
Bakan, B. et al. Specific adduction of plant lipid transfer protein by an allene oxide generated by 9-lipoxygenase and allene oxide synthase. J. Biol. Chem. 281, 38981–38988 (2006).
Broun, P., Poindexter, P., Osborne, E., Jiang, C. Z. & Riechmann, J. L. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 101, 4706–4711 (2004).
Shi, J. X. et al. SHINE transcription factors act redundantly to pattern the archetypal surface of Arabidopsis flower organs. PLoS Genet 7, e1001388 (2011).
Cernac, A. & Benning, C. WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 40, 575–585 (2004).
Masaki, T. et al. ACTIVATOR of Spomin::LUC1/WRINKLED1 of a Arabidopsis thaliana transactivates sugar-inducible promoters. Plant Cell Physiol 46, 547–556 (2005).
Baud, S. et al. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 50, 825–838 (2007).
Haslam, T. M., Mañas Fernández, A., Zhao, L. & Kunst, L. Arabidopsis ECERIFERUM2 is a component of the fatty acid elongation machinery required for fatty acid extension to exceptional lengths. Plant Physiol. 160, 1164–1174 (2012).
Marrs, K. A. The functions and regulation of Glutathione S-Transferases in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 127–158 (1996).
Deryabin, A. N. et al. Influence of yeast-derived invertase gene expression in potato plants on membrane lipid peroxidation at low temperature. J. Ther. Bio. 30, 73–77 (2005).
Slack, C. R., Roughan, P. G. & Balasingham, N. Labeling of Glycerolipids in Cotyledons of Developing Oilseeds by [1-C-14]Acetate and [2-H-3]Glycerol. Biochem J. 170, 421–433 (1978).
Maoyin, L., Welti, R. & Wang, X. Quantitative profiling of arabidopsis polar glycerolipids in response to Phosphorus starvation. Roles of Dξ1 and Dξ2 in phosphatidylcholine hydrolysis and digalactosyldiacylglycerol accumulation in phosphorus-starved plants. Plant Physiol. 142, 750–761.
Ganal, M. W. A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping and genetic mapping to compare with the B73 reference genome. PLOS ONE 6, e28334 (2011).
Grieder, C. et al. Determination of methane fermentation yield and its kinetics by near infrared spectroscopy and chemical composition in maize. JNIRS 19, 463–477 (2011).
Piepho, H. P. Data transformation in statistical analysis of field trials with changing treatment variance. Agronomy J. 101, 865–869 (2009).
Astle, W. & Balding, D. J. Population structure and cryptic relatedness in genetic association studies. Stat. Sci. 24, 451–471 (2009).
Sillanpää, M. J. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity 106, 511–519 (2011).
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208 (2006).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 100, 9440–9445 (2003).
Aulchenko, Y. S., Ripke, S., Isaacs, A. & van Duijn, C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007).
Sun, G. et al. Variation explained in mixed-model association mapping. Heredity 105, 333–340 (2010).
Piepho, H. P. Ridge regression and extensions for genomewide selection in maize. Crop Sci. 49, 1165–1176 (2009).
Acknowledgements
We thank the staff of the experimental research stations of the University of Hohenheim for assistance in conducting the field experiments and Aenne Eckardt and Gudrun Wolter for sample preparation. This research was supported by the Max Planck Society and the German Federal Ministry of Education and Research (BMBF) within the projects GABI-Energy (grant 0315045) and OPTIMAL (grant 03159580).
Author information
Authors and Affiliations
Contributions
C.R. performed leaf sampling, phenotypic analysis, genome-wide association and whole-lipidome prediction. C.R. and Y.B. carried out the annotation of the candidate genes. M.M. and Y.B. performed lipid profiling and M.M. accomplished the chemical annotation. C.R. and Y.B. wrote the manuscript. L.W. and A.E.M. designed the experiments and supervised the research.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Riedelsheimer, C., Brotman, Y., Méret, M. et al. The maize leaf lipidome shows multilevel genetic control and high predictive value for agronomic traits. Sci Rep 3, 2479 (2013). https://doi.org/10.1038/srep02479
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02479
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
