
Abstract
Due to the widespread prevalence of resistant parasites, chloroquine (CQ) was removed from front-line antimalarial chemotherapy in the 1990s despite its initial promise of disease eradication. Since then, resistance-conferring mutations have been identified in transporters such as the PfCRT, that allow for the efflux of CQ from its primary site of action, the parasite digestive vacuole. Chemosensitizing/chemoreversing compounds interfere with the function of these transporters thereby sensitizing parasites to CQ once again. However, compounds identified thus far have disappointing in vivo efficacy and screening for alternative candidates is required to revive this strategy. In this study, we propose a simple and direct means to rapidly screen for such compounds using a fluorescent-tagged CQ molecule. When this screen was applied to a small library, seven novel chemosensitizers (octoclothepin, methiothepin, metergoline, loperamide, chlorprothixene, L-703,606 and mibefradil) were quickly elucidated, including two which showed greater potency than the classical chemosensitizers verapamil and desipramine.
Similar content being viewed by others

Introduction
With the widespread distribution of drug and multi-drug resistant malaria parasites1 and the recent emergence of artemisinin-delayed clearance P. falciparum2, research into novel antimalarial approaches remains integral in the fight against this global disease. Although the recent discovery of spiroindolones as potential antimalarials holds great promise3, the cost of testing and developing such new chemical entities often restricts their numbers. Commendable initiatives have been made to fund new drug developments but the “out-dating” of drugs side-lined by resistant parasites is a bitter pill to swallow.
When resistance to a drug arises due to mutations in its biological target, as exemplified by atovaquone where changes in the cytochrome bc1-complex reduces drug-target affinity4, such drugs may only be useful in combinational therapy5. For resistance due to delayed clearance, as exemplified by artemisinin resistant parasites2, it seems conceivable to change the drug's disposition by altering the dosing regimen, formulation or chemical structure. This would reduce its elimination from the body and thereby prolong the duration of therapeutic blood concentrations of the drug. With transporter-mediated resistance, as in the case with chloroquine (CQ) resistance, augmentation of blood concentrations by adjusting drug disposition may also be effective if the kinetics is favourable and toxicity tolerable6. Another strategy to tackle transporter-mediated resistance is the co-administration of drugs that specifically inhibit these transporters or pumps. Indeed, the effectiveness of such chemosensitizing compounds in vitro has been long-known in the case of CQ resistance (CQR) in P. falciparum5,7.
Though CQ once held the promise of being able to eradicate malaria, its massive use (and misuse) has led to the emergence of CQR P. falciparum parasites8. Their rapid and almost global spread has dampened the clinical usefulness of the drug, relegating it almost exclusively to the treatment of P. vivax and even that use is being challenged by the emergence of CQR P. vivax9.
While numerous chemosensitizing compounds have been painstakingly identified in the hope of restoring the clinical usefulness of CQ, most of these display disappointing in vivo pharmacodynamics (poor potency owing to serum protein-binding10,11, poor pharmacokinetic properties such as rapid conversion into an inactive metabolite5) or have a poor safety profile and have been contraindicated against existing antimalarials12. The only chemosensitizer that has undergone clinical trials is chlorpheniramine13 and even then, research on its efficacy is scarce and the results disappointing. There is a clear need to identify novel ‘druggable’ candidates with suitable safety profiles and in vivo efficacy in an expedient manner.
In this study, we present a novel, direct, rapid and simple screening method to identify chemosensitizing compounds based on a new fluorophore-tagged CQ tool. Based on the increased fluorescence corresponding to the accumulation of this molecule in resistant parasites, we were able to differentiate between compounds that interfere with CQ resistance transporters from those that do not. After optimization of the assay, the screen was applied to a library of 1280 pharmacologically active compounds (LOPAC) and the validated hits are discussed in this report.
Results
Pre-screening validation of coumarin-tagged CQ (CM-CQ)
The pfcrt gene encodes for a putative amino acid transporter that is localized to the digestive vacuole membrane of the parasite and has been shown to be a major modulator of chloroquine resistance14. CQ resistant parasites accumulate less CQ than sensitive parasites via an efflux mechanism conferred by mutations in this gene15. As such, it was hypothesized that the accumulation of CM-CQ in resistant parasites would be similarly reduced but might be increased in the presence of chemosensitizers that inhibit the efflux of the drug.
To determine the functionality of CM-CQ in the identification of chemosensitizers, seven reported chemosensitizers12 were assayed for their effectiveness in CQ-resistant (CQR) K1 parasites. After 10 hrs co-treatment with CM-CQ and various concentrations of these compounds, flow cytometric analysis was carried out to determine if there was a detectable increase in CM-CQ fluorescence. Except for propranolol (PPL) and diltiazem (DTZ), there was a significant increase in the proportion of CM-CQ-positive K1 parasites when treated with verapamil (VPM), chlorpromazine (CPZ) and desipramine (DSP) at 5 μM and 10 μM (P < 0.001 for all) and also for promethazine (PMZ) and chlorpheniramine (CPR) at 10 μM (P < 0.001 and P < 0.05 respectively) (Fig. 1a and b). The concentration of chemosensitizers was set at 10 μM in subsequent screenings to reduce the incidence of false negatives.

Optimization of chemosensitizer detection assay.
(a) Representative flow cytometry histogram overlay of K1-infected erythrocytes (at 11.3% parasitemia) treated with 6 μM of CM-CQ and co-incubated with either vehicle control (Veh), 10 μM of propranolol (PPL) or 10 μM of verapamil (VPM) for 10 hrs. Total percentages of positively-stained erythrocytes in R2 are shown, with the relative proportion of parasite-infected cells indicated in brackets. (b) Proportion of CM-CQ-positive K1 parasites after co-incubation with between 0–10,000 nM of verapamil (VPM), propranolol (PPL), diltiazem (DTZ), chlorpromazine (CPZ), desipramine (DSP), promethazine (PMZ) and chlorpheniramine (CPR). (**, P < 0.001 and *, P < 0.05 compared to negative control) Figures show mean ± SEMs; n = 3. (c) Fluorescence plate measurements of 3D7 (CQ-sensitive) or K1 (CQ-resistant) cultures treated with CM-CQ in the absence or presence of 10 μM verapamil (VPM), propranolol (PPL), diltiazem (DTZ), chlorpromazine (CPZ), desipramine (DSP), promethazine (PMZ) and chlorpheniramine (CPR). (***, P < 0.001; **, P < 0.01 and *, P < 0.05 compared to negative control) Figures show mean ± SEMs; n = 3. (d) Fluorescence plate measurements of 3D7 (CQ-sensitive), 7G8 (CQ-intermediate resistance) or K1 (CQ-resistant) cultures treated with CM-CQ in the absence or presence of 10 μM verapamil (VPM), chlorpromazine (CPZ) or desipramine (DSP). (***, P < 0.001; **, P < 0.01 and *, P < 0.05 compared to negative control) Figures show mean ± SEM; n = 4.
The strictly standardized mean difference (SSMD) for this assay was calculated to be 5.87, a score suggesting that this would make an “excellent” screening tool with 10 μM VPM as a positive control. However, the lack of a high-throughput cytometer led us to consider using fluorescence-plate measurements to detect the changes in CM-CQ accumulation instead.
To ensure the suitability of the fluorescence plate reader, the same staining procedure was repeated with the five chemosensitizers which showed a significant increase in CM-CQ fluorescence by flow cytometry and these also demonstrated a significant increase in relative fluorescence intensity as determined by the plate reader (Fig. 1c). Although less robust than using flow cytometry, the SSMD for the plate readings was calculated to be 2.81, indicating that this was a “good” assay with 10 μM VPM acting as control.
Additionally, VPM, CPZ and DSP were tested for their ability to increase CM-CQ accumulation in CQ-sensitive (CQS) 3D7 and CQ-intermediate resistance (CQIR) 7G8 parasites. The high fluorescence levels in 3D7 compared to 7 G8 and K1 suggest that the drug is behaving like its parent molecule (unlabelled CQ) in showing reduced accumulation in parasites harboring the mutant PfCRT (Fig. 1d). While none of these compounds significantly augmented the fluorescence readings of 3D7 parasites, an increase was observed in 7G8 after co-incubation with CPZ (P < 0.05) and DSP (P < 0.01) but not with VPM (Fig. 1d). This is in line with literature that has demonstrated the reduced sensitivity of 7 G8 parasites to VPM chemosensitization11. Having sufficiently validated the assay's performance, we proceeded to screen the LOPAC library for novel chemosensitizers.
Screening of LOPAC Library and Hit Selection
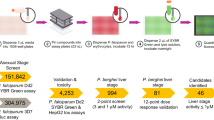
The entire library (supplied on sixteen 96-well plates) was screened five times and hits were selected that were at least the mean of VPM positive controls in each plate plus one standard deviation ( Positive Control + 1SD). Out of the 1280 compounds in the LOPAC library, 46 tested positive in at least three of the five screens (Supplementary Table 1).
Positive Control + 1SD). Out of the 1280 compounds in the LOPAC library, 46 tested positive in at least three of the five screens (Supplementary Table 1).
Two separate screens were then performed (with 10 μM of library compounds and 6 μM of unlabelled CQ) to detect for false-positive hits that possessed inherent fluorescence. Hits that surfaced in either of these auto-fluorescence screens were discarded leaving only 16 hits (Supplementary Table 2). Of these, a further two were discarded: compound WIN62577 was discarded because it had uncharacteristically high fluorescence measurements; also, chlorpromazine because it was already used in the optimization of the screen.
Validation of primary hits
To ensure that results were free from contamination or mix-up during the full-library screen, individual samples of hit compounds were purchased directly for subsequent validation. Due to product discontinuation, clemastine fumarate was omitted from further validation leaving only 13 hits for validation.
Flow cytometry was used to confirm the increase in CM-CQ fluorescence in hit-sensitized K1 and 10 of the 13 hits were validated as having significantly increased the proportion of CM-CQ-positive parasites (Fig. 2, P < 0.001 for cyproheptadine, loperamide, metergoline, L765314, L703606, mibefradil and xanthine amine; P < 0.01 for prochlorperazine and chlorprothixene; P < 0.05 for methiothepin). It was also demonstrated that CM-CQ accumulation was not augmented in CQS 3D7 parasites treated with the same compounds (Supplementary Figure 1). Flow cytometry was also performed to determine if these compounds were auto-fluorescent (Supplementary Figure 2a and 2b). It was shown that L765314 had a higher level of background fluorescence in both 3D7 and K1 (P < 0.001) and prochlorperazine had a higher background level in 3D7 only (P < 0.05). On the basis that their fluorescence levels were not excessively high, we decided to continue with the validation of these hits.

Confirmation of screening hits via flow cytometry.
Proportion of CM-CQ-positive K1 parasites after treatment with 6 μM of CM-CQ and 10 μM of verapamil, desipramine, chlorpromazine, cyproheptadine, prochlorperazine, chlorprothixene, cyclosporine A, loperamide, metergoline, L765314, L703606, mibefradil, methiothepin, octoclothepin, tamoxifen, xanthine amine or vehicle control (Veh). (***, P < 0.001; **, P < 0.01 and * P = 0.05) Figures show mean ± SEM; n = 3.
To exclude the interference of a compound's possible intrinsic antimalarial activity from its bona fide chemosensitizing effect, the approximate IC50 of all compounds against K1 were determined by plotting a 12-point curve (Table 1). Based on these values, the potency of each hit in reducing CQ IC50 in K1 was assayed up to 50% of each hit's IC50 or 10,000 nM, whichever was lower. The modulation of CQ IC50 was indicated by a response modification index (RMI) that has been developed by others11. Of the 13 compounds, four compounds (cyclosporin A, xanthine amine, tamoxifen and L765314) did not appear to modulate CQ IC50 even at 1 μM (Fig. 3). In the case of xanthine amine and L765314, these hits were probably false-positive due to their earlier noted inherent fluorescence (Supplementary Figure 2 and Supplementary Table 2 respectively). Four compounds, octoclothepin, methiothepin, metergoline and loperamide, appeared to have a moderate effect on reducing CQ IC50 while another three compounds, prochlorperazine, chlorprothixene and cyproheptadine, appear to have potencies similar to the VPM and DSP controls. Notably, L703606 and mibefradil appear to have superior potencies to the classical chemosensitizers, considerably reducing CQ IC50 even at concentrations below 100 nM. A summary of the potency of all 13 compounds (described by their corresponding EC50), their novelty as chemosensitizers and current applications are summarized in Table 1.

Dose-dependent effects of hits on the Response Modification Index (RMI).
Chloroquine (CQ) IC50 was determined by 8 dilutions of CQ by the reinvasion assay in of varying concentrations of the respective controls/hits (up to half each compounds' approximate IC50 value or 10,000 nM, whichever was lower, see Table 1). To reduce inter-experiment variability, the derived IC50 values were normalized against each day's vehicle (CQ-only) control IC50 to get the respective RMI value. Although the experiment was not repeated due to the large number of samples needed for each run, each curve was drawn with between 6 to 16 points that were constructed based on IC50 derived from the experiments performed on between 2 to 5 separate days. Constraints for regression were set with a maximum of 1.0 and a minimum of 0.1.
Focussing only on the two most potent hits, more accurate IC50 values for mibefradil and L703606 in K1 were established with a 96-point IC50 curve to be 108 nM and 529 nM respectively (Supple Fig. 3). In order to be stringent in excluding the effect of their intrinsic antimalarial activities, only a quarter of these concentrations (27 nM and 132 nM respectively) were used on subsequent validation experiments on multiple lab strains/field isolates.
Both compounds had negligible effects on the CQ IC50 of CQS 3D7 and HB3 and CQIR 7G8 (Fig. 4a and Supplementary Table 3). However, both compounds reduced the CQ IC50 values of all tested CQR parasites significantly (P < 0.05 for all). Compared to the DSP control, L703606 was similarly effective against 8 of 8 CQR parasites, while mibefradil was only as effective as DSP in 3 of 8 CQR parasites. As an alternative means of assessing the two hits, their ability to half the CQ IC50 value in the tested strains/isolates was evaluated (Fig. 4b). Once again, L703606 was able to achieve a RMI below 0.5 for all 8 of 8 CQR parasites while mibefradil only accomplished this in 3 of 8 of the tested strains/isolates, suggesting that L703606 is more suitable than mibefradil as a chemosensitizer owing to its widespread efficacy.

Effect of L703606, mibefradil and desipramine on multiple lab strains and field isolates.
(a) Chloroquine (CQ) IC50 in CQ-sensitive 3D7 and HB3, CQ-intermediate resistant 7G8 and CQ-resistant K1, Dd2, CS2, T9/94, SMRU0233, SMRU0272, SMRU1093 and SMRU1116 parasites in the presence of vehicle control (Veh), 132 nM of L703606, 27 nM of mibefradil or 500 nM of desipramine. P-values are in Supplementary Table 3 and show that all three compounds are effective in reducing the respective IC50, but that mibefradil is less effective than desipramine in CS2, SMRU0233, SMRU0272, SMRU1093 and SMRU1116. Figures show mean ± SEM; n = 3. (b) Response modification index shows the relative reduction of CQ IC50 between the untreated and treated cultures.
Discussion
Traditionally, the discovery of chemosensitizing activity has relied heavily on the detection of changes in CQ IC50 values or isobologram analysis. Such assays have routinely been performed by tedious microscopic examination of Giemsa-stained slides or radioactive assays, such as the hypoxanthine incorporation assay to measure parasite development16. Although the utility of fluorescent nucleic acid stains (like dihydroethidium, Hoechst33342 and SYBR Green I16,17) and the availability of detection tools (like fluorescence plate readers and flow cytometers) has made IC50 determination less tedious, the screening for chemosensitizers remains hampered by the sheer number of drug dilutions that need to be made and the consequential number of wells that need to be read. In this study, we present a novel, simple and rapid method of screening for new CQ chemosensitizers. To validate the assay, a preliminary screen on the LOPAC library was performed, which subsequently uncovered seven novel chemosensitizers with pre-established drug-like properties.
By utilizing a newly-developed, fluorescent-labelled CQ tool18,19, we have developed screening assays to quickly, easily and directly detect changes in the uptake and retention of CQ within CQR K1 parasites. Since CQR parasites possess transporters like the CQ-resistance transporter (PfCRT) and p-glycoprotein homolog 1 (Pgh-1), these transporters are believed to efflux the protonated form of CQ from the parasite's digestive vacuole, the drug's primary site of action, thereby giving rise to a low fluorescence reading. Chemosensitizing compounds block these transporters and increase the concentration of CQ within the parasite12 leading to an increase in fluorescence readings. Though this difference in fluorescence levels can be measured though flow cytometry sensitively, limitations in throughput led us to utilize a fluorescent plate reader instead.
While chemosensitizers are generally believed to function by interfering with the PfCRT, our assay is not designed to be target-specific. Whether via interference with PfCRT, Pgh-1, multi-drug resistance protein (PfMRP) or other transporters12, it is the net increase in CM-CQ concentration that is being used to detect the actual functional hits. This detection however, does not discount the possibility of alternative mechanisms for CM-CQ accumulation: beyond transporter interference, compounds which facilitate the import of CM-CQ into the parasite or increase the acidity of the DV leading to greater levels of CM-CQ retention20,21,22 would also be detected. Though this lack of specificity may lead to questions about the precise molecular target of any hits generated, it does allow for the identification of compounds that have the correct functional properties to achieve an increase of CM-CQ accumulation in whole-cell environments. The elucidation of the various possible mechanisms would be clearly the subject of further detailed studies.
After excluding for auto-fluorescent compounds and validating the effect of the compounds in enhancing CM-CQ accumulation in K1 via flow cytometry, further efforts were made to ensure that the effect on altering the CQ IC50 was primarily a result of the compounds' chemosensitizing effect and not a synergistic effect. This was achieved by performing a dose-dependent potency study, limited to only half the IC50 value of the hit. As a result, seven novel chemosensitizers of varying potencies were validated and these displayed overall structural similarity to other known chemosensitizers.
A three-dimensional QSAR pharmacophore model for CQ-resistance chemosensitizers has previously been characterized23 using the CATALYST software from SPARTAN based on a training set of 17 compounds including imipramine and its analogues. This model describes two aromatic hydrophobic interaction sites and a hydrogen bond acceptor site at a side chain, preferably a nitrogen atom (Fig. 5, top). The distances between these features are not always conserved, but the secondary or tertiary aliphatic nitrogen atom is linked to the aromatic rings by a two or three carbon side-chain for most of the examples.

Schematic representation of the pharmacophore model developed by Bhattacharjee et al. and three CQ-resistance reversal agents with their “Best-Fit” Scores.
(top); chemical structure of the seven novel chemosensitizers presented in this report (bottom).
The seven novel chemosensitizers described in this report also possess similar characteristics (Fig. 5, bottom) with some minor differences. With mibefradil, the distance between the fluoro-aromatic system and the nitrogen atom is four carbon atoms. For L-703,606, the three aromatic rings and two nitrogen atoms mean that several combinations are possible for the role of the two hydrophobic interaction sites and the hydrogen bond acceptor. Similarly, loperamide also possesses two nitrogen atoms and three aromatic rings, but the amide-nitrogen is less likely to play the role of the hydrogen bond acceptor. Chlorprothixene is structurally similar to imipramine, thus having the same distance of the aromatic hydrophobic interaction sites to the nitrogen atom. The structure of metergoline is more complex yet bearing all the salient features of the pharmacophore. For this compound, the basic centre in the polycycle is probably the better hydrogen bond acceptor, compared to the carbamate group. Methiothepin and octoclothepin also conform to the pharmacophore and differ from each other only in the substituent on the C8 position (methylthio and chloro, respectively).
Of these seven novel chemosensitizers, only L703606 and mibefradil were observed to be considerably more potent than classical chemosensitizers. Unlike the classical chemosensitizers VPM and DSP which required concentrations of about 291 nM and 152 nM respectively to halve the IC50 of CQ in K1, L703606 and mibefradil required only about 98 nM and 38 nM, respectively.
L703606 is a potent and selective non-peptide neurokinin-1 (NK1) tachykinin receptor antagonist24 but little is known about its pharmacokinetics or toxicity profile. Mibefradil is a long-acting calcium antagonist used in the treatment of hypertension and chronic stable angina pectoris25. More recently and independent to our current findings, the synergistic antimalarial effect of mibefradil with CQ has been identified, though its effect as a chemosensitizer was not determined26. As a reference point, the pharmacokinetics of a single oral 320 mg dose is briefly as follows: Cmax is about 2700 nM, Tmax is about 2 hrs and T½ is about 16 hrs25. However, due to inhibition of the CYP3A4 pathway, numerous clinically relevant drug interactions were predicted and this led to its voluntary withdrawal from the market. Since mibefradil appears to be effective in K1 at below 100 nM, it is tempting to propose mibefradil as a chemosensitizing candidate. However, it should be noted that CYP3A4 is important in the metabolism of many antimalarial drugs27 and future clinical studies involving the combination of antimalarial drugs with potential CYP3A4 inhibitors should proceed cautiously.
As a final round of validation, the effectiveness of L703606 and mibefradil to sensitize multiple CQR laboratory and field isolates were assayed at minimally toxic levels of ¼ their IC50. While mibefradil was more potent than L703606 in K1, its effectiveness was not consistent across the tested parasite strains isolates. Although a higher level of mibefradil might compensate for this discrepancy, such heterogeneity may be suggestive of existing genotypic variations that modulate susceptibility to mibefradil chemosensitization. In turn, this suggestion raises concerns about the long-term clinical usefulness of mibefradil as a chemosensitizer. In contrast, the effectiveness of L703606 was consistent in all CQR strains, suggesting that no existing resistance traits appear to exist. This potency even in artemisinin-resistant SMRU0233 and SMRU0272 field isolates is indicative of the clinical relevance of L703606. However, further studies are needed to determine if selection pressure would readily give rise to resistant mutants.
Although assaying for the effectiveness of mibefradil and L703606 in murine models of malaria would be the next logical step, the mechanism of CQ-resistance for murine parasites remains to be well characterized and may or may not be similar to P. falciparum. For example, CQ-resistant P. chabaudi mutants selected by drug-pressure had no mutations in their respective orthologs of pfcrt and pfmdr128. In P. berghei recombinant clones expressing the PfCRT from CQIR 7G8, the transporter was shown to be correctly expressed and localized to the DV of parasites but failed to confer CQ resistance ex vivo or in vivo29. In P. yoelii, innate CQ-resistance has long been known to exist in wild-type parasites without prior drug pressure but mechanisms of resistance have yet to be well understood. More recently however, a study using a novel high-resolution quantitative whole-genome re-sequencing approach revealed four point mutations on three other genes that were predicted to confer CQ resistance in P. chabaudi30. Of these, a mutation in aat1, a putative amino acid transporter, was shown to be crucial for resistance and has many similarities with resistance-conferring PfCRT mutations such as a shifting of the negative charge away from the start of the first transmembrane helix. Though the mechanisms of CQ resistance in mice models may yet to be clear, such models have been successfully used to validate some chemosensitizers31,32,33. As such, future studies in these useful model organisms may be pursued to determine if mibefradil and L703606 will have a sensitizing effect on resistant murine parasites in addition to validating the safety profile of such combinations in a pre-clinical model.
While the screening process for novel chemosensitizers was found to be both rapid (each screen of the LOPAC library was accomplished within 15 hrs) and simple to perform (three steps: staining, washing and taking readings), future work to further improve the screen includes the development and use of a more highly-fluorescent CQ molecule (to increase the resolution between high and low fluorescence levels and reduce false negatives) and the utilization of a high-throughput flow cytometer (which has a greater sensitivity than the plate reader). In addition, the development of a counter-screen using a complimentary (non-overlapping) fluorophore-emission is another possibility (so as to minimize the exclusion of true positives that possess inherent blue fluorescences).
Notwithstanding, the identification of seven novel chemosensitizers from the LOPAC library of 1280 compounds is now revealed. Two of these hits possess greater potency than classical chemosensitizers. Such results clearly demonstrate the usefulness of our assay in the fight against malaria. In a high throughput context, extensive screening campaigns of larger drug-like compound libraries are conceivable. Such endeavours should help uncover a greater set of novel chemosensitizers with suitable efficacy and safety profiles. Arguably, in combination with CQ, this straightforward screening method has the potential to return CQ to the arsenal of effective antimalarial treatments in the clinic.
Methods
Parasite culture
The seven laboratory strains of P. falciparum, used include 3D7 (MRA-102), HB3 (MRA-155), 7G8 (MRA-154), K1 (MRA-159), CS2 (MRA-96), Dd2 (MRA-156) and T9/94 (MRA-153), all of which were obtained from MR4, ATCC Manassas Virginia. Four P. falciparum field isolates were collected from malaria patients attending the clinics of the Shoklo Malaria Research Unit (SMRU) Mae Sod region of Tak Province in the northwest of Thailand from January 2009 to December 2010 and designated as SMRU0233, SMRU0272, SMRU1093 and SMRU1116. All P. falciparum cultures were cultured continuously and synchronized weekly as described previously18.
Preparation of drugs & library
CM-CQ was synthesized, stored and prepared as described previously18.
All other drugs were purchased from Sigma-Aldrich including the Library of Pharmacologically Active Compounds (LOPAC1280), chloroquine diphosphate (CQ), (±)-verapamil hydrochloride (VPM), chlorpromazine hydrochloride (CPZ), desipramine hydrochloride (DSP), promethazine hydrochloride (PMZ), (±)-chlorpheniramine maleate salt (CPR), (±)-propranolol hydrochloride (PPL), (+)-cis-diltiazem hydrochloride (DTZ), cyproheptadine hydrochloride sesquihydrate, prochlorperazine dimaleate salt, cyclosporine A (CsA), loperamide hydrochloride, metergoline, L-765,314 hydrate (L765314), L-703,606 oxalate salt hydrate (L703606), mibefradil dihydrochloride hydrate, methiothepin mesylate salt, octoclothepin maleate salt, tamoxifen citrate and xanthine amine congener.
CQ was dissolved in PBS at the beginning of each experiment and filter-sterilized prior to use. All individually purchased compounds were dissolved in DMSO, stored in aliquots at −20°C and diluted to working concentrations with PBS. All compounds were protected from light.
LOPAC compounds were provided in 96-well format and dissolved in 10 mM DMSO. Working plates were made by dilution to 100 μM in PBS and immediately aliquoted to experimental plates (i.e. experimental plates were pre-plated with LOPAC compounds) which were stored at −20°C and thawed on the day of the experiment.
CM-CQ staining
CM-CQ is a blue-fluorescent labeled CQ moiety that has close resemblance to its parent CQ molecule as described previously18 . Parasite cultures at 2.5% hematocrit and between 7–12% parasitemia (late-ring stage 22–26 hrs post-invasion) were incubated in a flat-bottom 96-well plate at a final staining concentration of 6 μM of CM-CQ for 10 hrs in a perfused dark humidified incubation chamber at 37°C. After which, cultures were resuspended in fresh PBS using either a 12-channel (Eppendorf) or a 96-channel (VIAFLO-96) pipette. Cells were then either diluted further for flow cytometry analysis using LSRII or transferred to black flat-bottom 96-well plates and left to settle for 1 hr prior to fluorescence plate measurements.
Hoechst staining
Hoechst 33342 (Invitrogen, Eugene, OR, USA) is a lipophilic DNA-binding fluorescent stain that is excited by ultraviolet light (350 nm) and emits a blue fluorescent signal (461 nm). Cultures at 1.25% to 2.5% hematocrit and varying parasitemia and stages were stained with Hoechst at a final concentration of 1 μg/ml for 30 min at 37°C. After which, cells were washed once with PBS prior to flow cytometry (LSRII or Quanta).
8-point, 12-point and 96-point IC50
Parasitemia of each strain was determined by flow cytometry and diluted with fresh RBC to 1% parasitemia and hematocrit adjusted to 1.25% at the start of each IC50 assay. A 10× working solution of each drug concentration was made on a separate plate and 20 μl was added per well. Plates were kept away from light and stored for 48 hrs at 37°C in a humidified and perfused incubation chamber. After which, cells were stained with Hoechst and analyzed by flow cytometry.
In most cases, a 12-point curve was used to derive an estimated IC50 value. However, this was reduced to an 8-point curve for IC50 used in Fig. 3 due to the large number of compounds at different concentrations being tested. Conversely, this was expanded to a 96-point curve in Supplementary Figure 3 so as to accurately determine the respective IC50 values in K1.
Flow cytometry
Cell numbers and fluorescence intensity was assayed using either the BD LSRII Special Order System (BD, San Jose, CA, USA) or the Beckman Coulter Cell LabQuanta SC, both of which are equipped with automated 96-well plate samplers.
LSRII was used for CM-CQ and Hoechst-stained cells. Both were excited with a 355 nm UV laser while detection was by a 450LP 450/50BP filter (380 V). Forward and side scatter was adjusted as described elsewhere19 and at least 100,000 erythrocytes were analyzed. In determining the proportion of CM-CQ parasites, Hoechst-stained duplicate wells were used to determine parasitemia.
LabQuanta was used for Hoechst-stained parasites in IC50 assays only. Excitation was through a 355/37 nm arc lamp while detection used a 550DLP 465BP filter. Electronic voltage was adjusted to accommodate erythrocytes and to exclude debris. Initial experiments were run simultaneously on both LSRII and LabQuanta to ensure suitability of LabQuanta in IC50 determination. At least 50,000 erythrocytes were analyzed from each sample.
Fluorescence plate measurements
After cells were stained with CM-CQ, washed and transferred to black flat-bottom 96-well plates (NUNC), cells were allowed to settle for 1 hr before measurements. Fluorescence measurements were taken with a TECAN Infinite M200 plate reader (top-reading), with excitation at 350/9 nm, emission at 500/20 nm, number of flashes default at 25 and integration time default at 20 μs.
When multiple strains were used (Fig. 1c & d), parasitemia between strains was equilibrated by addition of uninfected erythrocytes prior to staining.
Statistics
All data shown are means ± SEM. Statistical difference was analyzed using ANOVA and post-hoc comparison using Tukey's test. Significantly different results (P < 0.05) were highlighted. IC50 curve-plotting was done using GraphPad Prism 5 (v5.04) using a four-parameter logistic curve (variable slope).
Ethics
The blood collection protocol for malaria in vitro culture was approved by the Institutional Review Board (IRB) (NUS-IRB Reference Code: 11-383, Approval Number: NUS-1475) of the National University of Singapore (NUS). Written informed consent was obtained from all participants involved in the study.
The clinical field isolates used in this study were collected under the following ethical guidelines in the approved protocols: OXTREC Reference Number 29-09 (Center for Clinical Vaccinology and Tropical Medicine, University of Oxford, Oxford, United Kingdom).
Permission to use the field isolates for research within NUS was in accordance with NUS IRB (Reference Code: 12-369E).
References
Mita, T., Tanabe, K. & Kita, K. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int 58, 201–209 (2009).
Dondorp, A. M. et al. Artemisinin Resistance in Plasmodium falciparum Malaria. New England Journal of Medicine 361, 455–467 (2009).
Rottmann, M. et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180 (2010).
Vaidya, A. B. & Mather, M. W. Atovaquone resistance in malaria parasites. Drug Resist Updat 3, 283–287 (2000).
Egan, T. J. & Kaschula, C. H. Strategies to reverse drug resistance in malaria. Curr Opin Infect Dis 20, 598–604 (2007).
Ch'ng, J. H., Renia, L., Nosten, F. & Tan, K. S. Can we teach an old drug new tricks? Trends Parasitol 28, 220–224 (2012).
van Schalkwyk, D. A. & Egan, T. J. Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist Updat 9, 211–226 (2006).
Dondorp, A. M. et al. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol 8, 272–280 (2010).
Baird, J. K. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother 48, 4075–4083 (2004).
Gross, A. S., Heuer, B. & Eichelbaum, M. Stereoselective protein binding of verapamil enantiomers. Biochem Pharmacol 37, 4623–4627 (1988).
Pereira, M. R. et al. In vivo and in vitro antimalarial properties of azithromycin-chloroquine combinations that include the resistance reversal agent amlodipine. Antimicrob Agents Chemother 55, 3115–3124 (2011).
Henry, M., Alibert, S., Rogier, C., Barbe, J. & Pradines, B. Inhibition of efflux of quinolines as new therapeutic strategy in malaria. Curr Top Med Chem 8, 563–578 (2008).
Gbotosho, G. O. et al. Comparative study of interactions between chloroquine and chlorpheniramine or promethazine in healthy volunteers: a potential combination-therapy phenomenon for resuscitating chloroquine for malaria treatment in Africa. Ann Trop Med Parasitol 102, 3–9 (2008).
Fidock, D. A. et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6, 861–871 (2000).
Krogstad, D. J., Schlesinger, P. H. & Herwaldt, B. L. Antimalarial agents: mechanism of chloroquine resistance. Antimicrob Agents Chemother 32, 799–801 (1988).
Karl, S., Wong, R. P., St Pierre, T. G. & Davis, T. M. A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malar J 8, 294 (2009).
Malleret, B. et al. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci Rep 1, 118 (2011).
Ch'ng, J. H., Kotturi, S. R., Chong, A. G., Lear, M. J. & Tan, K. S. A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis 1, e26 (2010).
Ch'ng, J. H., Liew, K., Goh, A. S., Sidhartha, E. & Tan, K. S. Drug-induced permeabilization of parasite's digestive vacuole is a key trigger of programmed cell death in Plasmodium falciparum. Cell Death Dis 2, e216 (2011).
Spiller, D. G., Bray, P. G., Hughes, R. H., Ward, S. A. & White, M. R. The pH of the Plasmodium falciparum digestive vacuole: holy grail or dead-end trail? Trends Parasitol 18, 441–444 (2002).
Ursos, L. M. & Roepe, P. D. Chloroquine resistance in the malarial parasite, Plasmodium falciparum. Med Res Rev 22, 465–491 (2002).
Ursos, L. M., Dzekunov, S. M. & Roepe, P. D. The effects of chloroquine and verapamil on digestive vacuolar pH of P. falciparum either sensitive or resistant to chloroquine. Mol Biochem Parasitol 110, 125–134 (2000).
Bhattacharjee, A. K., Kyle, D. E., Vennerstrom, J. L. & Milhous, W. K. A 3D QSAR pharmacophore model and quantum chemical structure-activity analysis of chloroquine(CQ)-resistance reversal. J Chem Inf Comp Sci 42, 1212–1220 (2002).
Fong, T. M., Huang, R. R. & Strader, C. D. Localization of agonist and antagonist binding domains of the human neurokinin-1 receptor. J Biol Chem 267, 25664–25667 (1992).
Welker, H. A., Wiltshire, H. & Bullingham, R. Clinical pharmacokinetics of mibefradil. Clin Pharmacokinet 35, 405–423 (1998).
Yuan, J. et al. Chemical genomic profiling for antimalarial therapies, response signatures and molecular targets. Science 333, 724–729 (2011).
Giao, P. T. & de Vries, P. J. Pharmacokinetic interactions of antimalarial agents. Clin Pharmacokinet 40, 343–373 (2001).
Hunt, P., Cravo, P. V., Donleavy, P., Carlton, J. M. & Walliker, D. Chloroquine resistance in Plasmodium chabaudi: are chloroquine-resistance transporter (crt) and multi-drug resistance (mdr1) orthologues involved? Mol Biochem Parasitol 133, 27–35 (2004).
Ecker, A., Lakshmanan, V., Sinnis, P., Coppens, I. & Fidock, D. A. Evidence that mutant PfCRT facilitates the transmission to mosquitoes of chloroquine-treated Plasmodium gametocytes. J Infect Dis 203, 228–236 (2011).
Kinga Modrzynska, K. et al. Quantitative genome re-sequencing defines multiple mutations conferring chloroquine resistance in rodent malaria. BMC Genomics 13, 106 (2012).
Miki, A., Tanabe, K., Nakayama, T., Kiryon, C. & Ohsawa, K. Plasmodium chabaudi: association of reversal of chloroquine resistance with increased accumulation of chloroquine in resistant parasites. Exp Parasitol 74, 134–142 (1992).
Singh, N. & Puri, S. K. Interaction between chloroquine and diverse pharmacological agents in chloroquine resistant Plasmodium yoelii nigeriensis. Acta Trop 77, 185–193 (2000).
Osa, Y. et al. Structural properties of dibenzosuberanylpiperazine derivatives for efficient reversal of chloroquine resistance in Plasmodium chabaudi. J Med Chem 46, 1948–1956 (2003).
Acknowledgements
The authors wish to thank Laurent Renia, Thierry Diagana, Thomas Dick and Brian Dymock for their critical input to the project design. The authors thank all of the patients and staff of the SMRU for their contribution to this study. SMRU is sponsored by the Wellcome Trust of Great Britain, as part of the Oxford Tropical Medicine Research Program of Wellcome Trust–Mahidol University. The authors are also thankful for the following reagents which were obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: Plasmodium falciparum 3D7, MRA-102, deposited by DJ Carucci; P. falciparum HB3, MRA-155, deposited by TE Wellems; P. falciparum 7G8, MRA-154, deposited by DE Kyle; P. falciparum K1, MRA-159, deposited by DE Kyle; P. falciparum Dd2, MRA-156, deposited by TE Wellems; P. falciparum CS2, MRA-96, deposited by SJ Rogerson; P. falciparum T9/94, MRA-153, deposited by D Walliker. Research from Kevin Tan's and Martin Lear's laboratories has been generously supported by grants from the National Research Foundation (NRF2009NRF-POC002-102) and the National Medical Research Council (NMRC/1310/2011).
Author information
Authors and Affiliations
Contributions
Study design: K.S.W.T., J.H.C.; assay optimization: J.H.C., S.M., Z.B.; screening and validation: J.H.C.; pharmacophore comparisons: A.B.; fluorescent-tagged chloroquine design: M.J.L.; field isolates: B.M.R. and F.N.; manuscript preparation: all authors.
Ethics declarations
Competing interests
K.S.W.T. and M.J.L. are founding directors of BioLynx Technologies, a private company that focuses on fluorophore-conjugated antimalarials. However, the tool (CM-CQ) used in this study is currently not a commercial entity. Other authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ch'ng, JH., Mok, S., Bozdech, Z. et al. A Whole Cell Pathway Screen Reveals Seven Novel Chemosensitizers to Combat Chloroquine Resistant Malaria. Sci Rep 3, 1734 (2013). https://doi.org/10.1038/srep01734
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01734
This article is cited by
-
Uptake of a fluorescently tagged chloroquine analogue is reduced in CQ-resistant compared to CQ-sensitive Plasmodium falciparum parasites
Malaria Journal (2019)
-
Current scenario and future strategies to fight artemisinin resistance
Parasitology Research (2019)
-
ω-Tbo-IT1–New Inhibitor of Insect Calcium Channels Isolated from Spider Venom
Scientific Reports (2015)
-
Validation of a chloroquine-induced cell death mechanism for clinical use against malaria
Cell Death & Disease (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
