
Abstract
Glioblastoma multiforme (GBM) is a highly invasive and chemoradioresistant brain malignancy. Temozolomide (TMZ), a DNA-alkylating agent, is effective against GBM and has become the standard first-line drug. However, the mechanism by which TMZ regulates the progression of GBM remains elusive. Here, we demonstrate that TMZ targets TAp63, a p53 family member, inducing its expression to suppress the progression of human GBM. High levels of TAp63 expression in GBM tissues after TMZ treatment was an indicator of favourable prognosis. In human GBM cells, TMZ-induced TAp63 directly repressed MYC transcription. Activation of this TAp63-MYC pathway by TMZ inhibited human GBM progression both in vitro and in vivo. Furthermore, downregulation of MYC mRNA levels in recurrent GBMs after TMZ treatment correlated with better patient survival. Therefore, our results suggest that the TAp63-mediated transcriptional repression of MYC is a novel pathway regulating TMZ efficacy in GBM.
Similar content being viewed by others

Introduction
Glioblastoma multiforme (GBM) has been resistant to a variety of chemotherapeutic regimens for the last half-century and the average survival duration of patients was previously about 12 months1,2. In 2005, however, temozolomide (TMZ) was reported to be effective against GBM and to increase the survival period by an average of three months3. The survival advantage conferred by TMZ is largely restricted to GBMs with a methylated promoter status of the DNA-repair gene O6-methylguanine-DNA methyltransferase (MGMT)4. MGMT repairs cytotoxic O6-methylguanine adducts induced by TMZ and thus the expression of MGMT contributes to TMZ-resistance in GBM4,5. Nevertheless, the molecular mechanism by which TMZ achieves its effect on GBM has remained elusive2,6.
The p53 tumour suppressor family includes p63 and p73 members7,8,9. The TAp63 and TAp73 isoforms, which bear transactivation domain, function similarly to the p53 tumour suppressor by inducing apoptosis following DNA damage10, while ΔNp63 and ΔNp73, which lack this domain, act as oncogenes11. The p53 signalling pathway is often altered in human GBM12; however, there have been no reports on the function of p63 and p73 in GBM. Therefore, we focused on the roles of p63 and p73 in GBM to investigate the mechanisms of DNA damage-induced activation of the other p53 family members following TMZ treatment. In this study, we show that TMZ activates TAp63 to repress MYC transcription and that the downregulation of MYC by TAp63 augments the efficacy of TMZ in human GBM by suppressing proliferation and invasion.
Results
High TAp63 expression correlates with favorable prognosis in human GBM
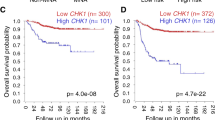
To investigate the expression levels of p63 and p73 in human GBM, 69 newly diagnosed malignant gliomas (59 GBMs; 7 anaplastic astrocytomas; 3 anaplastic oligoastrocytomas) were selected for RNA and DNA analyses (Supplementary Table S1). Forty-nine cases had received procarbazine, ACNU and vincristine (PAV) therapy and twenty had received TMZ therapy. First, our clinical data showed that TMZ treatment significantly prolonged the survival period of patients with malignant glioma (P = 0.0394; Supplementary Fig. S1a) and that high levels of MGMT expression were significantly associated with unfavourable prognoses (P = 0.0015; Supplementary Fig. S1b). Following quantitative RT-PCR for the analysis of TAp63 and TAp73 expression, the samples were divided into groups with high or low levels of TAp63 (Fig. 1a) and TAp73 (Supplementary Fig. S2) based on their respective expression levels in a normal adult brain. Kaplan–Meier cumulative survival curves showed that high TAp63 expression was significantly associated with prolonged survival (P = 0.0373; Fig. 1b). In contrast, the levels of TAp73 expression showed no prognostic potential (Supplementary Fig. S2). Therefore, from here on we focused on TAp63 and selected the 20 TMZ-treated GBM samples to perform immunohistochemical staining for p63. We found that p63 immunopositivity (Fig. 1c) was significantly associated with better recurrence-free survival (P = 0.0066, Fig. 1d) and as ΔNp63 mRNA was not detectable in these samples (data not shown), the positive p63 immunohistochemical staining may largely reflect the expression levels of TAp63 protein. Therefore, a high expression level of TAp63 is a favourable prognostic indicator of human GBM for cases that undergo TMZ treatment.

High TAp63 expression correlates with favourable prognoses in human GBM.
(a) TAp63 mRNA expression detected by quantitative RT-PCR (qRT-PCR) in 69 newly diagnosed malignant glioma samples (59 GBM; 7 anaplastic astrocytoma; 3 anaplastic oligoastrocytoma). TAp63 expression was normalised to ACTB mRNA and designated high (n = 37) or low (n = 32) based on the normal human brain expression (dashed red line). (b) Overall survival of subjects with newly diagnosed malignant gliomas according to relative TAp63 expression levels before chemotherapy (n = 69; high, n = 37; low, n = 32). P value by log-rank test. (c) p63 immunohistochemical staining in human GBM. Scale bar, 50 μm. (d) Recurrence-free survival of 20 TMZ-treated GBM subjects according to p63 immunohistochemical staining in newly diagnosed samples. P values by log-rank test.
TAp63 directly represses MYC transcription in response to TMZ
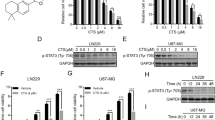
Since other DNA damaging agents activate TAp63 by inducing its mRNA expression13, we next investigated the role of TMZ in TAp63 expression. The steady-state mRNA levels of TAp63 in four human GBM cell lines were analysed (Supplementary Fig. S3a) and TAp63 isoforms was the major TAp63 isoform expressed (Supplementary Fig. S3b). TMZ stimulated TAp63 expression in both TP53 mutant (YH-13) and wild-type (U87MG) cells and induced the expression of TAp63 target genes, including BAX, CDKN1A (p21) and MDM2 (Fig. 2a; Supplementary Fig. S4). In recent reports on functional network modelling analyses of GBM, a significant association between the p53 and MYC pathways has been suggested14,15. In addition, deletions of TP53 and PTEN in murine brains caused the spontaneous development of GBM with a simultaneous induction of MYC16. Therefore, we examined the expression of MYC in TMZ-treated GBM. Interestingly, TMZ induced TAp63 expression and suppresses MYC expression in a dose-dependent manner in both U87MG and YH-13 cells (Fig. 2a). Immunoblotting also showed an increase in TAp63 with suppression of MYC (Fig. 2b). Since p53 is reported to directly repress MYC transcription17,18, we investigated whether TAp63 could regulate the transcription of MYC. Regardless of the status of TP53, siRNA knockdown of TAp63 induced MYC expression at both the mRNA (Fig. 2c; Supplementary Fig. S5a) and protein (Fig. 2d) levels, whereas TAp63 overexpression suppressed MYC (Fig. 2e; Supplementary Fig. S5b) and induced BAX, CDKN1A and MDM2 (Supplementary Fig. S6a,b). MYCN levels remained unaffected in GBM cell lines (Supplementary Fig. S7a,b). To further confirm if TAp63-mediated suppression of MYC occurred in primary GBM cells, we introduced the use of cancer tissue-originated spheroids (CTOS) composed of pure tumor cells derived directly from the GBM tissue19. As shown in Supplementary Fig. S8, the TAp63-MYC regulatory pathway was intact in the CTOS. To examine whether TAp63 directly represses MYC transcription, we performed a chromatin immunoprecipitation (ChIP) assay. We designed four primer sets (R1, R2, R3 and R4) to amplify the indicated genomic regions of the putative TP53 binding sequence (Fig. 2f). Our results showed that endogenous TAp63 was recruited to the upstream promoter and the intron 1 region of the MYC gene in YH-13 cells, but not intron 2, which has been previously shown as a p53-binding site18 (Fig. 2g). In response to TMZ treatment, the amount of TAp63 recruited onto the MYC promoter was significantly increased (Fig. 2h,i), however, there was no increase in TAp63 recruitment onto intron 1 (Supplementary Fig. S9). TAp63 overexpression in YH-13 and U87MG cells inhibited MYC promoter activity (Fig. 2j; Supplementary Fig. S10), suggesting that TMZ stimulates the TAp63-mediated repression of MYC transcription in GBM cells.

TMZ-induced TAp63 suppresses MYC expression in human GBM cells.
(a) TAp63 and MYC mRNA levels in GBM cells with increasing TMZ concentrations, 24 h. (b) Immunoblot of TAp63 and MYC from GBM cells treated with increasing concentrations of TMZ, 24 h. (c) RT-PCR analyses of relative MYC expression in U87MG and YH-13 cells following siTAp63 transfection, normalised to ACTB mRNA. (d) Analysis of MYC expression in YH-13 cells transfected with siControl or siTAp63 by western blotting. (e) RT-PCR analyses showing MYC suppression after TAp63α overexpression in GBM cell lines. (f) Positions of PCR primer sets R1, R2, R3 and R4 for the chromatin immunoprecipitation (ChIP) assays. (g) Identification of the TAp63-binding region in the MYC promoter by ChIP assays. YH-13 cells were transfected with or without indicated siRNAs. Genomic DNA was amplified by PCR using the indicated primers. (h) Semi-quantitative PCR and (i) quantitative PCR of ChIP assays showing endogenous TAp63 recruitment onto the MYC promoter after 24 h TMZ treatment, 150 μM. *P < 0.005 (two-tailed t-test). (j) Luciferase activity of MYC reporters after lentiviral TAp63α or GFP infection of U87MS cells. Data shown as the fold change in the luciferase activity compared with control cells.
The TAp63-MYC pathway regulates sphere formation and invasion in GBM
MYC is a regulator of stemness in glioma and its expression is required for glioma cell neurosphere formation20. Consistent with these notions, TAp63 knockdown increased both the number and size of YH-13 neurospheres, while knockdown of MYC suppressed the sphere-forming ability (Fig. 3a,b). Moreover, the number of spheres formed by YH-13 cells co-transfected with both siTAp63 and siMYC was less than that of cells transfected with siTAp63 alone, suggesting that MYC induction by TAp63 knockdown contributes to the sphere-forming ability of tumour cells.

TAp63-MYC pathway is critical for glioblastoma sphere-forming ability and invasion in vitro.
(a) Sphere formation assay of YH-13 cells, showing induction of sphere-forming activity after knockdown of TAp63 and MYC. (b) The graph indicates the differences in the sphere numbers per microscopic field at 400× magnification. The values represent the mean ± SD of triplicate samples from a single representative experiment (n = 9). **P < 0.0005, ***P < 0.00005 (two-tailed t-test). (c) Cell viability assay of TAp63 and MYC knockdown in YH-13 cells. (e) The graph indicates the percentage of YH-13 cells invading the Matrigel relative to control migration, following TAp63 and MYC knockdown. *P < 0.05, **P < 0.0005 (two-tailed t-test).
TAp63 also plays a critical role in the regulation of cancer invasion21,22. To assess whether the TAp63-MYC pathway also regulates invasion, TAp63 and MYC were knocked down in YH-13 cells with siRNA for 24 and 48 h. Although knockdown of endogenous TAp63 and MYC had no effect on cell proliferation by day 2 of culture (Fig. 3c), knockdown of TAp63 alone significantly promoted the cellular invasion of YH-13 cells. Additionally, co-transfection of siTAp63 and siMYC decreased the percentage of invading cells compared to siControl-transfected cells (Fig. 3d), suggesting that TAp63 downregulates GBM invasion via MYC suppression.
Temozolomide suppresses tumour cell growth and invasion via TAp63 induction
We assessed whether TMZ affects the invasive property of GBM cells via the TAp63-MYC pathway. The treatment of U87MG cells with 75 μM TMZ inhibited cell proliferation and significantly inhibited cellular invasion on day 5 of exposure (Fig. 4a,b). Since apoptotic cell death was not observed under these experimental conditions (Supplementary Fig. S11), TMZ may have anti-invasive properties. Moreover, U87MG cells were transfected with the indicated siRNAs for TAp63 and MYC and subjected to Boyden chamber invasion assays. TAp63 knockdown in U87MG cells induced MYC mRNA expression and rescued the TMZ-inhibited cellular invasion, compared to the control (Fig. 4c). Next, to assess the antitumour effects of TAp63 in vivo, we knocked down TAp63 in the tumors of U87MG xenograft mouse models. Our results showed that TAp63 knockdown promotes tumour cell proliferation (Supplementary Fig. S12a,b). To investigate the role of TAp63 in TMZ efficacy, we implanted U87MG cells into the hind legs of mice (n = 4) and, 7 days later, injected siControl and siTAp63 into the palpable tumours, which were then treated with TMZ (15 mg/kg) intraperitoneally on the same day. At day 14, tumours subjected to siTAp63 knockdown with TMZ treatment were considerably larger compared to the control tumours, indicating a drug-resistant phenotype (Fig. 4d). Finally, we assessed whether the TMZ-induced activation of the TAp63-MYC pathway is associated with the clinical outcome of patients. To this end, we analysed the RNA from 20 paired (initially diagnosed tumour and recurrent tumour from the same patient) malignant glioma samples (17 GBMs, three anaplastic oligoastrocytomas) from TMZ-treated patients. The expression levels of MYC mRNA were significantly decreased after the treatment (Fig. 4e). Furthermore, the subgroup with decreased MYC expression after TMZ treatment showed significantly better overall survival (Fig. 4f). We performed a similar analysis using only the GBM mRNA data and both TAp63 expression and MYC suppression indicated good prognoses (Fig S13a–f). Taken together, these findings suggest that the TMZ-mediated suppression of MYC via TAp63 activation is a key pathway for the drug's efficacy against GBM.

TMZ inhibits GBM progression via the TAp63-MYC pathway.
(a) Cell viability assay showing suppression of proliferation in cells treated with TMZ, 75 μM. (b) Cellular invasion was suppressed by day 5, at which point the treated and control cultures were adjusted to 5 × 104 cells/500 μl and subjected to Boyden chamber invasion assays. *P < 0.05 (two-tailed t-test). (c) Percentage of U87MG cells invading the Matrigel relative to control migration, following TAp63 knockdown and TMZ treatment. Corresponding mRNA analysis. *P < 0.05 (two-tailed t-test). (d) Effect of TAp63 knockdown on the U87MG cells treated with TMZ treatment at 14 days after subcutaneous transplantation in nude mice. The end volumes were compared to the volumes at implantation. Tumour growth was measured with callipers and calculated by the formula: volume = length (A) × width (B) × width (B) × 0.5, where A and B are the long and short axes respectively. TMZ in PBS was administered intraperitoneally at 15 mg/kg once a week. Data are representative of five independent experiments (n = 4). *P < 0.05 by two-tailed t-test. Photographs in (d) are representative of n = 4 mice. Bar, 10 mm. (e) MYC downregulation in recurrent tumours after TMZ plus radiotherapy (n = 20; solid line: mean difference; dashed lines: 95% confidence interval. P value by paired t-test. (f) Overall survival according to MYC downregulation (≥1.5-fold decrease in primary/recurrent (p/r) MYC mRNA), post-TMZ treatment. MYC decrease: n = 11, mean p/r = 2.65 ± 0.31 s.d.; no change: n = 9, mean p/r = 0.9 ± 0.13 s.d. P value by log-rank test.
Discussion
In this study, we report that TAp63 regulates MYC transcription and tumor progression in TMZ-treated GBM. The study highlighted four points. First, TAp63 expression is a favourable prognostic factor in TMZ-treated GBM. Second, TMZ induces TAp63 to suppress growth and invasion. Third, TAp63 directly represses MYC expression in response to TMZ treatment and fourth, MYC downregulation correlates with TMZ efficacy.
A possible mechanism of TMZ action could be that TMZ induces TAp63 expression in GBM cells and in turn, activated TAp63 inhibits cellular invasion via suppression of MYC expression. These findings are consistent with the previous reports in which TAp63 suppresses invasion through coordinated transcriptional regulation of its downstream target genes such as SERPINB5, CCNG2, BHLHE41 and DICER121,22,23,24.
A prior study on the regulatory mechanisms of p53 upon its downstream targets showed that in response to hypoxic stress, p53 is recruited onto intron 2 of the MYC gene to directly inhibit MYC transcription18. Our work has identified a critical role for TAp63 as an alternative regulator of MYC in human GBM. Distinct from the p53-MYC regulatory mechanism, TAp63 is activated in response to TMZ-induced stress and does not bind to intron 2 but is recruited onto the upstream promoter region of MYC to directly repress transcription. In addition, the interaction between the p53 and MYC signaling networks are important for cellular proliferation and differentiation in the prevention of GBM pathogenesis14,15,17. However p53 is functionally inactivated in many aggressive GBMs12. Therefore, the newly identified TAp63-MYC regulatory pathway may serve as an alternative activator of the p53-regulated tumour suppressive pathways10,21,25,26.
Recently, there have been several reports on the role of p53 in TMZ resistance in GBM. TMZ activates p53 to induce apoptosis in GBM cells27,28 and functional inactivation of the p53 pathway by overexpression of the α5β1 integrin contributes to TMZ resistance in high-grade glioma27. On the other hand, reports also suggest that p53 inactivation rather increases TMZ sensitivity in GBM cell lines29 and in vivo xenograft models30 and that the status of p53 is not a molecular predictor of the response to chemotherapy with TMZ31. These discrepancies indicate the existence of an alternative mechanism underlying TMZ resistance in GBM which is independent of the p53 status. Our present data show that the TAp63-MYC pathway contributes to TMZ sensitivity in both p53-wild type and -mutant GBM cells and that the expression levels of TAp63 and MYC are prognostic factors in TMZ-treated GBM.
In conclusion, our results clearly indicate that MYC is a novel TAp63 target gene and that the TAp63-MYC pathway has a crucial role in mediating suppression of GBM progression. Pharmacological targeting of the TAp63-MYC pathway may therefore provide new rational therapeutic strategies against TMZ-resistant GBMs.
Methods
Glioma tumour samples and tissue dissection
A total of 89 malignant glioma samples comprising of newly-diagnosed GBM (World Health Organization (WHO) grade 4, n = 59), newly-diagnosed anaplastic astrocytoma (WHO grade 3, n = 7), newly-diagnosed anaplastic oligoastrocytoma (WHO grade 3, n = 3) and 20 recurrent malignant gliomas (17 GBMs, 3 anaplastic oligoastrocytomas) were collected between 1994 and 2011 and obtained from the Chiba Cancer Center upon receiving informed consent under an institutional review board-approved protocol. The present project was approved by the Ethics Committee of the Faculty of Biology and Medicine at the Chiba Cancer Center (protocol 15–19). After surgery, the patients were treated according to previously described protocols32. Although procarbazine, ACNU and vincristine (PAV) were used for chemotherapy until 2006, TMZ was used from 2006 in our facility. The tumour samples were routinely processed using a BenchMark® XT automated slide-processing system (Ventana Medical Systems, USA) for optimization and performance evaluation of the immunohistochemical assay for p63 protein. Paraffin-embedded tissue sections on glass slides were baked prior to the deparaffinization step with EZ PrepTM (Ventana). The sections were then washed with a mixture of Immunoblock (Dainippon Sumitomo Pharma Co.) and reaction buffer, followed by incubation with a mouse anti-human p63 monoclonal antibody (clone 4A4, Dako). The tumour samples were divided into three specimens: immediately snap-frozen in liquid nitrogen; diagnostic frozen sections for the Department of Pathology; and formalin-fixed paraffin-embedded sections. The specimens were homogenised using a rotor/stator homogenizer. DNA and RNA were extracted from the patient samples as previously described33. Total RNA was extracted from three healthy human brain tissue samples purchased from separate sources, Clontech, Stratagene and Zyagen and the highest p63-expressing sample was used as the threshold standard. A newly diagnosed GBM specimen (supplied by Chiba Cancer Center) was used to generate cancer tissue-originated spheroid cultures as described previously19.
Cell lines and specimen collection
GBM cell lines were purchased from the American Type Culture Collection (U87MG) and Health Science Research Resources Bank (YH-13, KNS-42 and SF126). The cell lines were cultured in modified Eagle's medium containing 20% (YH-13), 5% (KNS42) or 10% (U87MG and SF126) heat-inactivated foetal bovine serum (Invitrogen).
Temozolomide (TMZ) treatment
TMZ was purchased from LKT Laboratories Inc. and freshly prepared for experimental use. TMZ was added at the final concentrations 24 h after cell seeding for each experiment.
RT-PCR and qRT-PCR
The primers are described in the SI Methods.
Gene knockdown assay
The following gene-specific siRNAs were purchased: siTAp63-1, sense 5′-CAGCUAUAUGUUCAGUUCTT-3′ and antisense 5′-GAACUGAACAUAUAGCUGTT-3′; siTAp63-3, sense 5′-CAGAAGAUGGUGCGACAAATT-3′ and antisense 5′-UUUGUCGCACCAUCUUCUGTT-3′ from Sigma; siMyc-26 for CGAUGUUGUUUCUGUGGAA and siMyc-29 for CUACCAGGCUGCGCGCAAA from Thermo Scientific. The control siRNA (Mission siRNA Universal Negative Control SIC-001) was purchased from Sigma. All transfections were performed using LipofectamineTM RNAiMax (Invitrogen). The transfections were carried out twice, with reverse transfection immediately following cell count to 1 × 105 cells/ml and forward transfection 24 h after reverse transfection.
Overexpression of TAp63 in human GBM cells
TAp63α was fused to the FLAG epitope at the NH2 terminus and cloned into the lentiviral pHR vector. The lentivirus was produced by cotransfecting pHR, pCMVR and pMDG plasmids into HEK293T cells using the FuGENE HD reagent (Roche). At 12 and 24 h after transfection, the viral supernatants were collected and mixed with GBM cells.
Western blot assay
We resolved cell proteins by SDS-PAGE prior to electroblotting onto a PVDF membrane. We incubated the membranes with the following primary antibodies overnight: anti-p63 (4A4) (1:1000; sc-8431, Santa Cruz Biotechnology), anti-Myc (N-262) (1:2000; sc-764, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-Actin (20-33) (1:4000; A5060, Sigma). The membranes were then incubated with a horseradish peroxidase-conjugated secondary antibody (anti-rabbit IgG #7074, 1:2000–1:4000 or anti-mouse IgG #7076, 1:2000; Cell Signaling Technology) and the bound proteins were visualized using a chemiluminescence-based detection kit (ECL and ECL pro kit; Amersham and PerkinElmer).
Chromatin immunoprecipitation (ChIP) assay
YH-13 cells transfected with siRNA or exposed to TMZ at increasing concentrations between 150–300 μM were harvested 24 h after the second transfection or after TMZ exposure. ChIP assays were performed with a ChIP assay kit (Millipore) according to the manufacturer's instructions using an anti-p63 antibody (clone (4A4) sc-8431, Santa Cruz Biotechnology) and normal mouse IgG (015-000-003, Jackson ImmunoResearch Laboratories Inc.). The primer sequences for the MYC promoter are described in the SI Methods.
Luciferase reporter assay
pBV-LUC Del1 (#16601) and Del2 (#16602) containing the MYC promoter regions at nucleotides −2268 to +525 and −1061 to +525 from the transcription start site, respectively, were obtained from Addgene. YH-13 and U87MG cells were seeded at 5 × 104 cells/well in a 24-well plate and allowed to adhere overnight. The cells were subjected to lentiviral infection on the following day with control pHR vector or pHR-TAp63α vector. At 54 h after infection, the cells were seeded in triplicate on 12-well plates at 1 × 105 cells/well and cultured for 24 h. They were then cotransfected with 400 ng of MYC luciferase reporter construct and 40 ng of Renilla TK with Lipofectamine 2000 (Invitrogen). At 18 h after the second transfection, the cells were harvested and the luciferase activity was determined using a dual-luciferase assay system (Promega) according to the manufacturer's instructions.
Migration and invasion assay
The invasive potential of GBM cells in vitro was measured by evaluating the number of invading cells using Matrigel-coated Transwell inserts (BD Biosciences) according to the manufacturer's instructions. YH-13 and U87MG cells transfected with siTAp63 and siMyc were seeded onto an insert with 8 μm pores (BD Biosciences) in a 24-well plate at 2 × 105 cells/ml. The cells were treated with 150 μM TMZ and counted 18 h after siRNA knockdown. Cells on the lower side of the membrane were fixed with 4% paraformaldehyde and stained using a Diff Quick Staining Kit (Sysmex).
Cell viability assay (MTT assay)
Cell viability was quantified by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) method. Cells were collected and seeded in 96-well plates at 1 × 105 cells/well. After addition of 10 μl of MTT tetrazolium salt (Sigma) solution to each well, the plates were incubated in a CO2 incubator. The absorbance of each well was measured using a Dynatech MR5000 plate reader with a test wavelength of 450 nm and a reference wavelength of 630 nm.
Sphere formation assay
We evaluated neurospheres derived from YH-13 cells transfected with the appropriate siRNA. After performing cell counts, we plated single cells in 60 mm non-coated dishes (2.5 × 105 cells/dish; Iwaki) to check the sphere morphology and used a 96-well ultra-low cluster plate (2.5 × 104 cells/well; Costar) to count the spheres. The cells were allowed to proliferate in serum-free DMEM (Sigma) and F12 medium (Invitrogen) containing epidermal growth factor (Sigma) and 20 ng/ml basic fibroblast growth factor (Invitrogen) with 2% B27 supplement (Invitrogen). Half of the medium was replaced with fresh culture medium every 7 days.
Mouse xenograft models
Six- or seven-week-old male athymic BALB/c nu/nu mice were obtained from Japan SLC, Inc. The mice were anesthetized with intraperitoneal tribromoethanol (Wako) at 20 mg/kg body weight. U87MG cells mixed with an equal volume of Matrigel were implanted into the right and left hind legs. One week after tumour cell implantation, we injected 50 μl of Atelogene (Koken) with either control siRNA or siTAp63 (10 μM) into the U87MG xenografts. A total of 15 mg/kg TMZ in PBS was administered intraperitoneally once per week. Tumour growth was measured with calipers and calculated by the formula: volume (V) = length (A) × width (B) × width (B) × 0.5. These studies were approved by the Committee for Animal Care at the Chiba Cancer Center Research Institute.
Statistical analysis
Data is presented as the mean ± the standard deviation. Statistical significances in the clinical data were calculated using Kaplan-Meier survival curves. Statistical analyses were performed with JMP® 10 (SAS institute Japan).
References
Chen, J., McKay, R. M. & Parada, L. F. Malignant glioma: lessons from genomics, mouse models and stem cells. Cell 149, 36–47 (2012).
Mrugala, M. M. & Chamberlain, M. C. Mechanisms of disease: temozolomide and glioblastoma - look to the future. Nat. Clin. Pract. Oncol. 5, 476–486 (2008).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Hegi, M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Shah, N. et al. Comprehensive analysis of MGMT promoter methylation: correlation with MGMT expression and clinical response in GBM. PLoS One 6, e16146 (2011).
Beier, D., Schulz, J. B. & Beier, C. P. Chemoresistance of glioblastoma cancer stem cells - much more complex than expected. Mol. Cancer 10, 128 (2011).
Osada, M. et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 4, 839–843 (1998).
Yang, A. et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing and dominant-negative activities. Mol. Cell 2, 305–316 (1998).
Kaghad, M. et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90, 809–819 (1997).
Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 7, 165–168 (2007).
Deyoung, M. P. & Ellisen, L. W. p63 and p73 in human cancer: defining the network. Oncogene 26, 5169–5183 (2007).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Suenaga, Y. et al. TATA-binding protein (TBP)-like protein is engaged in etoposide-induced apoptosis through transcriptional activation of human TAp63 gene. J. Biol. Chem. 284, 35433–35440 (2009).
Aguda, B. D., Kim, Y., Kim, H. S., Friedman, A. & Fine, H. A. Qualitative network modeling of the Myc-p53 control system of cell proliferation and differentiation. Biophys. J. 101, 2082–2091 (2011).
Bredel, M. et al. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 65, 8679–8689 (2005).
Zheng, H. et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455, 1129–1133 (2008).
Ho, J. S., Ma, W., Mao, D. Y. & Benchimol, S. p53-dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell Biol. 25, 7423–7431 (2005).
Krieg, A. J., Hammond, E. M. & Giaccia, A. J. Functional analysis of p53 binding under differential stresses. Mol. Cell Biol. 26, 7030–7045 (2006).
Kondo, J. et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc. Natl. Acad. Sci. USA 108, 6235–6240 (2011).
Wang, J. et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 3, e3769 (2008).
Su, X. et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467, 986–990 (2010).
Melino, G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 18, 1487–1499 (2011).
Kim, S., Han, J., Kim, J. & Park, C. Maspin expression is transactivated by p63 and is critical for the modulation of lung cancer progression. Cancer Res. 64, 6900–6905 (2004).
Adorno, M. et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98 (2009).
Sachdeva, M. et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 106, 3207–3212 (2009).
Guo, X. et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 11, 1451–1457 (2009).
Janouskova, H. et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 72, 3463–3470 (2012).
Zhang, W. B. et al. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 285, 40461–40471 (2010).
Blough, M. D. et al. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neurooncol. 102, 1–7 (2011).
Dinca, E. B. et al. p53 small-molecule inhibitor enhances temozolomide cytotoxic activity against intracranial glioblastoma xenografts. Cancer Res. 68, 10034–10039 (2008).
Weller, M. et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J. Clin. Oncol. 27, 5743-5750 (2009).
Iuchi, T. et al. Hypofractionated high-dose irradiation for the treatment of malignant astrocytomas using simultaneous integrated boost technique by IMRT. Int. J. Radiat. Oncol. Biol. Phys. 64, 1317–1324 (2006).
Ohira, M. et al. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell 7, 337–350 (2005).
Acknowledgements
We would like to thank R. I. Selim, Y. Kaneko, D. Matsumoto and K. Ando for technical support, A. Sada and N. Kitabayashi for DNA and RNA extractions, Y. Nakamura for sequencing support, R. Takano for lentivirally expressed TAp63α and E. Isogai for helpful discussions and experimental assistance with mice throughout the course of this study. This work was supported in part by a grant-in-aid from the Ministry of Health, Labour and Welfare for the Third Term Comprehensive Control Research for Cancer, Japan and a grant-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Contributions
T.Y. and Y.S. designed, performed and analysed cellular and animal experiments and wrote the manuscript. T.I. collected clinical samples and analysed clinical data. J.A. and A.T. performed and analysed cellular and animal experiments and wrote the manuscript. M.I. and A.A. performed pathological analyses of surgical tissue samples. M.O. assisted with figures and experimental design. M.I. supported the generation of cancer tissue originated-spheroid cultures. H.K. and S.Y. performed genomic mutation search experiments. N.S. assisted in experiments and associated clinical annotations. A.N. designed and supervised the experiments, analysed data and wrote and edited the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yamaki, T., Suenaga, Y., Iuchi, T. et al. Temozolomide suppresses MYC via activation of TAp63 to inhibit progression of human glioblastoma. Sci Rep 3, 1160 (2013). https://doi.org/10.1038/srep01160
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01160
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
