
Abstract
Marine oxygen minimum zones (OMZs) are widespread regions of the ocean that are currently expanding due to global warming. While inhospitable to most metazoans, OMZs are hotspots for microbial mediated biogeochemical cycling of carbon, nitrogen and sulphur, contributing disproportionately to marine nitrogen loss and climate active trace gas production. Our current understanding of microbial community responses to OMZ expansion is limited by a lack of time-resolved data sets linking multi-omic sequence information (DNA, RNA, protein) to geochemical parameters and process rates. Here, we present six years of time-resolved multi-omic observations in Saanich Inlet, a seasonally anoxic fjord on the coast of Vancouver Island, British Columbia, Canada that undergoes recurring changes in water column oxygenation status. This compendium provides a unique multi-omic framework for studying microbial community responses to ocean deoxygenation along defined geochemical gradients in OMZ waters.
Design Type(s) | time series design • observation design • data integration objective |
Measurement Type(s) | metagenomics analysis • transcription profiling assay • ribosomal RNA • protein sequencing by tandem mass spectrometry assay |
Technology Type(s) | DNA sequencing • RNA sequencing • liquid chromatography-tandem mass spectrometry |
Factor Type(s) | sampling depth • temporal_interval • protocol |
Sample Characteristic(s) | marine metagenome • Saanich Inlet • coastal sea water |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Marine oxygen minimum zones (OMZs), areas of low dissolved oxygen (O2) in subsurface waters, result from a combination of microbial respiration of organic matter raining down from surface waters and increased water column stratification1–3. As O2 becomes limiting, microbial communities shift their energy metabolism to use alternative terminal electron receptors in a thermodynamically defined order resulting in increased nitrogen loss and the production of climate active trace gases including nitrous oxide (N2O) and methane (CH4)4–9. Currently, OMZs are expanding throughout the global ocean3,10–14 making it increasingly important to define the microbial metabolic networks driving nutrient and energy cycling under changing levels of water column O2-deficiency2,10,15.
Advances in sequencing technology are enabling the study of microbial communities at unprecedented scales16. Tag sequencing uses primers to amplify specific target genes and subsequently sequence them on high-throughput platforms, generating molecular barcodes that can be used to study microbial community structure and function depending on the marker. The small subunit ribosomal RNA (SSU rRNA) gene is a universally conserved marker commonly used to compare microbial communities among and between samples in a quantitative manner17. Metagenomic sequencing enables reconstruction of microbial community metabolic potential at the level of genes and pathways over relatively long time scales reflecting persistent information storage in the environment. However, metabolic potential does not necessarily indicate active processes as different genes may be expressed under changing environmental conditions18. Metatranscriptomic sequencing opens a window into microbial community gene expression patterns on relatively short time scales reflecting environmental response patterns18. Similarly, metaproteomics identifies proteins present in the microbial community on intermediate times scales, providing an alternative perspective on post-translational regulation and catalysis18. Collectively, multi-omic methods (DNA, RNA and protein) can be used to chart microbial metabolism at individual, population and community levels along defined geochemical gradients in OMZs19–26.
Saanich Inlet, a seasonally anoxic fjord on the coast of Vancouver Island, British Columbia is a model ecosystem for studying microbial community responses to changing levels of water column O2-deficiency8,20–22,27–33. As microbial communities shift their energy metabolism to use alternative electron acceptors within the Saanich Inlet water column, differential modes of metabolic coupling can be observed including a modular denitrification pathway coupled to sulphide oxidation20,21,28. Such metabolic coupling is reminiscent of symbiotic associations and is likely widespread in OMZ ecosystems2. Although current research efforts are increasingly focused on defining co-metabolic innovations among and between ubiquitous OMZ microorganisms, many open questions remain regarding the regulatory and ecological dynamics modulating microbial community responses to OMZ expansion2,5,20–22,34.
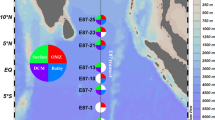
Here we present a compendium of multi-omic sequence information from the Saanich Inlet water column (Fig. 1) encompassing 412 SSU rRNA pyrotag (V6-V8 region) samples (Data Citation 1), 82 SSU rRNA iTag (V4 region) samples (Data Citation 1) (Table 1), 90 metagenomes (Data Citation 1) (totalling 4.1 TB of cleaned reads or 16.2 GB of assembled data), 62 metatranscriptomes (Data Citation 1) (including 46 unique samples and 16 replicates, totalling 1.7 TB of cleaned reads or 2.88 GB of assembled data) and 68 metaproteomes (64 unique samples, totalling 5.2 million unique proteins) (Data Citation 2) (Table 2). Together sequence read data is approximately 5.9 TB of data, comparable to nearly 2,000 human genome equivalents. These data sets, in combination with a cognate geochemical compendium35 comprise a unique time-resolved framework for reconstructing microbial community metabolism along defined geochemical gradients and promoting the development of models to predict microbial community responses to changing levels of water column oxygen-deficiency11,22,36.

(a) Oxygen concentration contour for CTD data (February 2008 onward)35 indicating 16 sampling depths for water column geochemistry and high-resolution (HR) DNA samples for SSU libraries (small black dots) and six major depths for large volume (LV) samples for meta-genomics, -transcriptomics, -proteomics and LV SSU libraries (large black dots). (b) Sample inventory from February 2006 to October 2014 indicating multi-omic datasets included in this manuscript (solid black), in previous publications (gray) and accompanying datasets currently undergoing processing and analysis (open gray).
Methods
Environmental sampling
Time-series monitoring in Saanich Inlet was conducted on a monthly basis aboard the MSV John Strickland at station S3 (48°35.500 N, 123°30.300 W) as described in ref. 8. Samples for large volume (LV) SSU rRNA gene tags, metagenomics, metatranscriptomics, and metaproteomics were taken from six major depths spanning the oxycline (10, 100, 120, 135, 150, and 200 m). Samples for high-resolution (HR) SSU rRNA gene tag sequencing were taken at 16 depths along the oxycline (10, 20, 40, 60, 75, 80, 90, 97, 100, 110, 120, 135, 150, 165, 185 and 200 meters). A detailed seawater sampling video protocol can be found online at http://www.jove.com/video/1159/seawater-sampling-and-collection37.
During sampling procedure, large volume waters were collected in 2×12 l Go-Flow bottles on a wire separated by less than one meter. Waters for metatranscriptomics and metaproteomics (2 l each) were collected consecutively from the Go-Flow into 2 l Nalgene bottles with sterile silicon tubing immediately following sampling for dissolved gases to minimise changes in microbial gene expression. Waters for metatranscriptomics were filtered on-board within 8 min of collection on deck, followed immediately by filtering waters for metaproteomics. For both metatranscriptomics and metaproteomics, a peristaltic pump was used to filter waters through a 0.22 μm Sterivex filter with an in-line 2.7 μm GDF pre-filter. Following removal of residual seawater by extrusion using a 10 cc or 60 cc syringe, 1.8 ml of RNAlater (Ambion) was added to metatranscriptomic sample filters and 1.8 ml of sucrose lysis buffer was added to metaproteomic sample filters. Filters were placed on dry ice, returned to lab and stored at −80 °C until processing. A detailed small volume filtration protocol can be found online at http://www.jove.com/video/1163/small-volume-1-3l-filtration-of-coastal-seawater-samples37.
Waters for LV SSU rRNA gene tags and metagenomics were combined from two Go-Flows into a 20 l carboy using sterile silicon tubing. Carboys were then transported back to the lab for filtration approximately 6–10 h after collection. Approximately 10 l was filtered through 2 Sterivex filters per depth as described above. Following removal of residual seawater by extrusion, 1.8 ml of sucrose lysis buffer was added to LV sample filters. Filters were placed on dry ice, returned to lab and stored at −80 °C until processing. A detailed large volume filtration protocol can be found online at http://www.jove.com/video/1161/large-volume-20l-filtration-of-coastal-seawater-samples38.
Waters for high resolution (HR) SSU rRNA gene tags were collected in 12 l Go-Flow or 8 l Niskin bottles and transferred into 1 l (February 2006—September 2009) or 2 l (September 2009 onward) Nalgene bottle with sterile silicon tubing and stored on ice. Bottles were then transported back to the lab for filtration approximately 12–16 h after collection. A peristaltic pump was used to filter waters through a 0.22 μm Sterivex filter without a pre-filter. Following removal of residual seawater by extrusion, 1.8 ml of sucrose lysis buffer was added to HV sample filters. Filters were placed on dry ice, returned to lab and stored at −80 °C until processing. A detailed small volume filtration protocol can be found online at http://www.jove.com/video/1163/small-volume-1-3l-filtration-of-coastal-seawater-samples37.
Environmental DNA, RNA and protein extraction
Genomic DNA (HR and LV) was extracted from Sterivex filters as described in27. Video protocols describing the extraction process in detail can be found online at http://www.jove.com/video/1352/dna-extraction-from-022-m-sterivex-filters-cesium-chloride-density39. Briefly, after thawing Sterivex filters on ice, lysozyme (Sigma) was added and incubated at 37 °C for 1 h with rotation followed by addition of Proteinase K (Sigma) and 20% SDS and incubation at 55 °C for 2 h with rotation. Lysate was removed by extrusion and then filters were rinsed with sucrose lysis buffer. Combined lysate was extracted with phenol:chloroform followed by chloroform and the aqueous layer collected and loaded onto a 10 K Amicon filter (Millipore) cartridge, washed three times with TE buffer (pH 8.0) and concentrated to a final volume between 150–400 μl by centrifugation.
Total RNA was extracted from Sterivex filters using the mirVana miRNA Isolation kit (Ambion)19,25 modified for Sterivex filters. Briefly, after thawing filters on ice, RNAlater was removed by extrusion, followed by rinsing with Ringer’s solution (Sigma) and incubation at room temperature for 20 min with rotation. Ringer’s solution was removed by extrusion, followed by addition of lysozyme and incubation at 37 °C for 30 min with rotation. Lysate was removed by extrusion into 15 ml tube and subjected to organic extraction as described in the mirVana kit protocol, adjusting for total volume of lysate. Removal of DNA and purification of total RNA were conducted using the TURBO DNA-free kit (Thermofisher) and the RNeasy MinElute Cleanup kit (Qiagen) protocols respectively.
Total protein was extracted from Sterivex as described in Hawley et al.40. Briefly, after thawing Sterivex filters on ice, Bugbuster (Novagen) was added and incubated at room temperature for 20–30 min with rotation. Lysate was removed by extrusion and filters were rinsed with 1 ml lysis buffer. Buffer exchange was carried out on combined lysate using Amicon Ultra 10 K (Millipore) with 100 mM NH4HCO3 a total of three times with a final volume between 200–500 μl. Protein concentration was determined using the 2-(4-carboxyquinolin-2-yl) quinoline-4-carboxylic acid (Bicinchoninic acid or BCA) assay. Urea was added to a final concentration of 8 M and dithiothreitol added to a final concentration of 5 mM and incubated at 60 °C for 30 min, followed by 10-fold dilution with 100 mM NH4HCO3. Samples were then subject to trypsin digest at 37 °C for 6 h followed by C18 solid phase extraction and strong cation exchange.
Environmental DNA and RNA sequencing
Extracted genomic DNA was used to generate small subunit ribosomal RNA (SSU or 16S/18S rRNA) gene pyrotag libraries20,41. Pyrotag datasets from HR and LV samples were generated by PCR amplification using universal three-domain forward and reverse bar-coded primers targeting the V6-V8 hypervariable region of the SSU rRNA gene42: 926F (5′- AAA CTY AAA KGA ATT GRC GG-3′) and 1392R (5′- ACG GGC GGT GTG TRC-3′). Samples were purified using the QIAquick PCR Purification Kit (Qiagen), and sequenced by 454-pyrosequencing43 at the DOE Joint Genome Institute (JGI), or Génome Québec Innovation Centre at McGill University (Table 3 and Supplementary Table 1). iTag datasets from HR and LV samples were generated by PCR amplification using forward and reverse bar-coded primers targeting the V4-V5 hypervariable region of the bacterial and archaeal SSU rRNA gene: 515F (5′- Y GTG YCA GCM GCC GCG GTAA—3′) and 806R (5′- CCG YCA ATT YMT TTR AGT TT -3′)44,45. Samples were sequenced according to the standard operating protocol on an Illumina MiSeq platform at the JGI46.
Ilumina metagenomic shotgun libraries from LV samples were generated at the JGI and paired end sequenced on the Illumina HiSeq platform43,47–51. In addition to the datasets described above, specific methods for full-length SSU rRNA gene, large-insert genomic (fosmid) libraries, Sanger shotgun metagenomes and pyrotags generated from 2006 to 2008 samples have been previously reported20,27,52.
Extracted environmental total RNA was used to generate paired end sequenced Illumina metatranscriptome libraries53 at the JGI on the HiSeq and (Table 4 and Supplementary Table 2).
Environmental protein sequencing
Extracted environmental protein was sequenced using tandem mass spectrometry (MS/MS) as described previously40. Samples were analysed by capillary liquid chromatography-tandem mass spectrometry (Thermo, LTQ ion trap mass spectrometer or Thermo LTQ-Orbitrap mass spectrometer) in data-dependent mode. Spectra were matched to peptide sequences using the search tool MSGFDBPlus54. The amino acid sequence database used for matching spectra to protein sequences was constructed from metagenomic information from Saanich Inlet and the Line P transect in the Northeastern subarctic Pacific Ocean with additional full-length fosmid libraries20, and single cell genomes from Saanich Inlet55 totalling over 23 million protein sequences.
Data Records
A Table unifying all multi-omic samples and Saanich Inlet geochemical samples from Torres-Beltrán et al.35 is summarised in Table 2 and detailed in Supplementary Table 2.
Small subunit rRNA gene tag sequences
Small subunit rRNA gene pyrotag and iTag sequences are available from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (Data Citation 1). A summary of Pyrotag and iTag samples can be located in Table 1, key to the Pyrotag and iTag data files is located in Table 3 and NCBI BioSample IDs and sequencing centre information for individual samples are located in Supplementary Table 1. In addition, previously published full length small subunit rRNA gene libraries27 are archived at GenBank.
Metagenomes and Metatranscriptomes
Metagenomic and metatranscriptomic data sets are accessible through the JGI IMG/M portal (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) under the study name Marine microbial communities from expanding oxygen minimum zones in the northeastern subarctic Pacific Ocean (Data Citation 1). A unifying inventory of metagenomes, metatranscriptomes and metaproteomes sequenced for the Saanich Inlet time-series is summarised in Table 2, with key in Supplementary Table 2 (Data Citation 1). Sanger sequenced fosmid library28, 454 and Illumina sequenced metagenomic libraries20,21 and 454 sequenced viral metagenomes33 have been previously published.
Metaproteome
Metaproteomic datasets have been deposited in the ProteomeXchange Consortium via the PRIDE56 partner repository with the dataset identifier PXD004433 (Data Citation 2). The key detailing the file types available on the PRIDE repository is located in Table 5. Previously published metaproteomic data sets20 were archived at Mass Spectrometry Interactive Virtual Environment.
Technical Validation
Small subunit ribosomal RNA gene tag sequences
For SSU rRNA gene pyrotags and iTags the quality of extracted genomic DNA was verified on 0.8% agarose gels stained with ethidium bromide or SYBR Safe DNA dye (Thermofisher). Samples were run at 16 V overnight with molecular ladders (10 μl of 50 ngml−1 of 1 kb+; 2, 5 and 10 μl of 50 ngml−1 HindIII ladder) to determine size and estimate concentration of extracted sample DNA (5 μl of extract run on gel). After running, the gel was observed under a UV gel documentation system to check for approximate molecular weight (>36 kb) and evidence of shearing or degradation. In addition, DNA concentration was quantified and corrected to volume filtered using PicoGreen (Thermofisher) following the vendor’s protocol.
Quality control for 454-pyrosequencing entails accurate quantification of prepared library fragments to optimize the sequencing run output, therefore JGI has developed custom qPCR methods to quantify 454 libraries43. In addition, an optimized emulsion PCR protocol was developed to significantly improve the coverage in high GC regions that otherwise would be biased49. Génome Québec Innovation Centre quality control protocol for 454 pyrosequencing entails the use of the default parameters assigned to the signal processing software for Long Amplicon #3 pipeline as indicated in the Roche ‘454 Sequencing System Software Manual’ V3.0 Section 1.353. For produced 454 pyrotag datasets for both high resolution (HR) and large volume (LV) samples a histogram of raw read counts versus read length (Fig. 2a) was used to determine the success of a run e.g. a majority of reads exceeding 450 base pairs. A plot of read counts versus read length for all HR and LV samples is provided in Fig. 2a.

(a) 454 PyroTags for small subunit rRNA gene showing number of raw reads versus read length for large volume samples (99 samples in total) (left) and high resolution samples (311 samples in total) (right). (b) Metagenomic assemblies for two samples from different depths showing average fold coverage versus contig length and percentage GC versus average fold coverage for contigs.
Quality control protocol for iTag sequencing entails QC amplification with V4-V5 primers prior to sample submission to ensure that samples will be successful in JGI sample prep and that there are no contaminants present that will inhibit amplification. Amplified products are run on agarose gel or Bioanalyzer to ensure sample amplification occurred with the expected band size, ~450 bp for bacterial and archaeal SSU rRNA gene V4-V5 region (including primers). Samples showing proper amplification and sizing are passed for Amplification QC and approved to ship to the JGI for processing. All data from the sequencer were demultiplexed and stored in JGI’s archiving and metadata organizer system (JAMO). Read data was processed through JGI’s centralized rolling quality control system verifying that there were no sequencing issues and removing known contaminant reads using a kmer filter in bbduk. Quality controlled reads were processed by iTagger45,46.
Metagenomic and metatranscriptomic data validation
The quality of DNA used for metagenomic sequencing was determined at the same time as verification for SSU rRNA gene pyrotag and iTag amplification as described above. JGI quality control protocol for metagenomic sequences prior to assembly entails rolling QC, an in-house sequence QC pipeline that performs a set collection of analyses and produces a summary report for each lane of Illumina data produced by the sequencing group. This set of analyses calculates read quality, measures sequence uniqueness, and detects abnormal sequence motifs. An assembly, using Velvet, was used to measure coverage and detect contamination48. For individual sample assemblies the average fold coverage versus the contig length (Fig. 2b) was plotted and should have a distinct shape for different samples where peaks in contig length representing at a specific coverage represent a given closely related microbial population. Additionally, the percent GC versus average coverage can be plotted, again with distinct shapes for different samples and clusters representing divergent microbial populations.
The quality of purified RNA was verified on the Bioanalyzer using a RNA nano Analysis Kit (Agilent Technologies) in order to check on the RNA integrity and sample quantification before cDNA library production and sequencing at JGI. Due to variation inherent in environmental samples the RNA integrity number (RIN) varied between 5.5 and 9 for the sample sand averaged 7.3. JGI quality control protocol for metatranscriptomic sequencing preparation follows the ‘TruSeq Stranded Total RNA Sample Preparation Guide’ (Illumina). Briefly this protocol removed ribosomal rRNA with RiboZero, followed by RNA fragmentation for first strand cDNA synthesis. This was followed by second strand synthesis and subsequent ligation of adapters. After PCR amplification library quality was checked using Bioanalyzer for fragment size (260 bp) and purity. Indexed (barcoded) libraries were normalized to 10 nM and pooled in equal volumes. Transcriptomes were assembled de novo and or mapped to a corresponding metagenome. For Additional quality assessment of sequencing run for each sample, histograms of percentage of reads verse average read quality and of reads per percent GC were generated (Fig. 3a). Information on run quality for individual samples is available via the JGI genome portal.

(a) Metatranscriptomic reads for two samples from different depths showing distribution of reads over read quality (left) and percentage GC (right). (b) Metaproteome showing number of detected peptides (top) and detected proteins (bottom) for each depth sampled, colour coded by cruise ID. Higher number of detected proteins than peptides is due the sequence redundancy in the metagenomic database used to identify peptides.
Metaproteomic data validation
The quality of LC-MS runs were monitored visually by the instrument operator by individually inspecting each analytical run for instrument response, retention time characteristics, and background interference. QC was further monitored using a whole cell digest of Shewanella oneidensis that is prepared in bulk and routinely used across all LC-MS systems in the EMSL production labs. Analysis of this QC standard is fully automated once uploaded to an in-house data management system and reports back unique peptide identifications, chromatographic peak width, and mass error within ~1 h. Additionally, these data are subjected to dozens of other analytical metrics that can be further assessed as needed (i.e., when the first pass visual and 1 h results are in question). This QC standard was run at least once per week but generally more often, between sample batches (a single project), and sometimes between sample blocks (when a batch is relatively large and a blocking scheme has been utilized) due to the large diversity of samples analysed in-house. New LC columns were conditioned and tested prior to use on project samples by running a minimum of three QC standards. Blanks were always run between QC standards and between sample batches, but not necessarily between sample blocks. For peptide mapping to full length protein sequences the False Discovery Rate (FDR) was calculated using the spectra to peptide matches that resulted in reversed hits from the on-the-fly reversed database search and a filter on the MSGF value. Number of peptides and proteins detected varies between samples (Fig. 3b). Due to the large size of metagenomic dataset used and redundancy in protein sequences because of multiple sampling of the same environment in the Saanich Inlet time series most peptides mapped to multiple identical proteins, resulting in a greater number of proteins identified then peptides.
Usage Notes
Suggested modes of downstream data analysis
In brief we describe the main workflow used in the analysis of SSU rRNA gene pyrotag and iTag datasets. Tag sequences were clustered into operational taxonomic units (OTUs) using the Quantitative Insights Into Microbial Ecology (QIIME) software package57 and annotated using the SILVA or GreenGenes databases58,59. Community structure was determined using relative abundance and distribution of obtained microbial OTUs along with statistical analyses such as hierarchical clustering and indicator species analysis to identify characteristic groups of OTUs occurring under specific water column conditions such as different water column O2 concentrations.
Illumina metagenomes, metatranscriptomes and metaproteomes were analysed using MetaPathways (version 2.0 and 2.5), an open source pipeline for functional annotation and prediction of reactions and pathways in metagenomes and metatranscriptomes60. Direct link to software download and specifications can be found online on the Hallam Lab Github repository (https://github.com/hallamlab/metapathways2).
With respect to the metaproteomic datasets there is redundancy in the protein database used for peptide mapping in that amino acid sequences will have different names but identical sequences, and thus a given peptide may map to multiple sequences. To manage this one could cluster the proteins by sequence identity at the amino acid level prior to mapping. In previous publications we have calculated a normalised spectral abundance factor (NSAF) as a pseudo-quantative metric to compare abundance of the same proteins between samples40.
Additional information
How to cite this article: Hawley, A. K. et al. A compendium of multi-omic sequence information from the Saanich Inlet water column. Sci. Data 4:170160 doi: 10.1038/sdata.2017.160 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Ulloa, O., Canfield, D. E., Delong, E. F., Letelier, R. M. & Stewart, F. J. Microbial oceanography of anoxic oxygen minimum zones. Proc. Natl. Acad. Sci. U.S.A. 109, 15996–16003 (2012).
Wright, J. J., Konwar, K. M. & Hallam, S. J. Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394 (2012).
Paulmier, A. & Ruiz-Pino, D. Oxygen minimum zones (OMZs) in the modern ocean. Prog. Oceanogr. 80, 113–128 (2009).
Canfield, D. E. et al. A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean coast. Science 330, 1375–1378 (2010).
Lam, P. et al. Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc. Natl. Acad. Sci. U.S.A. 106, 4752–4757 (2009).
Ward, B. B. et al. Denitrification as the dominant nitrogen loss process in the Arabian Sea. Nature 461, 78–81 (2009).
Lam, P. & Kuypers, M. M. Microbial nitrogen cycling processes in oxygen minimum zones. Ann. Rev. Mar. Sci. 3, 317–345 (2011).
Torres-Beltrán, M. et al. Methanotrophic Community Dynamics in a Seasonally Anoxic Fjord: Saanich Inlet, British Columbia. Front. Mar. Sci. 3, 268. doi:10.3389/fmars.2016.00268 (2016).
Naqvi, S. W. A. et al. Marine hypoxia/anoxia as a source of CH4 and N2O. Biogeosciences 7, 2159–2190 (2010).
Diaz, R. J. & Rosenberg, R. Spreading dead zones and consequences for marine ecosystems. Science 321, 926–929 (2008).
Keeling, R. E., Kortzinger, A. & Gruber, N. Ocean deoxygenation in a warming world. Ann. Rev. Mar. Sci. 2, 199–229 (2010).
Arrigo, K. R. Marine Microorganisms and global nutrient cycles. Nature 437, 349–355 (2005).
Stramma, L., Johnson, G. C., Sprintall, J. & Mohrholz, V. Expanding Oxygen-Minimum Zones in the Tropical Oceans. Science 320, 655 (2008).
Whitney, F., Freeland, H. & Robert, M. Persistently declining oxygen levels in the interior waters of the eastern subarctic Pacific. Prog. Oceanogr. 75, 179–199 (2007).
Fuhrman, J. A., Cram, J. A. & Needham, D. M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 13, 133–146 (2015).
Hahn, A. S., Konwar, K. M., Louca, S., Hanson, N. W. & Hallam, S. J. The information science of microbial ecology. Curr Opin Microbiol 31, 209–216 (2016).
Huse, S. M., Welch, D. M., Morrison, H. G. & Sogin, M. L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898 (2010).
Moran, M. A. et al. Sizing up metatranscriptomics. ISME J. 7, 237–243 (2013).
Stewart, F. J., Ulloa, O. & DeLong, E. F. Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40 (2012).
Hawley, A. K., Brewer, H. M., Norbeck, A. D., Paša-Tolic, L. & Hallam, S. J. Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. U.S.A. 111, 11395–11400 (2014).
Hawley, A. K. et al. Diverse Marinimicrobia bacteria may mediate coupled biogeochemical cycles along eco-thermodynamic gradients. Nat. Commun. 8, doi:10.1038/s41467-017-01376-9 (2017).
Louca, S. et al. Integrating biogeochemistry with multi-omic sequence information in a model oxygen minimum zone. Proc. Natl. Acad. Sci. U.S.A. 113, E5925–E5933 (2016).
Ulloa, O. & Pantoja, S. The oxygen minimum zone of the eastern South Pacific. Deep Sea Res. Part 2 Top. Stud. Oceanogr. 56, 987–991 (2009).
DeLong, E. F. et al. Community genomics amoung stratified microbial assemblages in the Ocean's interior. Science 331 (2006).
Shi, Y., Tyson, G. W. & DeLong, E. F. Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature 459, 266–269 (2009).
Ram, R. J. et al. Community Proteomics of a Natural Microbial Biofilm. Science 208, 1915–1920 (2005).
Zaikova, E. et al. Microbial community dynamics in a seasonally anoxic fjord: Saanich Inlet, British Columbia. Environ. Microbiol. 12, 172–191 (2010).
Walsh, D. A. et al. Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 326, 578–582 (2009).
Anderson, J. J. & Devol, A. H. Deep Water Renewal in Saanich Inlet, an Intermittently Anoxic Basin. Estuar. Coast. Mar. Sci. 1, 1–10 (1973).
Carter, N. M. The oceanography of the fjords of southern British Columbia. Fish. Res. Bd. Canada Prog. Rept. Pacific Coast Sta. 12, 7–11 (1932).
Carter, N. M. Physiography and oceanography of some British Columbia fjords. Proc. Fifth. Pacific Sci. Cong. 1, 721 (1934).
Herlinveaux, R. H. Oceanography of Saanich Inlet in Vancouver Island, British Columbia. J. Fish. Res. Board Can. 19, 1–37 (1962).
Chow, C. E., Winget, D. M., White, R. A. 3rd, Hallam, S. J. & Suttle, C. A. Combining genomic sequencing methods to explore viral diversity and reveal potential virus-host interactions. Front. Microbiol. 6, 265 (2015).
Tsementzi, D. et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature 536, 179–183 (2016).
Torres-Beltrán, M. et al. A compendium of geochemical information from the Saanich Inlet water column. Sci. Data 4: 170159, doi:10.1038/sdata.2017.159 (2017).
Falkowski, P. G. et al. Ocean Deoxygenation: Past, Present, and Future. EOS, Trans AGU 92, 409–410 (2011).
Walsh, D. A., Zaikova, E. & Hallam, S. J. Small Volume (1-3L) Filtration of Coastal Seawater Samples. J. Vis. Exp. (28), e1163 (2009).
Walsh, D. A., Zaikova, E. & Hallam, S. J. Large Volume (20 L+) Filtration of Coastal Seawater Samples. J. Vis. Exp. (28), e1161 (2009).
Wright, J. J., Lee, S., Zaikova, E., Walsh, D. A. & Hallam, S. J. DNA Extraction from 0.22 µM Sterivex Filters and Cesium Chloride Density Gradient Centrifugation. J. Vis. Exp. (31), e1352 (2009).
Hawley, A. K. et al. Molecular tools for investigating microbial community structure and function in oxygen-deficient marine waters. Methods Enzymol. 531, 305–329 (2013).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Allers, E. et al. Diversity and population structure of Marine Group A bacteria in the Northeast subarctic Pacific Ocean. ISME J. 7, 256–268 (2013).
Daum, C. et al. Optimization of the Roche 454-Titanium & Illumina GAIIx Production Sequencing Pipelines at the DOE Joint Genome Institute (Advances in Genome Biology and Technology) https://jgi.doe.gov/news-publications/scientific-posters/ (2009).
Cram, J. A. et al. Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. The ISME journal 9, 563–580 (2015).
Tremblay, J. et al. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 6, 771 (2015).
Rivers, A. R. iTag amplicon sequencing for taxonomic identification at the Joint Genome Institute. http://1ofdmq2n8tc36m6i46scovo2e.wpengine.netdna-cdn.com/wp-content/uploads/2013/05/iTagger-methods-1.pdf (2016).
Agogue, H., Brink, M., Dinasquet, J. & Herndl, G. J. Major gradients in putatively nitrifying and non-nitrifying Archaea in the deep North Atlantic. Nature 456, 788–791 (2008).
Daum, C. et al. Illumina GA IIx & HiSeq 2000 Production Sequencing and QC Analysis Pipelines at the DOE Joint Genome Institute. http://jgi.doe.gov/wp-content/uploads/2013/11/AGBT-poster-Illumina-2011_FINAL-1.pdf (2011).
Daum, C. et al. Sanger, 454 and Illumina Production Lines (DOE BERAC review at the Joint Genome Institute). https://jgi.doe.gov/news-publications/scientific-posters/ (2009).
454 Sequencing System Software Manual Version 2.8. http://ftp.ccb.jhu.edu/pub/dpuiu/Wheat/USM-00058.08_454SeqSys_SWManual-v2.8_PartB.pdf (2014).
Castelle, C. J. et al. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 4, 2120 (2013).
Wright, J. J. et al. Genomic properties of Marine Group A bacteria indicate a role in the marine sulfur cycle. ISME J. 8, 455–468 (2014).
TruSeq RNA Sample Preparation Guide V2. Illuminahttps://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_truseq/truseqrna/truseq-rna-sample-prep-v2-guide-15026495-f.pdf (2014).
Kim, S. et al. The Generating Function of CID, ETD and CID/ETD Pairs of Tandem Mass Spectra: Applications to Database Search. Mol. Cell. Proteomics 9, 2840–2852 (2010).
Roux, S. et al. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 3, e03125 (2014).
Vizcaíno, J. et al. 2016 update of the PRIDE database and related tools. Nucleic. Acids Res. 44, D447–D456 (2016).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336 (2010).
DeSantis, T. Z. et al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic. Acids Res. 35, 7188–7196 (2007).
Konwar, K. M., Hanson, N. W., Page, A. P. & Hallam, S. J. MetaPathways: a modular pipeline for constructing pathway/genome databases from environmental sequence information. BMC Bioinformatics 14: 202, doi:10.1186/1471-2105-14-202 (2013).
Data Citations
Acknowledgements
We thank Captain Ken Brown and his crew for their engaged effort on every cruise aboard the RSV Strickland. We also thank past and present members of the Hallam lab and the many undergraduate trainees, aka minions, for contributions to cruise preparation and clean up, water filtration, DNA extraction and QC. We also thank Tortell lab members for logistical support. This work was performed under the auspices of the US Department of Energy (DOE) Joint Genome Institute an Office of Science User Facility, supported by the Office of Science of the U.S. Department of Energy under Contract DE-AC02- 05CH11231, the G. Unger Vetlesen and Ambrose Monell Foundations, the Tula Foundation-funded Centre for Microbial Diversity and Evolution, the Natural Sciences and Engineering Research Council of Canada, Genome British Columbia, the Canada Foundation for Innovation, and the Canadian Institute for Advanced Research through grants awarded to S.J.H. Ship time support was provided by NSERC between 2007-2014 through grants awarded to S.J.H. and P.D.T. Metaproteomics support came from the intramural research and development program of the W.R. Wiley Environmental Molecular Sciences Laboratory (EMSL). EMSL is a national scientific user facility sponsored by the US DOE’s Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory operated by Battelle for the US DOE. M.T-B. was funded by Consejo Nacional de Ciencia y Tecnología (CONACyT) and the Tula Foundation. A.K.H. was supported by the Tula Foundation.
Author information
Authors and Affiliations
Contributions
A.K.H. and M.T.-B.. provided extensive logistical support and planning for the time series and curated the datasets. A.K.H. extracted samples and carried out QC for metatranscriptomics and metaproteomics, M.T.-B. carried out QC for pyrotag samples. M.T.-B. aided in metagenomic and metatranscriptomic data curation and QC. E.Z., D.A.W. and S.J.H. provided additional technical and logistical support and developed field protocols. A.M., M.S., S.K., O.S., E.A.G., and D.F. provided technical support and contributed to data collection, sample extraction and curation. C.P. and L.P. provided technical and logistical support as sea-going technicians. S.M. and T.G. aided in metagenome and metatranscriptome sequencing and QC. S.T. managed the metagenomic and metatranscriptomic capability at JGI. H.B. aided in protein extraction and sequencing. A.N. conducted peptide matching and QC. L.P.T. managed the metaproteomics capability at EMSL. C.S. contributed materials and data and edited the manuscript. S.H. designed and initiated the time series with P.D.T. S.J.H. managed and directed the project. A.K.H., M.T.-B. and S.J.H. wrote the manuscript with editorial input from remaining co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Hawley, A., Torres-Beltrán, M., Zaikova, E. et al. A compendium of multi-omic sequence information from the Saanich Inlet water column. Sci Data 4, 170160 (2017). https://doi.org/10.1038/sdata.2017.160
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.160
This article is cited by
-
A compendium of bacterial and archaeal single-cell amplified genomes from oxygen deficient marine waters
Scientific Data (2023)
-
The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments
Scientific Data (2022)
-
Pangenomics reveals alternative environmental lifestyles among chlamydiae
Nature Communications (2021)
-
Mercury methylation by metabolically versatile and cosmopolitan marine bacteria
The ISME Journal (2021)
-
Monitoring microbial responses to ocean deoxygenation in a model oxygen minimum zone
Scientific Data (2017)
