
Abstract
Neurofibromin controls many cell processes, such as growth, learning, and memory. If neurofibromin is not working properly, it can lead to health problems, including issues with the nervous, skeletal, and cardiovascular systems and cancer. This review examines neurofibromin’s binding partners, signaling pathways and potential therapeutic targets. In addition, it summarizes the different post-translational modifications that can affect neurofibromin’s interactions with other molecules. It is essential to investigate the molecular mechanisms that underlie neurofibromin variants in order to provide with functional connections between neurofibromin and its associated proteins for possible therapeutic targets based on its biological function.
Similar content being viewed by others


Introduction
Neurofibromatosis type 1 (NF1) (OMIM#162200) is an autosomal dominant multisystemic disorder with a worldwide incidence of approximately 1 in 3000 individuals caused by germline mutations in the NF1 tumor suppressor gene1. The NF1 gene encodes neurofibromin, a multifunctional protein capable of regulating multiple signaling pathways including Ras/MAPK2, Raf/MEK/ERK3, PI3K/AKT/mTOR4, Rho/ROCK/LIMK2/cofilin5, PKA-Ena/VASP6 and cAMP/PKA7. As a consequence, neurofibromin regulates a wide variety of cellular processes such as proliferation, migration, differentiation, cytoskeletal dynamics, apoptosis and stress responses8. As neurofibromin regulates the Ras/MAPK pathway, NF1 is included among the RASopathies, a group of developmental disorders caused by germline mutations in genes encoding components of the Ras/MAPK pathway9.
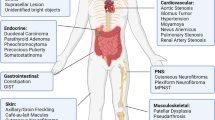
Neurofibromin can be found in human cells in two major isoforms: Isoform I (2818 residues) and Isoform II (2839 residues) and is ubiquitously expressed in most tissues; however, it is highest in the central nervous system, especially in neurons, oligodendrocytes, peripheral nerve trunks, glial cells, astrocytes, leukocytes, adrenal medulla and Schwann cells10,11. Despite the fact that the NF1 phenotype is variable, due to epigenetics, stochastic events and genetic modifiers12, it is characterized by café-au-lait macules (CALMS)13, skinfold freckling14, Lisch nodules15, optic pathway gliomas16 and neurofibromas17. Other symptoms include but are not limited to skeletal abnormalities, vascular injuries, learning disabilities, attention deficit, increased susceptibility to autism and social and behavioral problems18 and conferral of drug resistance in cancer therapy19. Hematopoietic neoplasms, such as juvenile myelomonocytic leukemia20 and the presence of pheochromocytoma21, are also associated with clinical manifestations in NF1 patients. The NF1 clinical observations suggest that the NF1 gene is a critical regulator of brain neuronal function22, embryonic development23, pneumothorax and cardiovascular defects23, as well as a common driver gene in several aggressive human sporadic malignancies not associated with NF124, including glioblastoma25, melanoma26, ovarian carcinoma27, lung cancer28, cholangiocarcinoma29, breast cancer30, lymphoblastic leukemia31 and other types of tumors32. To date, according to COSMIC database, more than 5524 different somatic variants in the NF1 gene have been identified in human tumors, whether they are pathogenic or benign is not known. The correlation between specific genetic variants and the manifestation of different tumors in NF1 has been extensively studied; The presence of NF1 microdeletions has been found to be associated with severe forms of NF1 and a higher risk of developing MPNST33, deletion of Met 992 (c.2970-2972 delAAT) has been identified as a causative factor for benign forms of NF134, and the R1809C mutation has also been shown to be associated with benign symptoms35. Missense mutations in the region between amino acids 844–848 of the cysteine/serine-rich domain (CSRD) have been linked to more severe phenotypes, including spinal plexiform neurofibromas, optic pathway gliomas and malignant tumors36, and missense mutations affecting R1276 and K1423 have been linked to severe phenotypes such as cardiovascular abnormalities and spinal plexiform neurofibromas, while those affecting Met 1149 are associated with more benign symptoms37. There are also other studies that provide a comprehensive overview of the direct correlation between specific variants in the NF1 gene and different tumors, and may be useful for further reading on the topic24,38,39,40. Also, an analysis was conducted on neurofibromin to identify regions that may be considered hotspots in neurofibromatosis type 1; the study found three regions within neurofibromin that were statistically significant including the RAS-GTPase domain, the CSRD, and the Armadillo141. In this context, elucidation of the canonical and non-canonical effector pathways downstream of Ras activation and their ultimate cell-specific consequences has identified promising therapeutic targets42. These findings demonstrate the crucial role that specific genetic variants play in the manifestation of tumors in NF1, and the importance of understanding the genetic basis of NF1 for the development of new therapeutic strategies.
For several years, only the Ras-GTPase-activating protein-related domain (GRD)43 and the Sec14-PH domain44 of neurofibromin were structurally characterized. Recently, the biggest breakthrough in the field consisted in the resolution of the structure of the neurofibromin dimer. Full-length neurofibromin 3D structures were solved by a series of biochemical and biophysical experiments including size-exclusion chromatography multi-angle light scattering (SEC-MALS), small-angle X-ray (SAXS), small-angle neutron scattering (SANS) and analytical ultracentrifugation45. These experiments showed that neurofibromin exists as a high-affinity dimer and identified the regions important for dimerization, suggesting that neurofibromin is highly sensitive to mutations that disturb its structure45. After this study, several detailed molecular structures of neurofibromin by cryo-electron microscopy (cryo-EM) were reported, suggesting that the dimeric architecture rest in an equilibrium between the closed and open conformation states46,47. Cryo-EM reveals domain organization and structural details of the isoform 2 in either a closed, self-inhibited, zinc-ion-binding site stabilized state, or an open state46. In the closed conformation, HEAT/ARM core domains shield the GRD so that Ras binding is sterically inhibited. In the open conformation, a large-scale movement of the GRD occurs, which is necessary to access Ras, whereas Sec14-PH reorients to allow interaction with the cellular membrane46. The transition between closed and open states provides guidance for targeted studies that decipher the complex molecular mechanism behind the widespread neurofibromatosis syndrome and neurofibromin dysfunction in carcinogenesis46. Furthermore, the homodimer is characterized by a central lemniscate-shaped core formed by the assembly of the N- and C-HEAT domains47. Three-dimensional variability analysis was captured by the GRD and Sec14-PH domains positioned against the core scaffold in a closed conformation, suggesting that interaction with the plasma membrane may release the closed conformation to promote Ras inactivation47. Mutation or deletion at a disproportionate number of sites is likely to result in improper assembly of the dimer, contributing to the acute sensitivity of NF1 gene to mutations in disease47. To date, the latest structural study that has been published using cryo-EM reveals an extended neurofibromin homodimer that has two conformational states: an auto-inhibited state with occluded Ras-binding site and an asymmetric open state with an exposed Ras-binding site48. This new model suggests that the GRD may interact simultaneously with two sets of Ras homodimers, but likely not a single homodimer48. While the occluded conformation is incompetent for Ras binding, both states of neurofibromin are compatible with the interaction of the SPRED1-EVH1 domain with the GAPex domain48. Also, the interaction of the Sec14-PH domain with membranes and lipids may have impacted the transition between occluded and open conformation, although the zinc-ion-binding site may be stripped out before the GRD-Sec14-PH linker is rearranged into an active conformation. It was found that nucleotide binding stimulates the active conformation of neurofibromin dimer and releases a lock that maintains an occluded inactive state, leading to the activation of the protein48. In addition, it was reported that a Zn2+ binding site stabilizes the dimer in a closed conformation, which is relevant to the overall conformational changes that neurofibromin undergoes upon activation46. These recent structures revealed a complex set of helical repeats throughout the protein, that if disrupted, are likely to affect the overall structure of the protein in a way that it interferes with the positioning of the GAP domain, probably leading to alteration of GAP activity. Despite these great advances, structural and functional insights into neurofibromin activation still remain incompletely defined. In this review, we provide functional connections between neurofibromin and its binding partners, signaling pathways and possible therapeutic targets.
Neurofibromin domains and known interacting partners
Neurofibromin contains several functional domains and regions allowing the interaction with many binding proteins and effectors (Fig. 1). The N-terminal region of neurofibromin contains several variants (R103K, D105N, M108I, L114M, E116*, A131S, and E225Rfs*6) that have been suggested to result in a non-functional protein leading to NF1 and tumor formation49 (Supplementary Table 1).

CSRD (cysteine-serine rich domain, residues 543–909), TBD (tubulin-binding domain, residues 1095–1197), GRD (GAP-related domain, residues 1198–1530), Sec14-PH (residues 1560–1816), LRD (leucine-rich domain, residues 1579–1971), the CTD (C-terminal domain, residues 2260–2818) including the NLS (bipartite nuclear localization signal domain, residues 2534–2550) and SBR (syndecan-binding region, residues 1357–1473 and 2619–2719). Domains are in bold whereas interacting partners are shown in colored boxes according to their cognate domains.
The cysteine/serine-rich domain (CSRD, residues 543–909) is an important allosteric activator of the adjacent GRD after PKC\(\alpha\)-dependent phosphorylation in neural cells50. In addition, it has been shown to increase the association of neurofibromin with actin upon phosphorylation, resulting in allosteric regulation of the GRD by increasing its Ras-GAP activity to arrest cell growth50. This region contains three cysteine pairs (622/632, 673/680, 714/721) presumably implicated in stabilizing the tridimensional structure of neurofibromin, and the highly probable palmitoylation site C84551 (Supplementary Table 2). Recent genetic analyses have pinpointed variants in CSRD as associated with a higher risk of developing optic pathway glioma36,52, plexiform and/or spinal neurofibromas, malignant neoplasm and osseous lesions36,51, and patients with pheochromocytoma53. Neurofibromin may interact with dimethylarginine dimethylaminohydrolase-1 (DDAH1), coinciding with the regions containing specific PKA phosphorylation sites54, whereas DDAH1 interacts with Ras55. DDAH1 is a nitric oxide regulator that degrades the endogenous nitric oxide inhibitor asymmetric dimethylarginine (ADMA) responsible for regulating cell proliferation56. Additionally, a microtubule-associated protein (MAP) domain resides within the CSRD and is thought to regulate the association of neurofibromin with microtubules57 (Fig. 1).
The tubulin-binding domain (TBD, residues 1095 to 1197) contains a series of 12 predicted HEAT-like repeats commonly involved in protein–protein interactions45 although it lacks the conserved four tandem C-terminal microtubule-binding repeats (KXGS motif)58 and contains sequences recognized by the proteasome (1095–1097 (KYF) and 1098–1100 (TLF))59. This domain interacts with \(\beta\)-tubulin60, cytoplasmic dynein heavy chain (DHC), and the leucine-rich pentatricopeptide repeat motif-containing (LRPPRC) protein61. The interaction with LRPPRC is of pathological interest in that it links neurofibromin to Leigh’s syndrome French Canadian (LSFC), an autosomal recessive neurodegenerative disorder that arises from variants in the LRPPRC gene62. Whereas some authors showed that TBD mediates neurofibromin dimerization45, this possibility has recently been ruled out46,47,48.
The GAP-related domain (GRD, residues 1198–1530) is responsible for the interaction with Ras, and it is the most well-studied functional domain of neurofibromin (Fig. 1). Neurofibromin-GRD, located central to the protein, consists of GAPex, including Nex (residues 1176 to 1247) and Cex (residues 1478–1573), and the Ras-GTPase-activating (Ras-GAP) region (1248 to 1477 residues)63. The Ras-GAP mediates the downregulation of the activity of all classical Ras proteins64, Ras-like RRAS and MRAS65. Indeed, when GRD is overexpressed it is sufficient to normalize the increased Ras activity and cell hyperproliferation in Nf1-deficient mouse cells and tissues66,67. It also partially restores normal phosphorylated cofilin levels and suppresses the accumulation of actin stress fibers68. The GRD’s Nex and Cex interact with the EVH1 domain of Spred163,69, and do not impact catalysis63. Mutations which reside on the protein surface in the vicinity of the GRD, are more likely to directly impair or abolish the binding of this domain with Ras, whereas mutations embedded deep within the protein structure (p.R1204G and p.R1204W) may impact upon protein structure stability and abolish Ras–GRD binding indirectly70. Other variants (R1276E, K1423E) abolished GAP activity without reducing protein stability48, whereas others are dispensable for GAP activity (Supplementary Table 1). A recent precise analysis provides insight into how the membrane targeting of neurofibromin by Spred1 allows simultaneous interaction with activated KRas71. Other GRD interacting proteins are FAF2, APP and kinesin-1. FAF2 interacts with neurofibromin fragments (372–1552, specially from 1176 to 1552)72, promoting its ubiquitin-dependent proteolysis72. APP interacts with neurofibromin residues 1357–1557 in human melanocytes, and both proteins colocalize with melanosomes73 and share their interaction with the molecular motor protein kinesin-1 (Fig. 1). Kinesin-1 is required for normal distribution of mitochondria and lysosomes, and also transports cargo such as ATP along microtubules, consisting of two 120-kDa heavy chains (KHC) and two 64-kDa light chains (KLC)74. Neurofibromin interacts with KHC whereas APP interacts with KLC75. There is a syndecan-binding region (SBR, residues 1357–1473), as well as one in the CTD (residues 2619–2719) (Fig. 1). The SBR was found to mediate the interaction of neurofibromin with the cytoplasmic tail of all four mammalian syndecans76, transmembrane proteins that regulate signaling pathways involved in cell adhesion and migration, cellular behavior, intracellular calcium regulation and homeostasis77. Finally, other potential interesting regions are the Poly-Ser (1352–1355), the putative palmitoylation site C1365 and a cholesterol motif (1364–1375)51.
The leucine-rich domain (LRD, residues 1579–1971) consists of a glycerophospholipid binding Sec14-like domain (1560–1698), a PH-like domain (1715–1816) and part of the HEAT-like repeats (HLR) (1825–2428)78 (Fig. 1). It is involved in learning disabilities, skeletal problems like tibia bone defects and scoliosis79, as well as in inhibiting tumor metastasis and invasion of human glioblastoma cells78. The LRD failed to hydrolyze Ras, suggesting that its suppressive function is independent of Ras signaling78, and binds to caveolin-1 (Cav-1) which may act as a scaffolding protein within caveolar membranes80. In particular, one of the most commonly mutated positions in neurofibromin (R1809G) would impair neurofibromin function, implying a Ras-independent mechanism48. The SecPH binds phospholipids44, it also interacts with LIMK2 and would specifically prevent LIMK2 activation by ROCK5. However, no structural data on the complexes are available and it is not known whether this proposed mechanism actually takes place with these or other partners81. The serotonin 5-HT6 receptor, one of the several GPCRs for serotonin, activates cAMP formation on agonist stimulation and was found to interact with neurofibromin PH and CTD domains82. The residues K1634 and K1731 within the SecPH domain have been identified as minor and major SUMO-conjugation sites, respectively, and are hypothesized to play a critical role in the function of neurofibromin83. Specifically, SUMOylation of K1731 has been shown to modulate the Ras-GAP activity of the GRD domain, which is located adjacent to the SecPH domain. Additionally, it has been observed that a K1731R mutation negatively impacts the Ras-GAP activity of GRD-SecPH fragment of neurofibromin, suggesting that this site is critical for the proper functioning of neurofibromin83.
The HEAT-Like Repeat (HLR, residues 1825–2428) contains the structurally related Armadillo (ARM) superfamily regions 1849–1886 and 1920–198478 (Fig. 1). These repeats are comprised of short hydrophobic α-helical hairpins that stack on top of each other to form long super-helical structures usually involved in protein–protein interactions78. Variants in this domain have been linked to NF1, suggesting an importance of the entire helical repeat scaffold for neurofibromin function84, including a lower risk for optic pathway glioma in NF1 patients52,85 (Supplementary Table 1).
The C-terminal domain (CTD, residues 2260–2818), contains a third Armadillo region (residues 2200–2571), the nuclear localization site (NLS) (residues 2534–2550)86 and the tyrosine kinase recognition sites (TRS) (residues 2549–2556)87 (Fig. 1). The CTD regulates cAMP via G-protein-dependent activation of adenylyl cyclase88, and has been shown to regulate the transition from metaphase to anaphase89, acting as a tubulin-binding domain, and to regulate nuclear localization through phosphorylation on S2808 a residue adjacent to a nuclear localization signal in the CTD by PKC-ε in glioblastoma cells, resulting in a predominantly nuclear localization60. Consequently, this phosphorylation could be responsible for neurofibromin translocation to the nucleus60, whereas phosphorylation by cAMP-dependent PKA (Supplementary Table 2) promotes association with 14-3-3 proteins, negatively regulating neurofibromin GAP activity55. It has been demonstrated a physical interaction between the C-terminal domain of neurofibromin and the N-terminal domain of FAK90, which also binds to the TPPKM motif present in the NLS (KRQEMESGITTPPKMRR) of neurofibromin90. The differential Microtubule-Associated-Protein (MAP) properties of NLS in both the assembly of the mitotic spindle as well as faithful genome transmission have been recently discussed91. Other CTD interacting proteins are CRMP-2 and CRMP-492 (Fig. 1). Neurofibromin directly regulates CRMP-2 phosphorylation accessibility and activity by suppressing CRMP-2-phosphorylating kinase cascades via its Ras-GAP function, regulating neurite outgrowth and dendritic filopodia formation93. CRMP-2 has been implicated in multiple neurological disorders, including the development of Alzheimer’s disease. In addition, neurofibromin and CRMP-2 also work together in non-neuronal cells and contribute to cell cycle control92.
Signaling pathways upstream of neurofibromin
A better understanding of the implications of neurofibromin signaling functions may help to explain the diverse clinical manifestations and the increased cancer risk observed in NF1 patients. Neurofibromin is under the regulation of several upstream signaling elements including several transmembrane receptors, kinases, and cytosolic proteins (Fig. 2). Transmembrane receptors include the cytokine receptor granulocyte-macrophage colony-stimulating factor (GM-CSFR)94, tyrosine kinases receptors (RTKs)95, anaplastic lymphoma kinase (ALK)96, vascular endothelial growth factor (VEGFR)97, epidermal growth factor (EGFR)98, platelet-derived growth factor (PDGFR)99, hepatocyte growth factor (MET), GPCRs endothelin B (EDNRB)100 and serotonin 5-HT6 receptor82 (Fig. 2).

The APP and GM-CSFR, several RTKs and several GPCRs have been involved in neurofibromin signaling and with different biological consequences. Main cytosolic upstream regulators of neurofibromin include 14-3-3, FAK, PKC, PKA, DDAH1 and Spred1. Neurofibromin posttranslational modifications include phosphorylation and ubiquitination.
Receptor tyrosine kinase APP is expressed in many tissues and concentrates in the synapses in neurons where it performs physiological functions relevant to neurite growth, neuronal adhesion and axonogenesis, including kinesin-mediated axonal transport of β-secretase and presenilin-1101. APP and neurofibromin colocalize with melanosomes73, perhaps, as part of a melanosome transport/biogenesis regulating mechanism (which could be related to the etiopathogenesis of pigment-cell-related manifestations in NF1) or a mechanism for sequestering neurofibromin from the plasma membrane73. Another receptor upstream of neurofibromin is the GM-CSFR, which is necessary to maintain JMML in Nf1-mutant mice and in neurofibroma formation after nerve injury94. ALK was identified as an upstream activator of neurofibromin-regulated Ras signaling in Drosophila and it is responsible for several dNf1 defects, including cognitive performance96. VEGFR is known to be expressed in cutaneous neurofibromas and MPNST, where it correlates with poor patient prognosis. The PDGFR is another RTK; overexpression of wild-type PDGFR associated with neurofibromin deficiency leads to aberrant activation of downstream Ras signaling and thus contributes importantly to MPNST development, indeed, it is overexpressed in Schwann cells derived from neurofibromas and MPNST99. Overexpression of PDGFR cooperates with loss-of neurofibromin and p53 to accelerate the molecular pathogenesis of MPNST99. MET has been reported activated in some MPNSTs, and implicated in resistance to Ras pathway inhibition in several cancers, including melanoma and colorectal cancer102. Also, the expression levels of MET and its ligand hepatocyte growth factor correlate with MPNST progression103.
Serum and growth factors trigger the rapid ubiquitination and complete proteasomal degradation of neurofibromin in many cell types59 (Fig. 2). Specifically, the literature has demonstrated that PKC plays a critical role in promoting Ras activation by destabilizing neurofibromin104. Additionally, research has indicated that PKCδ and α/β are essential components for maintaining the aberrant Ras signaling and promoting cell viability in neurofibromin-deficient cells105. Furthermore, PKCε-dependent H-Ras activation involves the recruitment of the RasGEF SOS1 and the Ras-GAP neurofibromin to the lipid rafts of embryonic neurons51. Lastly, neurofibromin has been shown to regulate atypical PKC activity in a RAS-dependent manner, implicating PKCz as a potentially novel effector of neurofibromin/Ras signaling in the brain22.
Other cytosolic upstream proteins include Spred169, DAAH155, the GPCR-activated protein Gβγ subunits106 and 5-HT6 receptor. Phosphorylation of S105 on Spred1 by an oncogenic RTK (EGFRL858R) disrupted the binding of Spred1 and neurofibromin, and as a consequence blocked negative regulation of Ras-GTP71. DDAH1 may interacts with CSRD and CTD and facilitates phosphorylation on these domains by PKA, T586, S818 and S876 on CSRD and some residues from 2620 to 2818 on CTD54. DDAH1 also exerts effects on cyclin D1 and cyclin E expression through multiple mechanisms, including VEGF, the NO/cGMP/PKG pathway, the Ras/PI3K/AKT pathway, and NF1 expression107. 14-3-3 interacts with CTD upon phosphorylation by PKA, an interaction that could decrease NF1-GAP activity55. Finally, neurofibromin promotes 5-HT6 constitutive activation of Gαs/AC pathway in striatal neurons82. Both NF1 silencing and NF1 patient variants within the PH domain inhibited constitutive receptor activity on the Gαs/AC pathway and reduced basal cAMP levels82.
Signaling pathways downstream of neurofibromin
One of the principal functions of neurofibromin consists in the modulation of the Ras/MAPK pathway2, although other signaling pathways have been studied including the Raf/MEK/ERK3, PI3K/AKT/mTOR4, Rho/ROCK/LIMK2/cofilin5, PKA-Ena/VASP6 and cAMP/PKA pathways7.
Ras signaling pathway
Neurofibromin enhances the rate at which the GTP-bound form of Ras is converted into the inactive GDP-bound form2,108. Therefore, loss-of-function mutations in neurofibromin result in the accumulation of Ras in the GTP-bound state. GTP-bound Ras proteins activate fundamental signaling pathways involved in several cellular processes such as cell polarity, proliferation, differentiation, adhesion, migration and apoptosis109. For instance, neurofibromin is the main Ras inactivator in dendritic spines of hippocampal pyramidal neurons110, long-term potentiation and hippocampal-dependent learning in interneurons111. Ras-GTP signals through three main molecular pathways, namely, the Raf/MEK/ERK, the Ral/NFkB and the PI3K/AKT/mTOR (Fig. 3).

Neurofibromin is involved in several cell signaling pathways, including the Ras/MAPK, Akt/mTOR, Ral, ROCK/LIMK/cofilin, and cAMP/PKA pathways. From the cytosol to the nucleus neurofibromin regulates many fundamental cellular processes, such as expression of estrogen response genes, proliferation, migration, cortex development, learning and memory (L/M).
The Raf/MEK/ERK pathway, as well as the PI3K/AKT/mTOR pathway, are both well-characterized pathways downstream Ras112,113. Both pathways are hyperactivated in neurofibromin-deficient cells, although the dependence on either MAPK or AKT signaling differs from cell type to cell type. For instance, neurofibromin-deficient hematopoietic cells are more dependent on neurofibromin/MAPK pathway growth control114, whereas the proliferation of neurofibromin-deficient glial cells (Schwann cells and astrocytes) primarily relies on neurofibromin/AKT signaling115. Hypo and hyperactivation of Ras/Rap signaling impair the capacity of synaptic plasticity, underscoring the importance of a “happy-medium” dynamic regulation of the signaling behavior116.
In the absence of neurofibromin, aberrant mTOR activation depends on Ras and PI3K, mediated by AKT phosphorylation and inactivation of tuberin4. The mechanism that involves AKT, the TSC complex and Rheb is the primary mechanism through which PI3K signaling activates mTORC1117. In lysosomes, neurofibromin negatively regulates mTOR in a LAMTOR1-dependent manner118. Ral-GEFs have also been implicated downstream of Ras as a novel cell signaling abnormality in MPNSTs119. Indeed, neurofibromin regulates EMT transition in a way that downregulation of neurofibromin encourages the EMT transition120. The EMT factors including SNAIL, SLUG, TWIST-1 and ZEB have been shown to be upregulated in MPNST deficient for neurofibromin120,121.
PKA-enabled/vasodilator-stimulated phosphoprotein (PKA-Ena/VASP)
Neurofibromin modulates PKA-Ena/VASP pathway, implicated in the formation of filopodia and dendritic spine6, axonal outgrowth122, actin polymerization along bundle formation123, and probably for proper differentiation of nerve cells6. In part due to neurofibromin association with microtubular and microfilamentous cytoskeleton91, FAK90 and syndecan-276, the normal development of the cerebral cortex may be affected74. FAK is one of the main proteins localized at focal adhesions, playing an essential role in regulating cell migration, adhesion, spreading, reorganization of the actin cytoskeleton, formation and disassembly of focal adhesions and cell protrusions, cell cycle progression, cell proliferation and apoptosis90. It also plays a role in osteogenesis and differentiation of osteoblasts124. Syndecan-2 interacts with neurofibromin and CASK, forming a multidomain scaffolding protein including also APP73 and neurexins125, while also having a role in synaptic transmembrane protein anchoring and ion channel trafficking. Indeed, it shows overlapping distribution at synaptic junctions, suggesting a potential role of neurofibromin in adhesion and signaling at neural synapses126. Therefore, deregulation of postsynaptic Ras signaling may explain learning disabilities associated with NF1110. Recent findings indicate a reciprocal regulation of Wnt/β-catenin signaling pathway and APP processing involving a physical interaction between APP and β-catenin127. Furthermore, APP is implicated in the pathogenesis of Alzheimer disease, as well as in cancer development in breast, pancreas, prostate and non–small cell lung cancers128.
Rho/ROCK/LIMK2/cofilin pathway
Neurofibromin enhances cell motility by regulating actin filament dynamics via the inhibition of the Rho/ROCK/LIMK2/cofilin pathway68 (Fig. 3). In neurofibromin-depleted cells, RhoA hyperactivates ROCK and its downstream effector LIMK1/2, which continued sustained phosphorylation and inactivation of cofilin, leading to enhanced cell migration and invasion68. Actin depolymerizing by cofilin is a mechanism that may be one of the responsible for the neurofibroma formation in patients with NF168.
Rac1/PAK1/LIMK1/cofilin pathway
Neurofibromin also acts as a negative regulator of the Rac1/PAK1/LIMK1/cofilin pathway independently of Ras signaling pathways68 (Fig. 3). LIMK1 is phosphorylated and activated by PAK1, downstream of Rac1 and Cdc42 GTPases68. Cells with constitutively activated Rac exhibit dramatic increase in membrane ruffling, with an increase in actin polymerization and formation of stress fibers129. Neurofibromin involvement in these signaling pathways has been established. However, most of its molecular targets are still unknown, and the molecular mechanisms remain in most cases to be elucidated64.
cAMP/PKA pathway
Neurofibromin is a positive regulator of intracellular cAMP levels (Fig. 3), which cannot be reversed by inhibition of Ras-MEK or Ras-PI3K downstream signaling130. The cAMP/PKA pathway is involved in many processes, such as inhibition of cell growth131, induction of apoptosis132, arrest of the rearrangement of the cytoskeleton133, neuropeptide responses134, development and functioning of the nervous system, synaptogenesis6, learning process and memory shaping135, sugar and lipid metabolism136 and cancer137. The stimulation of GPCR causes neurofibromin to regulate cAMP production in CNS neurons through different Gα activators, not Gαi. This regulation is dependent on Ras and results in interaction with target PKA22 (Fig. 3). Ras/cAMP regulation operates through the activation of atypical PKCz, leading to GRK2-driven Gα inactivation22. These observations highlight the regulation of a diverse number of GPCRs in distinct CNS cell population, suggesting a potential strategy to correct NF1-related CNS deficits22.
In NF1 melanocytes, the molecular mechanisms of melanin synthesis, if NF1 is inactivated, are linked to increased activity of cAMP-mediated PKA and ERK signaling pathways, which in turn leads to overexpression of the key transcription factor MITF and melanogenic enzymes, such as tyrosinase and TRP-2/dopachrome tautomerase, resulting in hyperpigmentation138. Moreover, the overexpression of cAMP-responsive element-binding protein (CREB) bound to the brain-enriched microRNA-9 promoter has been described to repress expression of NF1, and encourage cell migration139. Interestingly, miR-514a overexpression was correlated with increased melanoma cell resistance to BRAFi through decreased expression of NF1 and associated with pairing therapies involving target-based therapy140,141.
A Drosophila Nf1 model revealed that neurofibromin is essential for the cellular response to neuropeptides, like pituitary adenylate cyclase activating polypeptide-38 (PACAP38) at the neuromuscular junction, through activation of the cAMP/PKA pathway134. Human PACAP38 activates the receptor for insects PDF when co-expressed with neurofibromin, potentiating PDF action by coupling to AC142, and induces cell growth in astrocytes activating MAPK143. In neuroblastoma cells, PACAP38 regulation is mediated by PAC1 receptor through a cAMP-dependent but PKA-independent mechanism144,145.
The ubiquitin–proteasome pathway
Neurofibromin is dynamically regulated by the ubiquitin–proteasome pathway, triggered by several growth factors which reduced neurofibromin levels rapidly in a variety of cell lines59. This regulation appears to be independent of Ras activation, as exogenous expression of an activated Ras allele did not induce neurofibromin degradation, and inhibitors of MEK and PI3K did not prevent degradation59. Neurofibromin is a physiological substrate of the SAG E3 ubiquitin ligase during embryogenesis146. Also, the Cul E3 complex and the BTB adaptor protein KBTBD7 regulate PKC-mediated neurofibromin ubiquitination in normal and pathogenic settings147. The hypoxia-associated factor (HAF) might also promote ubiquitination and proteasomal degradation of neurofibromin, which may be significant during physiological hypoxia including embryonic development and wound healing, but may also play a driving role in the development and progression of hypoxia-associated cancers148.
Neurofibromin as an estrogen-receptor (ER) transcriptional co-repressor in breast cancer
The correlation between NF1 loss and upregulation of ER-associated pathways in human breast cancer149, increased tumor aggressiveness and poor patient prognosis, associate neurofibromin with breast cancer149. In addition, NF1 has been reported to be mutated more frequently in ER+ metastatic breast cancer, suggesting it is a driver of breast cancer progression150. Recently, it has been demonstrated that neurofibromin acts as a dual repressor for both Ras and ER signaling; co-targeting may treat neurofibromin-deficient ER+ breast tumors151.
Neurofibromin-HIPPO
HIPPO signaling is known to regulate a variety of cellular processes including cell cycle progression, apoptosis, tissue regeneration, cell differentiation, and the control of organ size and development. In this pathway MST1/2 kinases activate LATS1/2 kinases, which in turn phosphorylate and inhibit the nuclear translocation of transcriptional coactivators YAP and TAZ, regulating gene expression152 (Fig. 4a). Dysregulation of the HIPPO pathway contributes to cancer development through tumorigenesis153 and cutaneous neurofibromas from NF1 patients154,155. On the other hand, MPNSTs show an elevated HIPPO-YAP/TAZ expression156. Remarkably, the YAP signature is present in MPNSTs regardless of their NF1 genetic status, suggesting that activation of the HIPPO-YAP/TAZ pathway is common to both genetic and sporadic MPNSTs156. YAP and TAZ directly interact with JUNB and STAT3 via a WW domain important for transformation, and they stimulate transcriptional activation by AP-1 proteins, implicated in the development and maintenance of cancers157.

a The Hippo pathway is a master regulator of tissue homeostasis and organ size in which MST1/2, LATS1/2 and YAP/TAZ are major players. The JAK/STAT pathway regulates embryonic development and is involved in processes such as stem cell maintenance, hematopoietic and inflammatory response. These pathways also regulate gene expression implicated in tumorigenesis and cancer progression. b This pathway involves neurofibromin in regulating cAMP levels, important in neuronal connections in which dopamine travels to areas of the brain and body to convey important information such as executive thinking, cognition, feelings of reward and pleasure and voluntary motor movements. Dopamine is thought to guide learning via dynamic and differential modulation of PKA.
JAK2/STAT3 signaling pathway
This signaling pathway can influence the transcription and expression of multiple genes involved in biological processes such as cellular growth, metabolism, differentiation, degradation and angiogenesis (Fig. 4a). The JAK-signal transducer and STAT members, particularly STAT3, have been demonstrated to be very important for cancer progression158. EGFR is one of the receptor tyrosine kinases that mediates STAT3 phosphorylation and is expressed in most human MPNST159. In addition to the JAK-signal transducers and activators of transcription axis, JAKs can also affect other signaling pathways through intracellular crosstalk, highlighting the Ras pathway160.
Neurofibromin and dopamine
Neurofibromin is a positive regulator of dopamine homeostasis, as NF1 variants in human and mouse neurons lead to reduced levels of the neurotransmitter dopamine161,162. Using behavioral, electrophysiological and primary culture, it has been demonstrated that reduced dopamine signaling is responsible for cAMP-dependent defects, whereas pharmacological elevation of dopamine reverses in neuron function, learning, memory and attention deficits in NF1-mice163 (Fig. 4b). A dose-dependent relationship was identified between neurofibromin levels, dopamine signaling and cognitive deficits in the hippocampus and striatum on NF1 patients164.
Neurofibromin therapeutic targets
When altered, neurofibromin has been involved in neurofibromatosis type 1, highly aggressive malignant diseases and mechanisms of treatment resistance. In that context, several studies have reviewed the potential therapeutics, the mechanism of action, as well as the information on the status of how far a drug has progressed clinically42,165,166,167,168. In this work, we have summarized all therapies targeting the upstream/downstream molecules of neurofibromin with potential to become novel strategies for the treatment of NF1-related malignancies (Fig. 5). Attractive therapeutic targets include: the modulation of nitric oxide54,107, histone deacetylase inhibitors169, YAP and TAZ156, inhibition of VEGFR2/MET/RET by cabozantinib in MPNST102, the HIPPO pathway152,155 and Ral-GEFs for MPNSTs119,170, CRMP-2 for therapeutic intervention in patients with NF1 pain171, ALK172, inhibition of FAF272 and STAT3 on neurofibromas173, and VCP174 for NF1-cancer therapy, 5-HT6 receptor for cognitive impairment175 and neurofibromin-mediated cAMP production176.

The identification of key regulatory neurofibromin partners may have important clinical implications, in a strategy aimed at blocking its inactivation and/or upregulating the protein. These benefits may extend beyond therapies relevant to neurofibromin to serve as a potential clinical strategy to attenuate the Ras pathway in tumors harboring variants in genes that function upstream of Ras. Inhibitors and potential targets are shown in red.
MPNST therapies include nivolumab and ipilimumab (Phase II clinical trial)177, inhibition of BRD4 with CPI-0610 (Phase II clinical trial) triggering apoptosis through Bim178, and a combination of a BET, MEK, and PD-L1 receptor inhibitors166. Recently, a NF1 patient treated with tofacitinib, a selective JAK1/JAK3 inhibitor that regulated neurofibromas growth179, as well as their progression to MPNST, has been described159. On the other hand, the combination of everolimus and bevacizumab did not achieve favorable results in studies in patients with MPNST180. Likewise, neither targeting VEFG-A using ranibizumab, nor targeting VEFGR with sorafenib or imatinib, were effective for cutaneous neurofibromas or MPNST respectively181. EGFR is abundantly expressed in neurofibroma and MPNST cell lines, although unfortunately the EGFR inhibitor erlotinib failed to inhibit tumor growth in Phase 2 trials of patients with advanced-stage MPNST166. Forskolin and rolipram have also been used for impaired cAMP production, in particular, rolipram inhibited optic glioma growth and tumor size162,182. Everolimus is well suited for future consideration as initial therapy in patients with low-grade pediatric glioma183. Immune checkpoint inhibitors/therapies include targeting PD-1 and PD-L1184, although immunotherapy with monoclonal antibodies remain to be identified in NF1 preclinical models185. The proteasomal regulation of neurofibromin represents an important mechanism of controlling both the amplitude and duration of Ras-mediated signaling, which may be exploited therapeutically by promoting degradation of neurofibromin146,186,187.
Preclinical trials suggest that dopamine-target therapies, such as methylphenidate, a stimulant medication that increases dopaminergic and noradrenergic neurotransmission, may be useful treatments for children with NF1-associated cognitive abnormalities. However, the therapeutic effect on cognitive performance is unclear188. Nevertheless, targeting VEGF-A suggested a qualitative improvement in vision after bevacizumab-based treatment in children with OPGs189.
When expression of NF1 is inhibited, the resulting ER+ breast cancer cells were stimulated by tamoxifen (a drug commonly used to prevent relapses from ER+ breast cancer) instead of inhibited, and these cells became sensitive to a very low concentration of estradiol151. Breast cancer patients with NF1 sporadic mutations treated with the estrogen-receptor antagonist fulvestrant showed a good outcome190. Combination with CDK4/6 inhibitors, which target ER-independent cyclin D1 transcription, results in substantially enhanced efficacy of endocrine therapy in vitro190. These findings suggest that the prognosis of patients with pretreatment detection of NF1 mutation in the PALOMA- phase III trial191 could overcome the risk of early relapse via the investigation in an adjuvant setting in NF1-mutant cancers of combined fulvestrant and palbociclib192.
Inhibitors for the major signaling pathways related to neurofibromin have also been studied. Ras inhibitors include tipifarnib and pirfenidone, although they did not significantly prolong the time to progression compared with placebo in children and young adults with NF1 and progressive plexiform neurofibromas193. Inhibitors for the Raf/MEK/ERK pathway include the MEK inhibitor trametinib194 and selumetinib, which resulted in shrinkage in neurofibromin-related gliomas in the optic pathway195, becoming the first FDA-approved treatment for inoperable plexiform neurofibroma196. The mTOR pathway has been investigated as a potential therapeutic target180, including the inhibitor sirolimus (rapamycin) which did not provide any favorable results of shrinkage of the plexiform neurofibroma, although it could delay their growth in selected patients197.
Conclusions
Neurofibromin is ubiquitously expressed with enrichment in neurons, Schwann cells, oligodendrocytes, astrocytes, leukocytes and adrenal medulla, and it is highly conserved among species. Due to its high degree of conservation, several animal models can be used to identify potential effectors, partners, and promising therapeutic targets to complete the functional characterization of this protein. Neurofibromin is known to associate with a large number of proteins, including transmembrane receptors, soluble effectors, proteins at the cell surface, the cytoskeleton and the nucleus. Although the biological significance of these protein–protein interactions is largely unknown, there is profound evidence on neurofibromin roles in actin cytoskeleton remodeling, cell motility, cell adhesion, proliferation, differentiation, apoptosis, stress responses, learning and memory. Variants throughout the protein affecting different domains that interact with different binding partners may be associated with the vast array of clinical manifestations. Some neurofibromin effectors have been verified, whereas many reported interactions remain unsubstantiated and may be irrelevant. The diversity of protein associations does however emphasize the point that neurofibromin is likely to act through the canonical and non-canonical effector pathways downstream of Ras activation. Several groups have reviewed neurofibromin protein structure, putative interacting partners and therapeutic strategies42,198,199,200,201, but to date, a high-quality NF1 interactome has not been described yet. The fact that neurofibromin has been related to a variety of membrane receptors and that binding partners may be cell type-specific makes elucidating its additional binding partners and functions even more intriguing. Further studies on the regulation of neurofibromin in various model organisms and cell types are needed in order to identify the role of neurofibromin under pathological conditions.
References
Bergqvist, C. et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 15, 1–23 (2020).
Martin, G. A. et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63, 843–849 (1990).
Harrisingh, M. C. & Lloyd, A. C. Ras/Raf/ERK signalling and NF1: Implications for neurofibroma formation. Cell Cycle 3, 1255–1258 (2004).
Johannessen, C. M. et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl Acad. Sci. USA 102, 8573–8578 (2005).
Vallée, B. et al. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between ras and rho pathways. PLoS ONE 7, e47283 (2012).
Lin, Y. L., Lei, Y. T., Hong, C. J. & Hsueh, Y. P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 177, 829–841 (2007).
Tong, J., Hannan, F., Zhu, Y., Bernards, A. & Zhong, Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat. Neurosci. 5, 95–96 (2002).
Yap, Y. S. et al. The NF1 gene revisited -from bench to bedside. Oncotarget 5, 5873–5892 (2014).
Rauen, K. A. Defining RASopathy. DMM Dis. Model. Mech. 15, dmm049344 (2022).
Nishi, T. et al. Differential expression of two types of the neurofibromatosis type 1 (NF1) gene transcripts related to neuronal differentiation. Oncogene 6, 1555–1559 (1991).
Andersen, L. B. et al. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell. Biol. 13, 487–495 (1993).
Pemov, A. et al. Genetic modifiers of neurofibromatosis Type 1-associated Café-au-Lait macule count identified using multi-platform analysis. PLoS Genet. 10, e1004575 (2014).
Bernier, A., Larbrisseau, A. & Perreault, S. Café-au-lait macules and neurofibromatosis type 1: a review of the literature. Pediatr. Neurol. 60, 24–29.e1 (2016).
Gutmann, D. H. et al. Neurofibromatosis type 1. Nat. Rev. Prim. 3, 1–17 (2017).
Haddar, S. et al. Lisch nodules in neurofibromatosis type 1. J. Fr. Ophtalmol. 43, 559–560 (2020).
Campen, C. J. & Gutmann, D. H. Optic pathway gliomas in neurofibromatosis type 1. J. Child Neurol. 33, 73–81 (2018).
Gambarotti, M. Neurofibromas. in Diagnosis of Musculoskeletal Tumors and Tumor-like Conditions: Clinical, Radiological and Histological Correlations - the Rizzoli Case Archive 145–147 (StatPearls Publishing). https://doi.org/10.1007/978-3-030-29676-6_31.(2019).
Biotteau, M. et al. Sporadic and familial variants in NF1: an explanation of the wide variability in neurocognitive phenotype? Front. Neurol. 11, 368 (2020).
Hölzel, M. et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 142, 218–229 (2010).
Mayerhofer, C., Niemeyer, C. M. & Flotho, C. Current treatment of juvenile myelomonocytic leukemia. J. Clin. Med. 10, 3084 (2021).
Broadfoot, B. G. & Kumarapeli, A. R. Pheochromocytoma multisystem crisis and masquerading disseminated histoplasmosis in a neurofibromatosis type 1 patient with bilateral adrenal tumors. Acad. Forensic Pathol. 10, 62–68 (2020).
Anastasaki, C. & Gutmann, D. H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 23, 6712–6721 (2014).
Henkemeyer, M. et al. Vascular system defects and neuronal apoptosis in mice lacking Ras GTPase-activating protein. Nature 377, 695–701 (1995).
Philpott, C., Tovell, H., Frayling, I. M., Cooper, D. N. & Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genomics 11, 1–19 (2017).
Lobbous, M. et al. An update on neurofibromatosis type 1-associated gliomas. Cancers 12, 114 (2020).
Kiuru, M. & Busam, K. J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 97, 146–157 (2017).
Qiao, G., Jia, X., Zhang, Y. & Chen, B. Neurofibromin 1 expression is negatively correlated with malignancy and prognosis of epithelial ovarian cancer. Int. J. Clin. Exp. Pathol. 12, 1702–1712 (2019).
Tlemsani, C. et al. NF1 mutations identify molecular and clinical subtypes of lung adenocarcinomas. Cancer Med. 8, 4330–4337 (2019).
Farshidfar, F. et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 18, 2780–2794 (2017).
Suarez-Kelly, L. P. et al. Increased breast cancer risk in women with neurofibromatosis type 1: a meta-analysis and systematic review of the literature. Hered. Cancer Clin. Pract. 17, 1–13 (2019).
Parkin, B. et al. NF1 inactivation in adult acute myelogenous leukemia. Clin. Cancer Res. 16, 4135–4147 (2010).
Landry, J. P. et al. Comparison of cancer prevalence in patients with neurofibromatosis type 1 at an academic cancer center vs in the general population from 1985 to 2020. JAMA Netw. Open 4, e210945 (2021).
Venturin, M. et al. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J. Med. Genet. 41, 35–41 (2004).
Upadhyaya, M. et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 80, 140–151 (2007).
Pinna, V. et al. P.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 23, 1068–1071 (2015).
Koczkowska, M. et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 Codons 844–848. Am. J. Hum. Genet. 102, 69–87 (2018).
Koczkowska, M. et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affectingp.Met1149, p.Arg1276, andp.Lys1423: genotype–phenotype study in neurofibromatosis type 1. Hum. Mutat. 41, 299–315 (2020).
Peltonen, S., Kallionpää, R. A. & Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 26, 645–648 (2017).
Campos, B. et al. Germline mutations in NF1 and BRCA1 in a family with neurofibromatosis type 1 and early-onset breast cancer. Breast Cancer Res. Treat. 139, 597–602 (2013).
Canson, D. M., Mara, T. A. O., Spurdle, A. B. & Glubb, D. M. Splicing annotation of endometrial cancer GWAS risk loci reveals potentially causal variants and supports a role for NF1 and SKAP1 as susceptibility genes. Hum. Genet. Genomics Adv. 4, 100185 (2023).
Lois, S., Báez-Flores, J., Isidoro-García, M., Lacal, J. & Triviño, J. C. Identification of germinal neurofibromin hotspots. Biomedicines 10, 2044 (2022).
Walker, J. A. & Upadhyaya, M. Emerging therapeutic targets for neurofibromatosis type 1. Expert Opin. Ther. Targets 22, 419–437 (2018).
Scheffzek, K. et al. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 17, 4313–4327 (1998).
D’Angelo, I., Welti, S., Bonneau, F. & Scheffzek, K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 7, 174–179 (2006).
Sherekar, M. et al. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 295, 1105–1119 (2020).
Naschberger, A., Baradaran, R., Rupp, B. & Carroni, M. The structure of neurofibromin isoform 2 reveals different functional states. Nature 599, 315–319 (2021).
Lupton, C. J. et al. The cryo-EM structure of the human neurofibromin dimer reveals the molecular basis for neurofibromatosis type 1. Nat. Struct. Mol. Biol. 28, 982–988 (2021).
Chaker-Margot, M. et al. Structural basis of activation of the tumor suppressor protein neurofibromin. Mol. Cell 82, 1288–1296.e5 (2022).
Prasad, B. C. M., Chandra, V. V. R., Sudarsan, A., Kumar, P. S. & Sarma, P. V. G. K. Clinical characteristics and NF1 gene mutation analysis of three successive generations in three different Indian families with neurofibromatosis type 1 and peripheral nerve sheath tumours. J. Clin. Neurosci. 53, 62–68 (2018).
Mangoura, D. et al. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 25, 735–745 (2006).
Karouzaki, S., Peta, C., Tsirimonaki, E. & Mangoura, D. PKCε-dependent H-Ras activation encompasses the recruitment of the RasGEF SOS1 and of the RasGAP neurofibromin in the lipid rafts of embryonic neurons. Neurochem. Int. 131, 104582 (2019).
Xu, M. et al. Identification of mutation regions on NF1 responsible for high- and low-risk development of optic pathway glioma in neurofibromatosis type I. Front. Genet. 9, 270 (2018).
Bausch, B. et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 92, 2784–2792 (2007).
Tokuo, H. et al. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of NG,NG-dimethylarginine dimethylaminohydrolase. FEBS Lett. 494, 48–53 (2001).
Feng, L. et al. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 557, 275–282 (2004).
Zhang, P., Hu, X., Xu, X., Chen, Y. & Bache, R. J. Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and Akt. Arterioscler. Thromb. Vasc. Biol. 31, 890–897 (2011).
Gregory, P. E. et al. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat. Cell Mol. Genet. 19, 265–274 (1993).
Dehmelt, L. & Halpain, S. The MAP2/Tau family of microtubule-associated proteins Gene organization and evolutionary history. Genome Biol. 6, 204 (2004).
Cichowski, K., Santiago, S., Jardim, M., Johnson, B. W. & Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 17, 449–454 (2003).
Koliou, X., Fedonidis, C., Kalpachidou, T. & Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 136, 78–91 (2016).
Arun, V., Wiley, J. C., Kaur, H., Kaplan, D. R. & Guha, A. A novel neurofibromin (NF1) interaction with the leucine-rich pentatricopeptide repeat motif-containing protein links neurofibromatosis type 1 and the french canadian variant of Leigh’s syndrome in a common molecular complex. J. Neurosci. Res. 91, 494–505 (2013).
Cui, J., Wang, L., Ren, X., Zhang, Y. & Zhang, H. LRPPRC: a multifunctional protein involved in energy metabolism and human disease. Front. Physiol. 10, 595 (2019).
Dunzendorfer-Matt, T., Mercado, E. L., Maly, K., McCormick, F. & Scheffzek, K. The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation. Proc. Natl Acad. Sci. USA 113, 7497–7502 (2016).
Anastasaki, C., Orozco, P. & Gutmann, D. H. RAS and beyond: the many faces of the neurofibromatosis type 1 protein. Dis. Model. Mech. 15, dmm049362 (2022).
Weber, S. M. et al. R-Ras subfamily proteins elicit distinct physiologic effects and phosphoproteome alterations in neurofibromin-null MPNST cells. Cell Commun. Signal. 19, 1–21 (2021).
Hiatt, K. K., Ingram, D. A., Zhang, Y., Bollag, G. & Clapp, D. W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1-/- cells. J. Biol. Chem. 276, 7240–7245 (2001).
Ismat, F. A., Xu, J., Min, M. L. & Epstein, J. A. The neurofibromin GAP-related domain rescues endothelial but not neural crest development in Nf1-/- mice. J. Clin. Invest. 116, 2378–2384 (2006).
Ozawa, T. et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 280, 39524–39533 (2005).
Stowe, I. B. et al. A shared molecular mechanism underlies the human rasopathies legius syndrome and neurofibromatosis-1. Genes Dev. 26, 1421–1426 (2012).
Thomas, L. et al. Assessment of the potential pathogenicity of missense mutations identified in the GTPase-activating protein (GAP)-related domain of the neurofibromatosis type-1 (NF1) gene. Hum. Mutat. 33, 1687–1696 (2012).
Yan, W. et al. Structural insights into the SPRED1-neurofibromin-KRAS complex and disruption of SPRED1-neurofibromin interaction by oncogenic EGFR. Cell Rep. 32, 107909 (2020).
Phan, V. T. et al. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol. Cell. Biol. 30, 2264–2279 (2010).
De Schepper, S. et al. Neurofibromatosis type 1 protein and amyloid precursor protein interact in normal human melanocytes and colocalize with melanosomes. J. Invest. Dermatol. 126, 653–659 (2006).
Hakimi, M. A., Speicher, D. W. & Shiekhattar, R. The motor protein kinesin-1 links neurofibromin and merlin in a common cellular pathway of neurofibromatosis. J. Biol. Chem. 277, 36909–36912 (2002).
Kamal, A., Stokin, G. B., Yang, Z., Xia, C. H. & Goldstein, L. S. B. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28, 449–459 (2000).
Hsueh, Y. P., Roberts, A. M., Volta, M., Sheng, M. & Roberts, R. G. Bipartite interaction between neurofibromatosis type I protein (Neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J. Neurosci. 21, 3764–3770 (2001).
Afratis, N. A. et al. Syndecans – key regulators of cell signaling and biological functions. FEBS J. 284, 27–41 (2017).
Fadhlullah, S. F. B. et al. Pathogenic mutations in neurofibromin identifies a leucine-rich domain regulating glioma cell invasiveness. Oncogene 38, 5367–5380 (2019).
Wang, H. F. et al. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Invest. 121, 4820–4837 (2011).
Boyanapalli, M. et al. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem. Biophys. Res. Commun. 340, 1200–1208 (2006).
Bergoug, M. et al. Neurofibromin structure, functions and regulation. Cells 9, 2365 (2020).
Nadim, W. D. et al. Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl Acad. Sci. USA 113, 12310–12315 (2016).
Bergoug, M. et al. Noncanonical structural requirements of neurofibromin SUMOylation reveal a folding-deficiency of several pathogenic mutants. bioRxiv https://doi.org/10.1101/2021.12.09.471973 (2021).
Ko, J. M., Sohn, Y. B., Jeong, S. Y., Kim, H. J. & Messiaen, L. M. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis Type 1. Pediatr. Neurol. 48, 447–453 (2013).
Melloni, G. et al. Risk of optic pathway glioma in neurofibromatosis type 1: No evidence of genotype–phenotype correlations in a large independent cohort. Cancers (Basel). 11, 1838 (2019).
Vandenbroucke, I., Van Oostveldt, P., Coene, E., De Paepe, A. & Messiaen, L. Neurofibromin is actively transported to the nucleus. FEBS Lett. 560, 98–102 (2004).
Bianchessi, D. et al. 126 novel mutations in italian patients with neurofibromatosis type 1. Mol. Genet. Genom. Med. 3, 513–525 (2015).
Hannan, F. et al. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum. Mol. Genet. 15, 1087–1098 (2006).
Hinman, M. N., Sharma, A., Luo, G. & Lou, H. Neurofibromatosis type 1 alternative splicing is a key regulator of ras signaling in neurons. Mol. Cell. Biol. 34, 2188–2197 (2014).
Kweh, F. et al. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 48, 1005–1017 (2009).
Peta, C. et al. Two tails for neurofibromin: a tale of two microtubule-associated proteins. in Clinical and Basic Aspects of Neurofibromatosis Type 1 (IntechOpen). https://doi.org/10.5772/intechopen.97574 (2022).
Lin, Y. L. & Hsueh, Y. P. Neurofibromin interacts with CRMP-2 and CRMP-4 in rat brain. Biochem. Biophys. Res. Commun. 369, 747–752 (2008).
Patrakitkomjorn, S. et al. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J. Biol. Chem. 283, 9399–9413 (2008).
Birnbaum, R. A. et al. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol. Cell 5, 189–195 (2000).
Qin, A., Musket, A., Musich, P. R., Schweitzer, J. B. & Xie, Q. Receptor tyrosine kinases as druggable targets in glioblastoma: Do signaling pathways matter? Neuro-Oncol. Adv. 3, 1–12 (2021).
Gouzi, J. Y. et al. The receptor tyrosine kinase alk controls neurofibromin functions in drosophila growth and learning. PLoS Genet. 7, 1002281 (2011).
Kawachi, Y., Xu, X., Ichikawa, E., Imakado, S. & Otsuka, F. Expression of angiogenic factors in neurofibromas. Exp. Dermatol. 12, 412–417 (2003).
DeClue, J. E. et al. Epidermal growth factor receptor expression in neurofibromatosis type 1- related tumors and NF1 animal models. J. Clin. Invest. 105, 1233–1241 (2000).
Ki, D. H., He, S., Rodig, S. & Look, A. T. Overexpression of PDGFRA cooperates with loss of NF1 and p53 to accelerate the molecular pathogenesis of malignant peripheral nerve sheath tumors. Oncogene 36, 1058–1068 (2017).
Larribère, L. & Utikal, J. Nf1-dependent transcriptome regulation in the melanocyte lineage and in melanoma. J. Clin. Med. 10, 3350 (2021).
Kamal, A., Almenar-Queralt, A., LeBlanc, J. F., Roberts, E. A. & Goldstein, L. S. B. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414, 643–648 (2001).
Peacock, J. D. et al. Genomic status of MET potentiates sensitivity to MET and MEK inhibition in NF1-related malignant peripheral nerve sheath tumors. Cancer Res. 78, 3672–3687 (2018).
Rao, U. N. M., Sonmez-Alpan, E. & Michalopoulos, G. K. Hepatocyte growth factor and c-MET in benign and malignant peripheral nerve sheath tumors. Hum. Pathol. 28, 1066–1070 (1997).
McGillicuddy, L. T. et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 16, 44–54 (2009).
Zhou, X. et al. Regulation of the viability of Nf1 deficient cells by PKC isoforms. Oncotarget 5, 10709–10717 (2014).
Xie, K. et al. NF1 is a direct G protein effector essential for opioid signaling to Ras in the striatum. Curr. Biol. 26, 2992–3003 (2016).
Zhang, P. et al. DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PLoS One 8, 1–9 (2013).
Xu, G. et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62, 599–608 (1990).
Karnoub, A. E. & Weinberg, R. A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 (2008).
Oliveira, A. F. & Yasuda, R. Neurofibromin is the major ras inactivator in dendritic spines. J. Neurosci. 34, 776–783 (2014).
Molosh, A. I. et al. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat. Neurosci. 17, 1583–1590 (2014).
Roberts, P. J. & Der, C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 (2007).
Steelman, L. S. et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mtor pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 3, 192–222 (2011).
Zhang, Y. Y. et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J. Exp. Med. 187, 1893–1902 (1998).
Dasgupta, B., Yi, Y., Chen, D. Y., Weber, J. D. & Gutmann, D. H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 65, 2755–2760 (2005).
Stornetta, R. L. & Zhu, J. J. Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist 17, 54–78 (2011).
Dibble, C. C. & Cantley, L. C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 (2015).
Li, X. et al. Clustered, regularly interspaced short palindromic repeats (CRISPR)/Cas9-coupled affinity purification/mass spectrometry analysis revealed a novel role of neurofibromin in mTOR signaling. Mol. Cell. Proteom. 16, 594–607 (2017).
Richardson, D. S., Spehar, J. M., Han, D. T., Chakravarthy, P. A. & Sizemore, S. T. The RAL Enigma: Distinct Roles of RALA and RALB in Cancer. Cells 11, 1645 (2022).
Arima, Y. et al. Decreased expression of neurofibromin contributes to epithelial-mesenchymal transition in neurofibromatosis type 1. Exp. Dermatol. 19, e136–e141 (2010).
Miller, S. J. et al. Large-scale molecular comparison of human Schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 66, 2584–2591 (2006).
Drees, F. & Gertler, F. B. Ena/VASP: proteins at the tip of the nervous system. Curr. Opin. Neurobiol. 18, 53–59 (2008).
Hu, H. T., Shih, P. Y., Shih, Y. T. & Hsueh, Y. P. The involvement of neuron-specific factors in dendritic spinogenesis: Molecular regulation and association with neurological disorders. Neural Plast. 2016, 5136286 (2016).
Hu, P. et al. Fak silencing impairs osteogenic differentiation of bone mesenchymal stem cells induced by uniaxial mechanical stretch. J. Dent. Sci. 14, 225–233 (2019).
Biederer, T. & Südhof, T. C. CASK and protein 4.1 support F-actin nucleation on neurexins. J. Biol. Chem. 276, 47869–47876 (2001).
Hsueh, Y. P. et al. Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J. Cell Biol. 142, 139–151 (1998).
Zhang, N., Parr, C. J. C., Birch, A. M., Goldfinger, M. H. & Sastre, M. The amyloid precursor protein binds to β-catenin and modulates its cellular distribution. Neurosci. Lett. 685, 190–195 (2018).
Asiedu, M. K. et al. Pathways impacted by genomic alterations in pulmonary carcinoid tumors. Clin. Cancer Res. 24, 1691–1704 (2018).
Chung, C. Y., Lee, S., Briscoe, C., Ellsworth, C. & Firtel, R. A. Role of Rac in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc. Natl Acad. Sci. USA 97, 5225–5230 (2000).
Brown, J. A., Diggs-Andrews, K. A., Gianino, S. M. & Gutmann, D. H. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell. Neurosci. 49, 13–22 (2012).
Kim, H. A., Ratner, N., Roberts, T. M. & Stiles, C. D. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J. Neurosci. 21, 1110–1116 (2001).
Xu, F. et al. cAMP/PKA signaling pathway induces apoptosis by inhibited NF-κB in aluminum chloride-treated lymphocytes in vitro. Biol. Trace Elem. Res. 170, 424–431 (2016).
Yu, F. X. et al. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 27, 1223–1232 (2013).
Guo, H. F., The, I., Hannan, F., Bernards, A. & Zhong, Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276, 795–798 (1997).
Sutton, L. P. et al. NF1-cAMP signaling dissociates cell type–specific contributions of striatal medium spiny neurons to reward valuation and motor control. PLoS Biol. 17, e3000477 (2019).
Ravnskjaer, K., Madiraju, A. & Montminy, M. Role of the cAMP pathway in glucose and lipid metabolism. Handb. Exp. Pharmacol. 233, 29–49 (2015).
Ratner, N. & Miller, S. J. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 15, 290–301 (2015).
Allouche, J. et al. In vitro modeling of hyperpigmentation associated to neurofibromatosis type 1 using melanocytes derived from human embryonic stem cells. Proc. Natl Acad. Sci. USA 112, 9034–9039 (2015).
Tan, X. et al. The CREB-miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PLoS One 7, 31071203 (2012).
Varrone, F. & Caputo, E. The miRNAs role in melanoma and in its resistance to therapy. Int. J. Mol. Sci. 21, 878 (2020).
Stark, M. S. et al. The prognostic and predictive value of melanoma-related MicroRNAs using tissue and serum: a MicroRNA expression analysis. EBioMedicine 2, 671–680 (2015).
Cardoso, J. C. R., Garcia, M. G. & Power, D. M. Corrigendum: tracing the origins of the pituitary adenylate-cyclase activating polypeptide (PACAP). Front. Neurosci. 14, 366 (2020).
Moroo, I. et al. Pituitary adenylate cyclase activating polypeptide (PACAP) stimulates mitogen-activated protein kinase (MAPK) in cultured rat astrocytes. Brain Res. 795, 191–196 (1998).
Monaghan, T. K., MacKenzie, C. J., Plevin, R. & Lutz, E. M. PACAP-38 induces neuronal differentiation of human SH-SY5Y neuroblastoma cells via cAMP-mediated activation of ERK and p38 MAP kinases. J. Neurochem. 104, 74–88 (2008).
Georg, B., Falktoft, B. & Fahrenkrug, J. PKA, novel PKC isoforms, and ERK is mediating PACAP auto-regulation via PAC1R in human neuroblastoma NB-1 cells. Neuropeptides 60, 83–89 (2016).
Tan, M. et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev. Cell 21, 1062–1076 (2011).
Hollstein, P. E. & Cichowski, K. Identifying the ubiquitin ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 3, 880–893 (2013).
Green, Y. S. et al. Hypoxia-associated factor (HAF) mediates neurofibromin ubiquitination and degradation leading to Ras–ERK pathway activation in hypoxia. Mol. Cancer Res. 17, 1220–1232 (2019).
Dischinger, P. S. et al. NF1 deficiency correlates with estrogen receptor signaling and diminished survival in breast cancer. npj Breast Cancer 4, 29 (2018).
Bertucci, F. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
Zheng, Z. Y. et al. Neurofibromin is an estrogen receptor-α transcriptional co-repressor in breast cancer. Cancer Cell 37, 387–402.e7 (2020).
Feltri, M. L. & Poitelon, Y. HIPPO stampede in nerve sheath tumors. Cancer Cell 33, 160–161 (2018).
Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 17, 1–17 (2019).
Faden, D. L., Asthana, S., Tihan, T., De Risi, J. & Kliot, M. Whole exome sequencing of growing and non-growing cutaneous neurofibromas from a single patient with neurofibromatosis type 1. PLoS One 12, e0170348 (2017).
Chen, Z. et al. Spatiotemporal loss of NF1 in schwann cell lineage leads to different types of cutaneous neurofibroma susceptible to modification by the hippo pathway. Cancer Discov. 9, 114–129 (2019).
Wu, L. M. N. et al. Programming of schwann cells by lats1/2-TAZ/YAP signaling drives malignant peripheral nerve sheath tumorigenesis. Cancer Cell 33, 292–308.e7 (2018).
He, L. et al. Yap and taz are transcriptional co-activators of ap-1 proteins and stat3 during breast cellular transformation. Elife 10, 1–26 (2021).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 14, 736–746 (2014).
Wu, J. et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 33, 173–180 (2014).
Levine, R. L., Pardanani, A., Tefferi, A. & Gilliland, D. G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 7, 673–683 (2007).
Costa, R. M. et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530 (2002).
Brown, J. A., Gianino, S. M. & Gutmann, D. H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 30, 5579–5589 (2010).
Diggs-Andrews, K. A. et al. Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann. Neurol. 73, 309–315 (2013).
Anastasaki, C., Woo, A. S., Messiaen, L. M. & Gutmann, D. H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 24, 3518–3528 (2015).
Hebron, K. E., Hernandez, E. R. & Yohe, M. E. The RASopathies: from pathogenetics to therapeutics. DMM Dis. Models Mechanisms 15, dmm049107 (2022).
Brosseau, J. P., Liao, C. P. & Le, L. Q. Translating current basic research into future therapies for neurofibromatosis type 1. Br. J. Cancer 123, 178–186 (2020).
Rabab’h, O., Gharaibeh, A., Al-Ramadan, A., Ismail, M. & Shah, J. Pharmacological approaches in neurofibromatosis type 1-associated nervous system tumors. Cancers (Basel). 13, 1–18 (2021).
Wu, L. M. N. & Lu, Q. R. Therapeutic targets for malignant peripheral nerve sheath tumors. Future Neurol. 14,1 (2019).
Lopez, G. et al. Autophagic survival in resistance to histone deacetylase inhibitors: Novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 71, 185–196 (2011).
Neel, N. F. et al. The RalGEF-ral effector signaling network: the road less traveled for anti-ras drug discovery. Genes Cancer 2, 275–287 (2011).
Moutal, A. et al. Dissecting the role of the CRMP2-neurofibromin complex on pain behaviors. Pain 158, 2203–2221 (2017).
Weiss, J. B., Weber, S., Marzulla, T. & Raber, J. Pharmacological inhibition of anaplastic lymphoma kinase rescues spatial memory impairments in neurofibromatosis 1 mutant mice. Behav. Brain Res. 332, 337–342 (2017).
Fletcher, J. S. et al. STAT3 inhibition reduces macrophage number and tumor growth in neurofibroma. Oncogene 38, 2876–2884 (2019).
Costantini, S., Capone, F., Polo, A., Bagnara, P. & Budillon, A. Valosin-containing protein (VCP)/p97: a prognostic biomarker and therapeutic target in cancer. Int. J. Mol. Sci. 22, 10177 (2021).
Rosse, G. & Schaffhauser, H. 5-HT6 receptor antagonists as potential therapeutics for cognitive impairment. Curr. Top. Med. Chem. 10, 207–221 (2010).
Dasgupta, B., Dugan, L. L. & Gutmann, D. H. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J. Neurosci. 23, 8949–8954 (2003).
Martin, E., Flucke, U. E., Coert, J. H. & van Noesel, M. M. Treatment of malignant peripheral nerve sheath tumors in pediatric NF1 disease. Child’s Nerv. Syst. 36, 2453–2462 (2020).
Cooper, J. M. et al. Overcoming BET inhibitor resistance in malignant peripheral nerve sheath tumors. Clin. Cancer Res. 25, 3404–3416 (2019).
Rischin, A., De Silva, T. & Le Marshall, K. Reversible eruption of neurofibromatosis associated with tofacitinib therapy for rheumatoid arthritis. Rheumatol. 58, 1109–1111 (2019).
Widemann, B. C. et al. Targeting sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST) in a phase II study of everolimus in combination with bevacizumab (SARC016). Sarcoma 2019, 7656747 (2019).
Maki, R. G. et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 27, 3133–3140 (2009).
Costa, A. D. A. & Gutmann, D. H. Brain tumors in neurofibromatosis type 1. Neuro-Oncol. Adv. 2, I85–I97 (2020).
Ullrich, N. J. et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: a Neurofibromatosis Clinical Trials Consortium study. Neuro. Oncol. 22, 1527–1535 (2020).
Farschtschi, S. et al. Upregulated immuno-modulator PD-L1 in malignant peripheral nerve sheath tumors provides a potential biomarker and a therapeutic target. Cancer Immunol. Immunother. 69, 1307–1313 (2020).
Karmakar, S. & Reilly, K. M. The role of the immune system in neurofibromatosis type 1-associated nervous system tumors. CNS Oncol. 6, 45–60 (2017).
Swaroop, M. et al. Yeast homolog of human SAG/ROC2/Rbx2/Hrt2 is essential for cell growth, but not for germination: Chip profiling implicates its role in cell cycle regulation. Oncogene 19, 2855–2866 (2000).
Long, A. et al. Analysis of patient-specific NF1 variants leads to functional insights for Ras signaling that can impact personalized medicine. Hum. Mutat. 43, 30–41 (2022).
Pride, N. A. et al. Effects of methylphenidate on cognition and behaviour in children with neurofibromatosis type 1: A study protocol for a randomised placebo-controlled crossover trial. BMJ Open 8, 21800 (2018).
Avery, R. A., Hwang, E. I., Jakacki, R. I. & Packer, R. J. Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab. JAMA Ophthalmol. 132, 111–114 (2014).
Pearson, A. et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin. Cancer Res. 26, 608–622 (2020).
Turner, N. C. et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 373, 209–219 (2015).
Razavi, P. et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438.e6 (2018).
Widemann, B. C. et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro. Oncol. 16, 707–718 (2014).
Vaassen, P., Dürr, N., Röhrig, A., Willing, R. & Rosenbaum, T. Trametinib induces neurofibroma shrinkage and enables surgery. Neuropediatrics 50, 300–303 (2019).
Fangusaro, J. et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 20, 1011–1022 (2019).
Gross, A. M. et al. Selumetinib in children with inoperable plexiform neurofibromas. N. Engl. J. Med. 382, 1430–1442 (2020).
Weiss, B. et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: an NF clinical trials consortium phase II study. Pediatr. Blood Cancer 61, 982–986 (2014).
Sharafi, P. & Ayter, S. Possible modifier genes in the variation of neurofibromatosis type 1 clinical phenotypes. J. Neurogenet. 32, 65–77 (2018).
Rauen, K. A. et al. Recent developments in neurofibromatoses and RASopathies: Management, diagnosis and current and future therapeutic avenues. Am. J. Med. Genet. Part A 167, 1–10 (2015).
Carnes, R. M., Kesterson, R. A., Korf, B. R., Mobley, J. A. & Wallis, D. Affinity purification of NF1 protein–protein interactors identifies keratins and neurofibromin itself as binding partners. Genes (Basel). 10, 650 (2019).
Leier, A. et al. Mutation-directed therapeutics for neurofibromatosis type I. Mol. Ther. Nucleic Acids 20, 739–753 (2020).
Author information
Authors and Affiliations
Contributions
J.B.-F., M.R.-M., and J.L. all contributed to the conceptualization of the work, and the literature review. J.B.-F. and M.R.-M. were responsible for the initial drafting of the manuscript, with all authors contributing to subsequent revisions. J.B.-F. was responsible for creating Figs. 4 and 5, while M.R.-M. created Figs. 1–3 as well as Supplementary Tables 1 and 2. J.L. provided critical feedback throughout the writing process and supervised the manuscript as the principal investigator and corresponding author. All authors approved the final version of the manuscript for submission and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Karli Montague-Cardoso.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Báez-Flores, J., Rodríguez-Martín, M. & Lacal, J. The therapeutic potential of neurofibromin signaling pathways and binding partners. Commun Biol 6, 436 (2023). https://doi.org/10.1038/s42003-023-04815-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-04815-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
